
First-Principles Calculations to Investigate the Effect of Van der Waals Interactions on the Crystal and Electronic Structures of Tin-Based 0D Hybrid Perovskites



Abstract
:1. Introduction
2. Results and Discussion
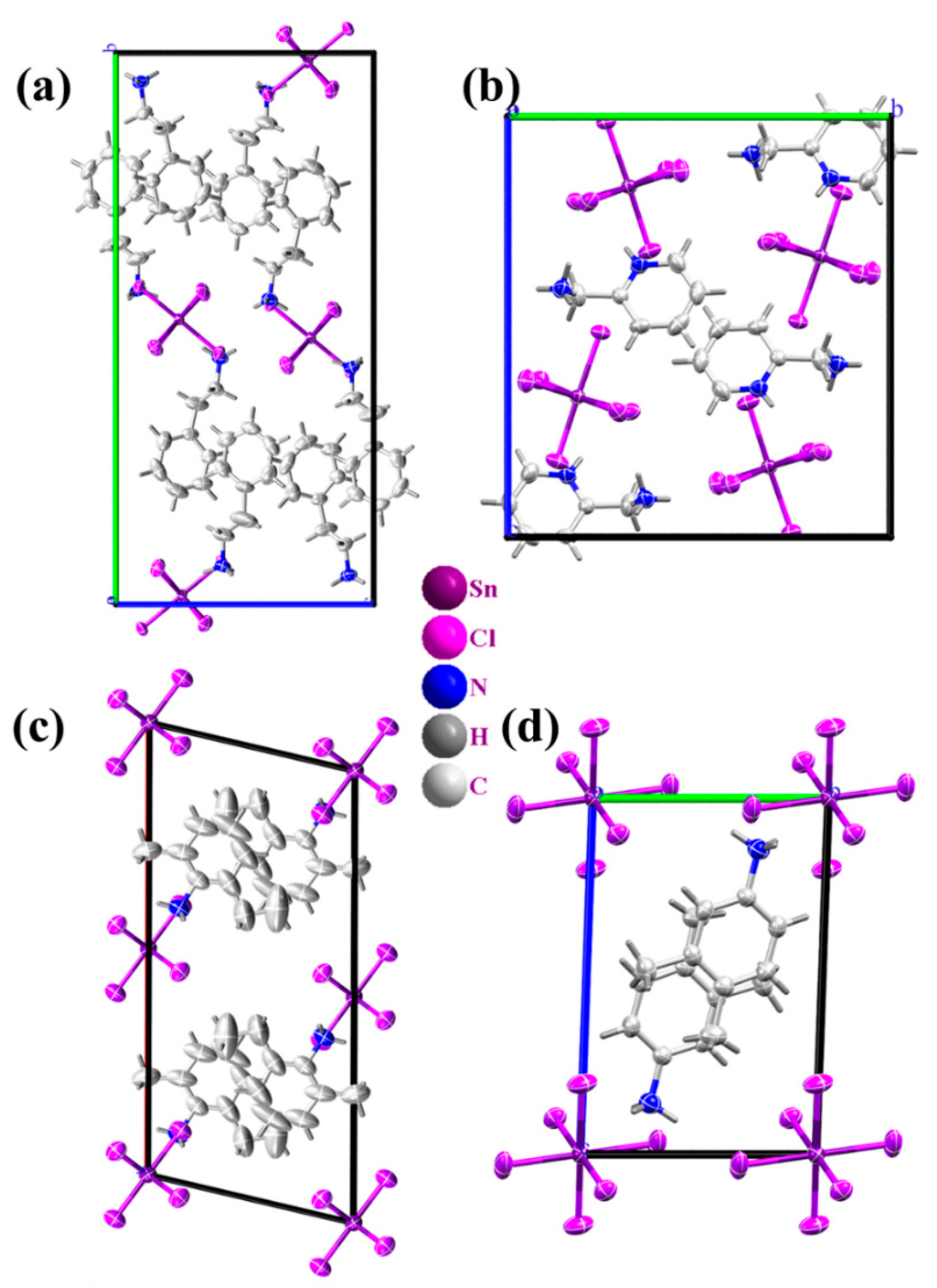
