
An LC-MS/MS Method for the Simultaneous Quantification of Insulin, Cortisol, Glucagon-like Peptide 1, Ghrelin, and Osteocalcin
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. LC-MS Analysis
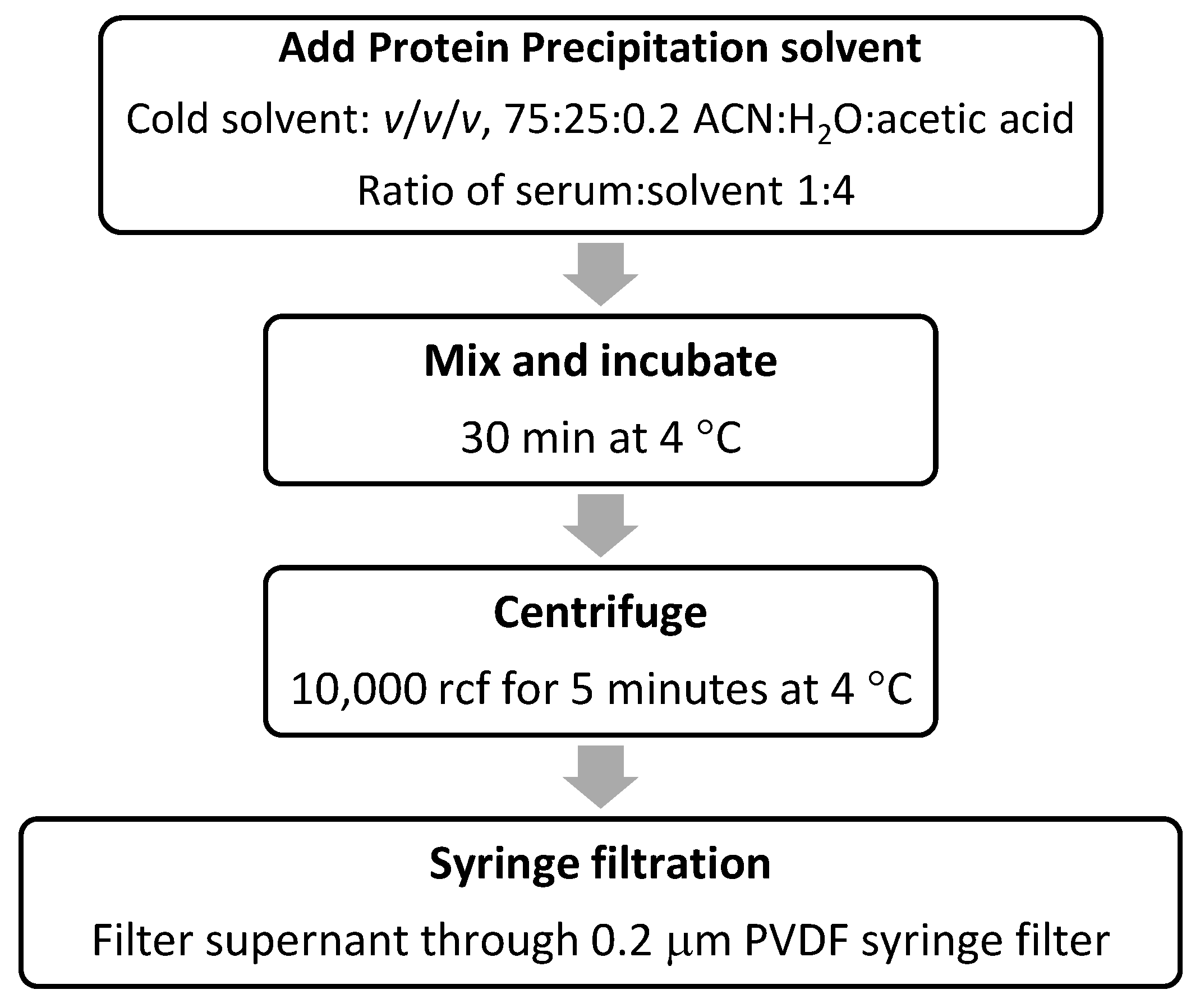
2.3. Optimization of Hormone Extraction from Serum
2.4. Calibration Curves
2.5. Quality Control (QC) Samples
2.6. Validation of the Method
2.7. Data Analysis
3. Results
3.1. Optimization of LC-MS/MS Parameters
3.2. Matrix Considerations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hiller-Sturmhofel, S.; Bartke, A. The endocrine system: An overview. Alcohol Health Res. World 1998, 22, 153–164. [Google Scholar]
- Kulkarni, J.; Gavrilidis, E.; Worsley, R. Chapter 27—Hormones and Schizophrenia. In Handbook of Behavioral Neuroscience; Mikhail, L.W., Pletnikov, V., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 23, pp. 463–480. [Google Scholar]
- Hughes, N.C.; Bajaj, N.; Fan, J.; Wong, E.Y. Assessing the matrix effects of hemolyzed samples in bioanalysis. Bioanalysis 2009, 1, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Foreman, R.E.; George, A.L.; Reimann, F.; Gribble, F.M.; Kay, R.G. Peptidomics: A Review of Clinical Applications and Methodologies. J. Proteome Res. 2021, 20, 3782–3797. [Google Scholar] [CrossRef]
- Chen, Z.; Caulfield, M.P.; McPhaul, M.J.; E Reitz, R.; Taylor, S.W.; Clarke, N.J. Quantitative insulin analysis using liquid chromatography-tandem mass spectrometry in a high-throughput clinical laboratory. Clin. Chem. 2013, 59, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Lapko, V.N.; Miller, P.S.; Brown, G.P.; Islam, R.; Peters, S.K.; Sukovaty, R.L.; Ruhn, P.F.; Kafonek, C.J. Sensitive glucagon quantification by immunochemical and LC–MS/MS methods. Bioanalysis 2013, 5, 2957–2972. [Google Scholar] [CrossRef] [PubMed]
- Rauh, M.; Gröschl, M.; Rascher, W. Simultaneous quantification of ghrelin and desacyl-ghrelin by liquid chromatography-tandem mass spectrometry in plasma, serum, and cell supernatants. Clin. Chem. 2007, 53, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Barton, C.; Kay, R.G.; Gentzer, W.; Vitzthum, F.; Pleasance, S. Development of High-Throughput Chemical Extraction Techniques and Quantitative HPLC-MS/MS (SRM) Assays for Clinically Relevant Plasma Proteins. J. Proteome Res. 2009, 9, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Hoffmann, T.; Rosche, F.; Demuth, H.-U. Simultaneous determination of incretin hormones and their truncated forms from human plasma by immunoprecipitation and liquid chromatography–mass spectrometry. J. Chromatogr. B 2004, 803, 91–99. [Google Scholar] [CrossRef]
- Delinsky, D.C.; Hill, K.T.; White, C.A.; Bartlett, M.G. Quantitation of the large polypeptide glucagon by protein precipitation and LC/MS. Biomed. Chromatogr. 2004, 18, 700–705. [Google Scholar] [CrossRef]
- Miyachi, A.; Kobayashi, M.; Mieno, E.; Goto, M.; Furusawa, K.; Inagaki, T.; Kitamura, T. Accurate analytical method for human plasma glucagon levels using liquid chromatography-high resolution mass spectrometry: Comparison with commercially available immunoassays. Anal. Bioanal. Chem. 2017, 409, 5911–5918. [Google Scholar] [CrossRef]
- Ziegler, A.; Gonzalez, L.; Blikslager, A. Large Animal Models: The Key to Translational Discovery in Digestive Disease Research. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 716–724. [Google Scholar] [CrossRef]
- Howard, J.W. The Development of Mass Spectrometry-Based Methodologies for the High Throughput Quantitation of Peptides in Biological Matrices, in Chemistry. Ph.D. Thesis, Loughborough University, Loughborough, UK, 2018. Epinal Way; LE11 3TU. [Google Scholar]
- Houghton, R.; Pita, C.H.; Ward, I.; Macarthur, R. Generic approach to validation of small-molecule LC–MS/MS biomarker assays. Bioanalysis 2009, 1, 1365–1374. [Google Scholar] [CrossRef]
- Sikora, M.J.; Johnson, M.D.; Lee, A.V.; Oesterreich, S. Endocrine Response Phenotypes Are Altered by Charcoal-Stripped Serum Variability. Endocrinology 2016, 157, 3760–3766. [Google Scholar] [CrossRef]
- Dixit, V.; Chang, T.M. In vitro and clinical studies of the removal of cortisol, thyroxine, insulin, and thyroid-stimulating hormone by coated charcoal haemoperfusion. Life Support Syst. 1984, 2, 238–244. [Google Scholar] [PubMed]
- Moon, M.J.; Park, S.; Kim, D.-K.; Cho, E.B.; Hwang, J.-I.; Vaudry, H.; Seong, J.Y. Structural and molecular conservation of glucagon-like Peptide-1 and its receptor confers selective ligand-receptor interaction. Front. Endocrinol. 2012, 3, 141. [Google Scholar] [CrossRef]
- Jenkins, R.; Duggan, J.X.; Aubry, A.-F.; Zeng, J.; Lee, J.W.; Cojocaru, L.; Dufield, D.; Garofolo, F.; Kaur, S.; Schultz, G.A.; et al. Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. AAPS J. 2014, 17, 1–16. [Google Scholar] [CrossRef]
- Olsen, C.; Wang, C.; Aizenshtadt, A.; Abadpour, S.; Lundanes, E.; Skottvoll, F.S.; Golovin, A.; Busek, M.; Krauss, S.; Scholz, H.; et al. Simultaneous LC-MS determination of glucose regulatory peptides secreted by stem cell-derived islet organoids. Electrophoresis 2023, 44, 1682–1697. [Google Scholar] [CrossRef]