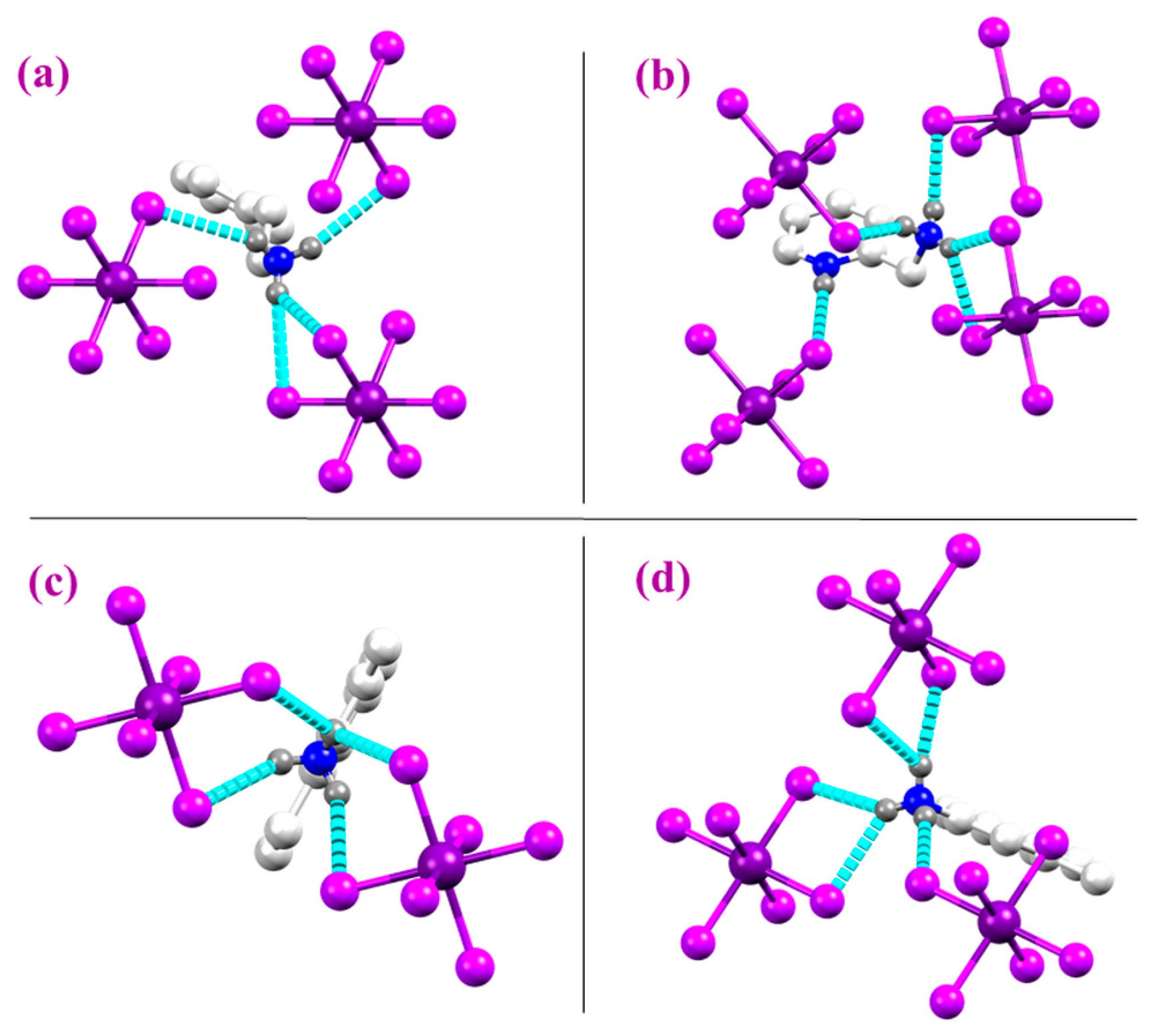
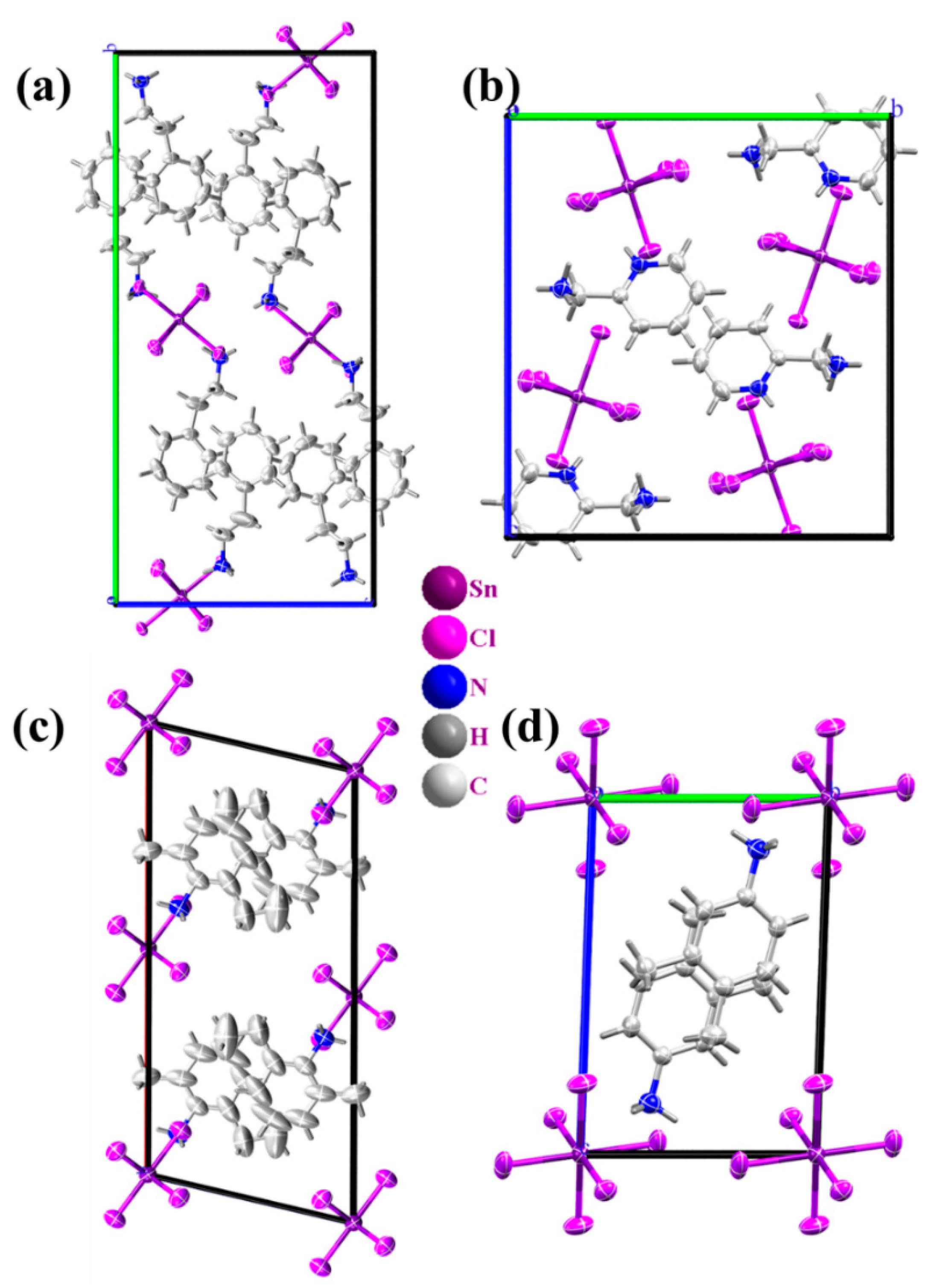