- Nouri-Nigjeh, E.; Zhang, M.; Ji, T.; Yu, H.; An, B.; Duan, X.; Balthasar, J.; Johnson, R.W.; Qu, J. Effects of calibration approaches on the accuracy for LC-MS targeted quantification of therapeutic protein. Anal. Chem. 2014, 86, 3575–3584. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Cromwell, G.L.; Combs, T.R.; Colombo, G.; Fanti, P. The determination of serum concentrations of osteocalcin in growing pigs and its relationship to end-measures of bone mineralization. J. Anim. Sci. 1996, 74, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Heng, J.; Song, H.; Shi, K.; Lin, X.; Chen, F.; Guan, W.; Zhang, S. Dietary Branched-Chain Amino Acids Regulate Food Intake Partly through Intestinal and Hypothalamic Amino Acid Receptors in Piglets. J. Agric. Food Chem. 2019, 67, 6809–6818. [Google Scholar] [CrossRef]
- Park, J.-H.; Bakheet, N.; Na, H.K.; Jeon, J.Y.; Yoon, S.H.; Kim, K.Y.; Zhe, W.; Kim, D.H.; Jung, H.-Y.; Song, H.-Y. A Novel Full Sense Device to Treat Obesity in a Porcine Model: Preliminary Results. Obes. Surg. 2019, 29, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Stull, C.L.; Kachulis, C.J.; Farley, J.L.; Koenig, G.J. The effect of age and teat order on alpha1-acid glycoprotein, neutrophil-to-lymphocyte ratio, cortisol, and average daily gain in commercial growing pigs. J. Anim. Sci. 1999, 77, 70–74. [Google Scholar] [CrossRef]
- El-Kadi, S.W.; Suryawan, A.; Gazzaneo, M.C.; Srivastava, N.; Orellana, R.A.; Nguyen, H.V.; Lobley, G.E.; Davis, T.A. Anabolic signaling and protein deposition are enhanced by intermittent compared with continuous feeding in skeletal muscle of neonates. Am. J. Physiol. Metab. 2012, 302, E674–E686. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, H.; Lin, A.; Su, Y. Antagonization of Ghrelin Suppresses Muscle Protein Deposition by Altering Gut Microbiota and Serum Amino Acid Composition in a Pig Model. Biology 2022, 11, 840. [Google Scholar] [CrossRef]
- Briegel, J.; Sprung, C.L.; Annane, D.; Singer, M.; Keh, D.; Moreno, R.; Möhnle, P.; Weiss, Y.; Avidan, A.; Michael Vogeser for the CORTICUS Study Group; et al. Multicenter comparison of cortisol as measured by different methods in samples of patients with septic shock. Intensiv. Care Med. 2009, 35, 2151–2156. [Google Scholar] [CrossRef]
- Cohen, J.; Ward, G.; Prins, J.; Jones, M.; Venkatesh, B. Variability of cortisol assays can confound the diagnosis of adrenal insufficiency in the critically ill population. Intensiv. Care Med. 2006, 32, 1901–1905. [Google Scholar] [CrossRef]
- Rosli, N.; Kwon, H.; Lim, J.; Yoon, Y.A.; Jeong, J. Measurement comparability of insulin assays using conventional immunoassay kits. J. Clin. Lab. Anal. 2022, 36, e24521. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H. Clinical and Technical Aspects in Free Cortisol Measurement. Endocrinol. Metab. 2022, 37, 599–607. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Time (min) | Flow Rate (mL/min) | %A (2% Formic Acid) | %B (ACN + 2% Formic Acid) |
|---|---|---|---|
| 0.00 | 0.200 | 99.0 | 1.0 |
| 5.00 | 0.200 | 95.0 | 5.0 |
| 10.00 | 0.200 | 79.0 | 21.0 |
| 15.00 | 0.200 | 75.5 | 24.5 |