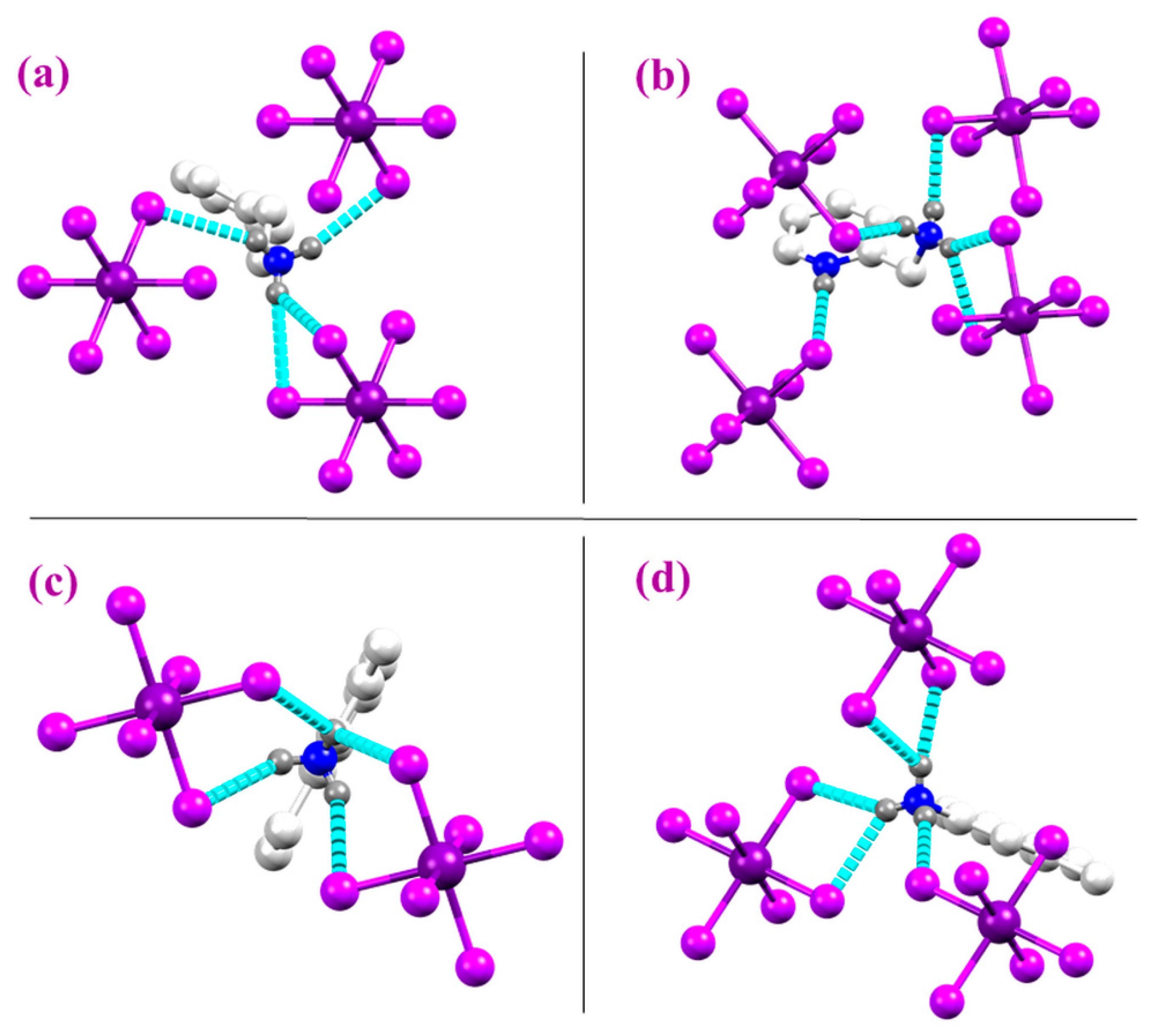
2.1. Structural Properties
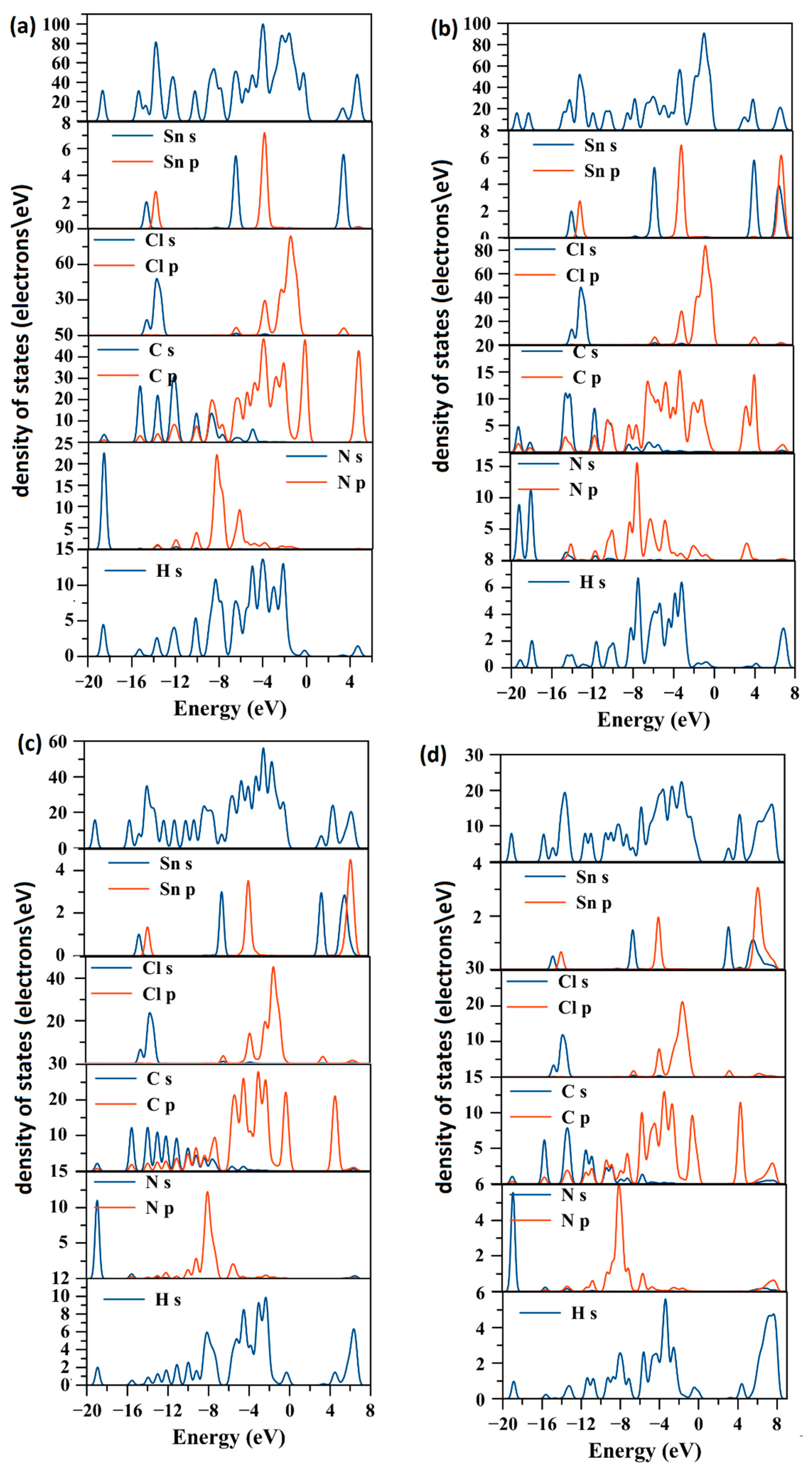
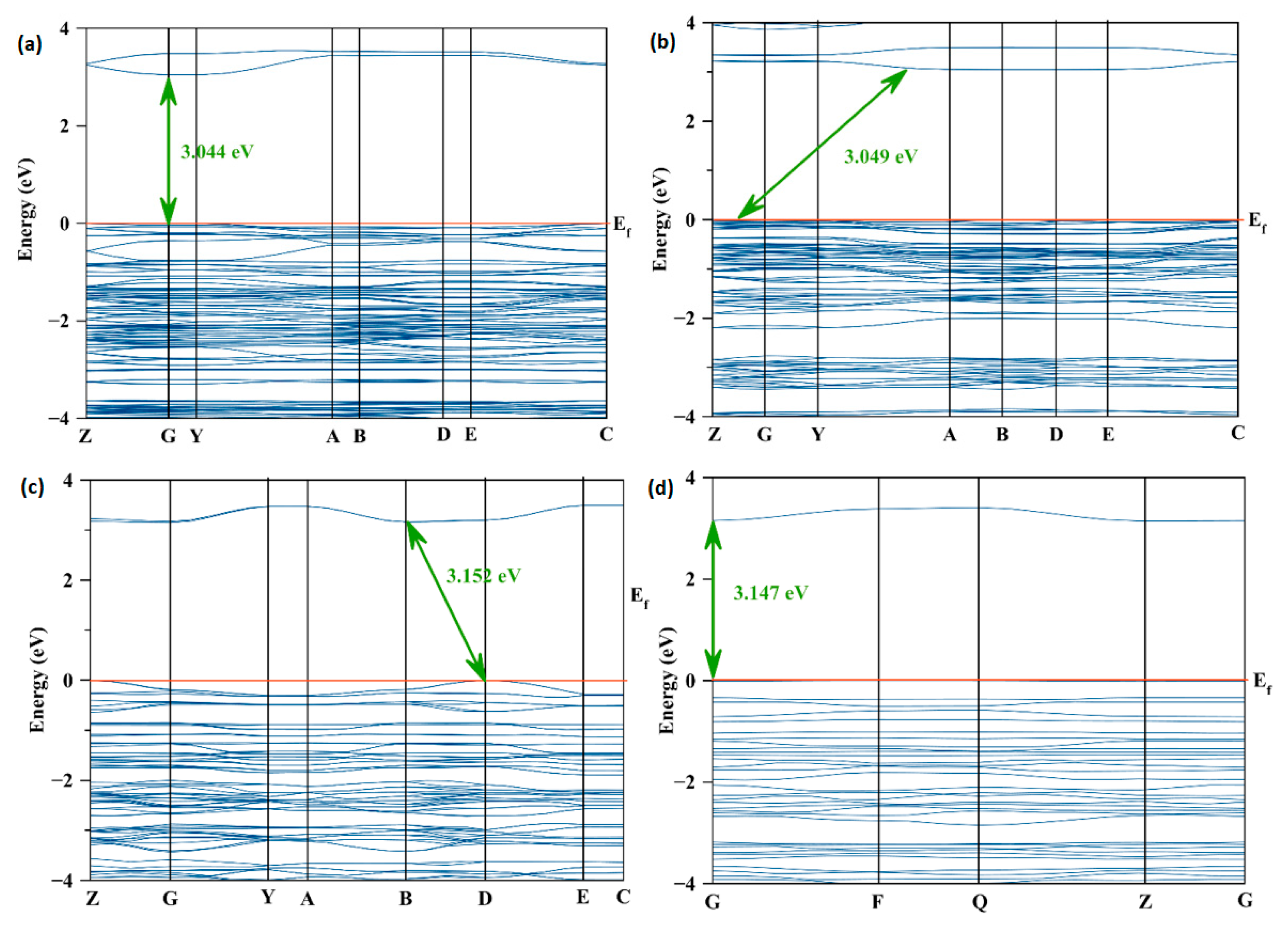
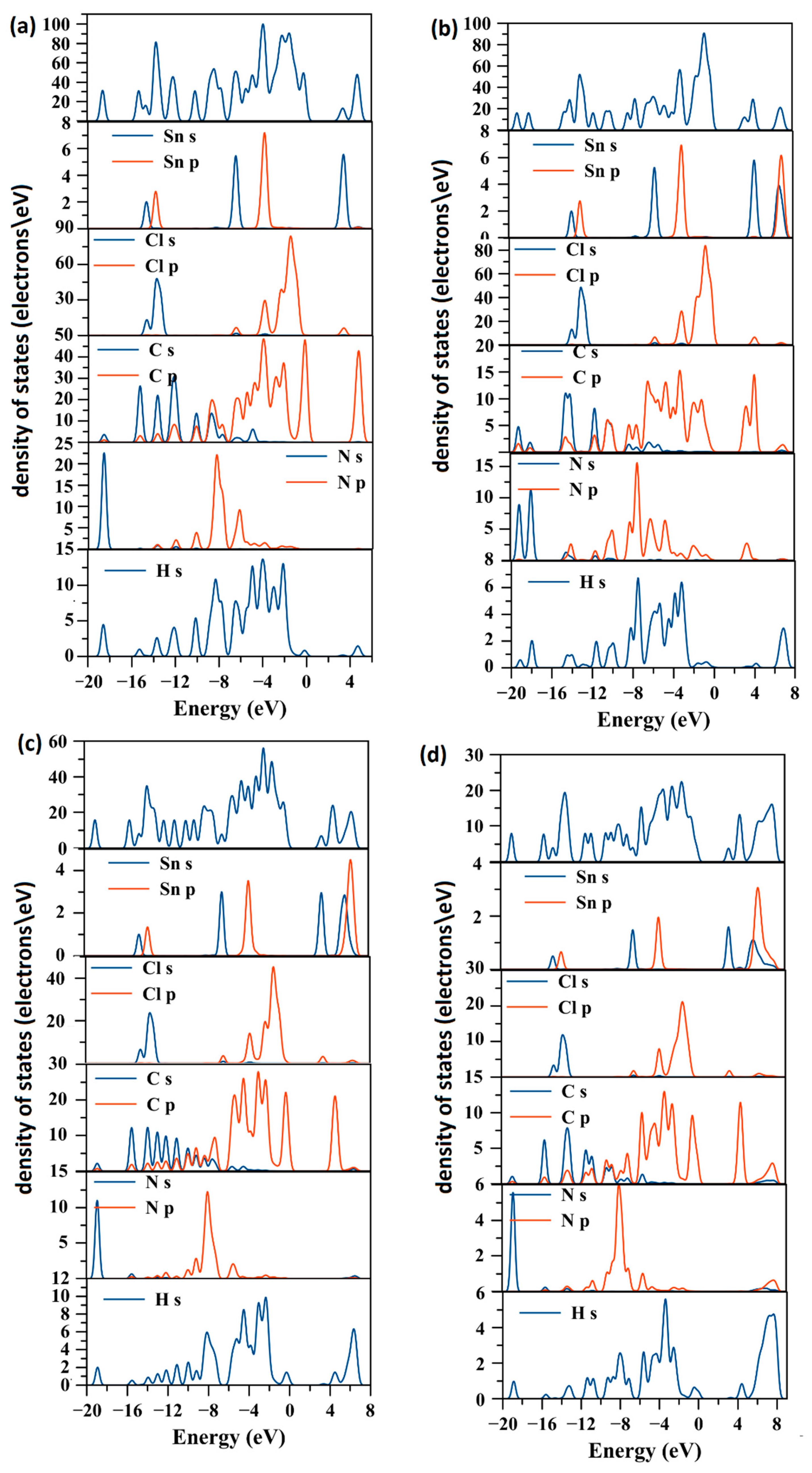