| 18.00 | 0.200 | 46.5 | 53.5 |
| 21.00 | 0.200 | 26.0 | 74.0 |
| 25.00 | 0.350 | 1.0 | 99.0 |
| 26.00 | 0.350 | 0.0 | 100.0 |
| 28.00 | 0.350 | 0.0 | 100.0 |
| 35.00 | 0.200 | 99.0 | 1.0 |
| 37.00 | 0.200 | 99.0 | 1.0 |
| Level | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 9 | 8 | 7 | 6 | 5 | 4 | 3 | 2 | 1 | 0 | |
| Pig Insulin | 0.6250 | 0.3125 | 0.1563 | 0.0781 | 0.0391 | 0.0195 | 0.0098 | 0.0049 | 0.0024 | 0.0012 | 0 |
| GLP-1 (7-36) | 0.6250 | 0.3125 | 0.1563 | 0.0781 | 0.0391 | 0.0195 | 0.0098 | 0.0049 | 0.0024 | 0.0012 | 0 |
| GLP-1 (7-37) | 0.6250 | 0.3125 | 0.1563 | 0.0781 | 0.0391 | 0.0195 | 0.0098 | 0.0049 | 0.0024 | 0.0012 | 0 |
| Pig Ghrelin | 0.6250 | 0.3125 | 0.1563 | 0.0781 | 0.0391 | 0.0195 | 0.0098 | 0.0049 | 0.0024 | 0.0012 | 0 |
| Pig Des-Acyl Ghrelin | 0.6250 | 0.3125 | 0.1563 | 0.0781 | 0.0391 | 0.0195 | 0.0098 | 0.0049 | 0.0024 | 0.0012 | 0 |
| Pig CarboxylatedOsteocalcin | 62.5000 | 31.2500 | 15.6300 | 7.8130 | 3.9060 | 1.9530 | 0.9766 | 0.4883 | 0.2441 | 0.1221 | 0 |
| Cortisol | 62.5000 | 31.2500 | 15.6300 | 7.8130 | 3.9060 | 1.9530 | 0.9766 | 0.4883 | 0.2441 | 0.1221 | 0 |
| Quantifiers | Qualifiers | |||||||
|---|---|---|---|---|---|---|---|---|
| External Standards | Internal Standards | External Standards | Internal Standards | |||||
| Transition | Retention Time (min) | Transition | Retention Time (min) | Transition | Retention Time (min) | Transition | Retention Time (min) | |
| Cortisol [M+H]+ | 363.2 → 121.0 | 20.1 | 367.2 → 121.0 | 20.1 | 363.2 → 327.2 | 20.1 | 367.2 → 331.2 | 20.1 |
| Des-acyl Ghrelin [M+5H]5+ | 639.1 → 84.0 | 10.1 | 541.5 → 70.0 | 10.6 | 639.1 → 129.1 | 10.1 | 541.5 → 620.9 | 10.6 |
| Ghrelin [M+6H]6+ | 553.9 → 84.0 | 13.5 | 562.5 → 70.0 | 13.9 | 553.9 → 129.1 | 13.5 | 562.5 → 689.0 | 13.9 |
| Carboxylated osteocalcin [M+5H]5+ | 1145.4 → 1115.3 | 21.2 | 1186.9 → 1186.9 | 19.9 | 1145.4 → 1118.9 | 21.2 | --* | 19.9 |
| GLP-1 (7-36) [M+5H]5+ | 660.6 → 660.6 | 19.7 | 663.5 → 72.1 | 19.7 | --* | -- | --* | 19.7 |
| GLP-1 (7-37) [M+5H]5+ | 672.1 → 84.1 | 20.1 | 674.9 → 84.1 | 20.1 | 672.1 → 136.1 | 20.1 | 674.9 →110.0 | 20.1 |
| Insulin [M+5H]5+ | 1155.7 → 86.0 | 20.2 | 1161.7 → 1158.5 | 19.7 | 1155.7 → 1152.4 | 20.2 | 1161.7 → 86.1 | 19.7 |
| Parameter | Setting |
|---|---|
| Gas Temp | 330 °C |
| Gas Flow | 10 L/min |
| Nebulizer | 30 psi |
| Sheath Gas Temp | 350 °C |
| Sheath Gas Flow | 11 L/min |
| Capillary | 3500 V |
| Nozzle Voltage | 500 V |
| Chamber current | 0.19 uA |
| Analyte | Quantitation Range |
|---|---|
| Pig insulin | 4.9–625 pg/mL |
| Pig des-acyl ghrelin | 1.2–625 pg/mL |
| Pig ghrelin | 1.2–625 pg/mL |
| Pig carboxylated osteocalcin | 0.98–62.5 ng/mL |
| GLP-1 (7-36) | 2.4–625 pg/mL |
| GLP-1 (7-37) | 2.4–625 pg/mL |