2.2. Electronic Structure
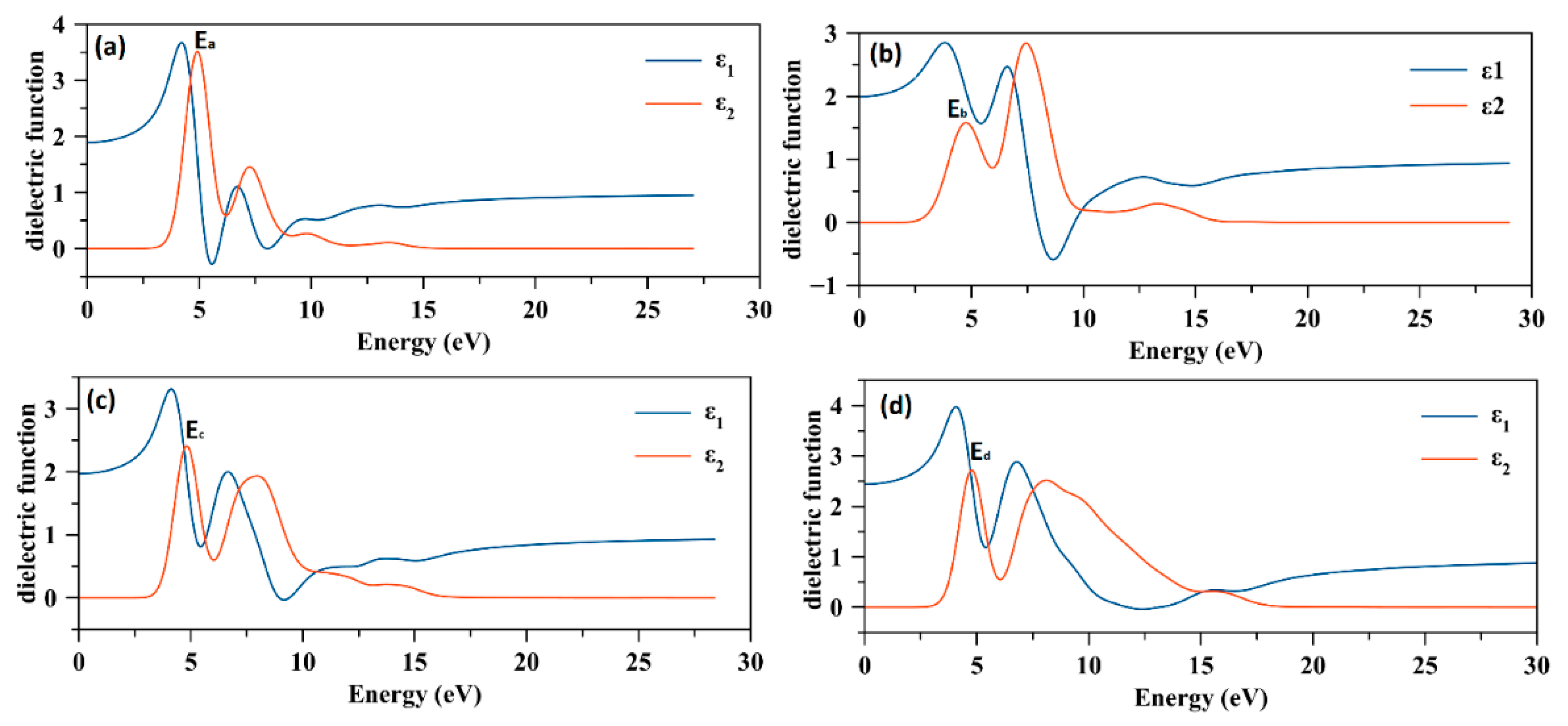
2.3. Dielectric