| Cortisol | 0.12–62.5 ng/mL |
| Analyte | Intra-Day (n = 3) | Inter-Day (n = 6) |
|---|---|---|
| Pig insulin | 10.1 | 23.1 |
| Pig des-acyl ghrelin | 1.0 | 15.0 |
| Pig acyl ghrelin | 7.2 | 7.2 |
| Pig carboxylated osteocalcin | 5.0 | 13.0 |
| GLP-1 (7-37) | 10.4 | 9.1 |
| GLP-1 (7-36) | 4.6 | 8.3 |
| Cortisol | 5.2 | 11.0 |
| External Analyte | %Recovery (n = 3) | Internal Analyte | %Recovery (n = 3) |
|---|---|---|---|
| Pig insulin | 115.3 ± 12.4 | Human insulin | 105 ± 10.8 |
| Pig des-acyl ghrelin | 83.7 ± 4.5 | Human des-acyl ghrelin | 88.9 ± 2.4 |
| Pig acyl ghrelin | 93 ± 3.5 | Human acyl ghrelin | 97.2 ± 3.3 |
| Pig carboxylated osteocalcin | 107.1 ± 8.9 | Human y-carboxy osteocalcin | 94.5 ± 14.0 |
| GLP-1 (7-37) | 97.0 ± 12.1 | Me-GLP-1 (7-37) | 99.6 ± 5.7 |
| GLP-1 (7-36) | 88.3 ± 7.0 | Me-GLP-1 (7-36) | 116.0 ± 17.7 |
| Cortisol | 87.9 ± 9.2 | D4-Cortisol | 90.2 ± 1.7 |
| QC1 | QC2 | QC3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Expected | Measured | Error (%) | Expected | Measured | Error (%) | Expected | Measured | Error (%) | |
| Insulin | 0.0125 | 0.0131 ± 0.0040 | 4.8 | 0.0625 | 0.0582 ± 0.0064 | 6.9 | 0.625 | 0.578 ± 0.028 | 7.5 |
| GLP-1 (7-36) | 0.0125 | 0.0177 ± 0.0007 | 41.6 | 0.0625 | 0.0701 ± 0.0064 | 12.2 | 0.625 | 0.670 ± 0.038 | 7.2 |
| GLP-1 (7-37) | 0.0125 | 0.0135 ± 0.0023 | 8.0 | 0.0625 | 0.0667 ± 0.0021 | 6.7 | 0.625 | 0.459 ± 0.018 | 26.6 |
| Des-acyl Ghrelin | 0.0125 | 0.0103 ± 0.0001 | 17.6 | 0.0625 | 0.0525 ± 0.0007 | 16.0 | 0.625 | 0.720 ± 0.013 | 15.2 |
| Ghrelin | 0.0125 | 0.0163 ± 0.0002 | 30.4 | 0.0625 | 0.0645 ± 0.0035 | 3.2 | 0.625 | 0.729 ± 0.004 | 16.6 |
| Osteocalcin | 1.25 | 1.56 ± 0.17 | 24.8 | 6.25 | 6.49 ± 0.23 | 3.8 | 62.5 | 58.4 ± 0.5 | 6.6 |
| Cortisol | 1.25 | 1.52 ± 0.01 | 21.6 | 6.25 | 6.46 ± 0.03 | 3.4 | 62.5 | 60.9 ± 0.5 | 2.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Siddiqi, H.; Huang, Y.-P.; McClorry, S.; Ji, P.; Barile, D.; Slupsky, C.M. An LC-MS/MS Method for the Simultaneous Quantification of Insulin, Cortisol, Glucagon-like Peptide 1, Ghrelin, and Osteocalcin. Separations 2024, 11, 41. https://doi.org/10.3390/separations11020041
Zhang Z, Siddiqi H, Huang Y-P, McClorry S, Ji P, Barile D, Slupsky CM. An LC-MS/MS Method for the Simultaneous Quantification of Insulin, Cortisol, Glucagon-like Peptide 1, Ghrelin, and Osteocalcin. Separations. 2024; 11(2):41. https://doi.org/10.3390/separations11020041
Chicago/Turabian StyleZhang, Zhichao, Hareem Siddiqi, Yu-Ping Huang, Shannon McClorry, Peng Ji, Daniela Barile, and Carolyn M. Slupsky. 2024. "An LC-MS/MS Method for the Simultaneous Quantification of Insulin, Cortisol, Glucagon-like Peptide 1, Ghrelin, and Osteocalcin" Separations 11, no. 2: 41. https://doi.org/10.3390/separations11020041