Function and Determination of the First Transition
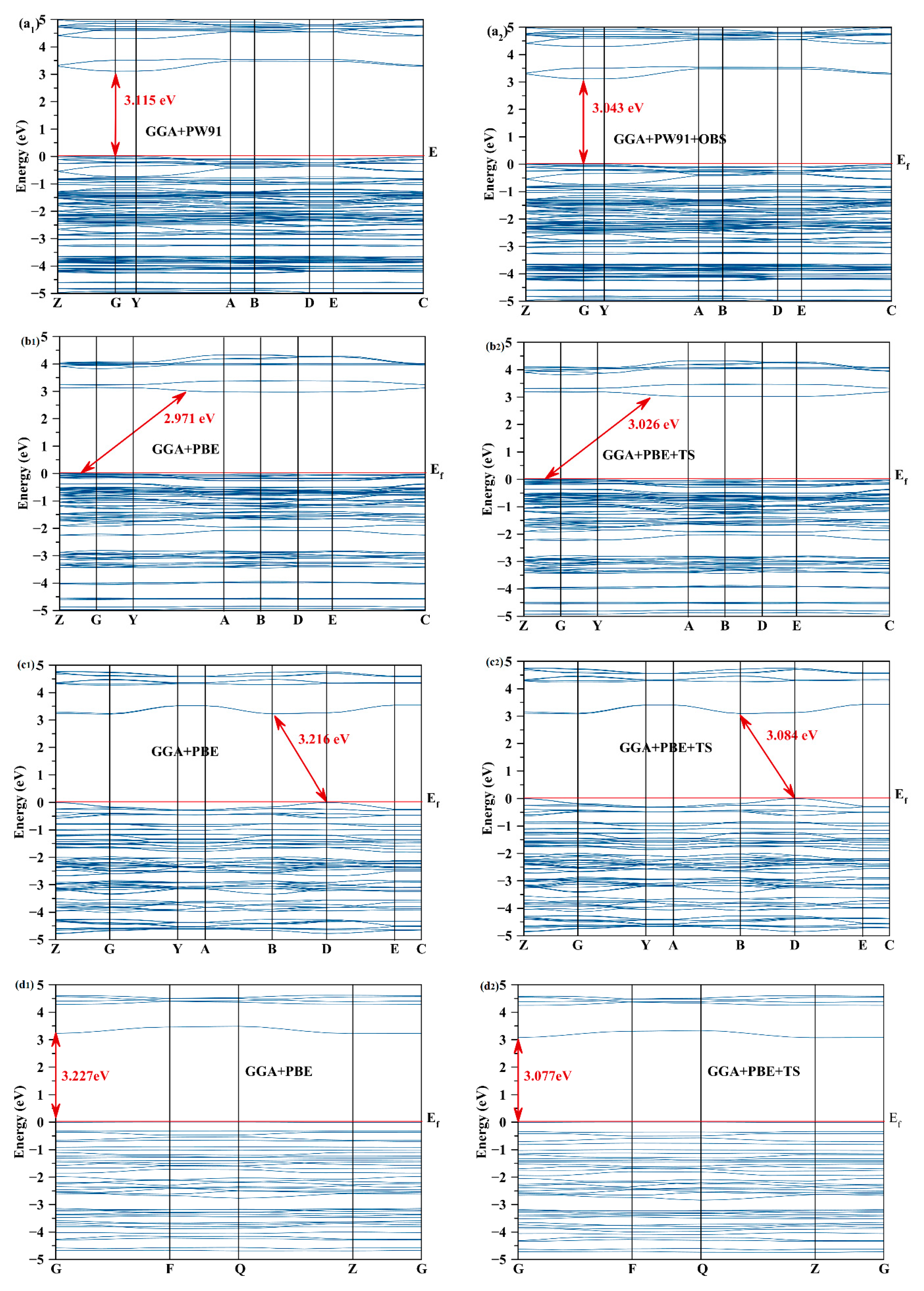
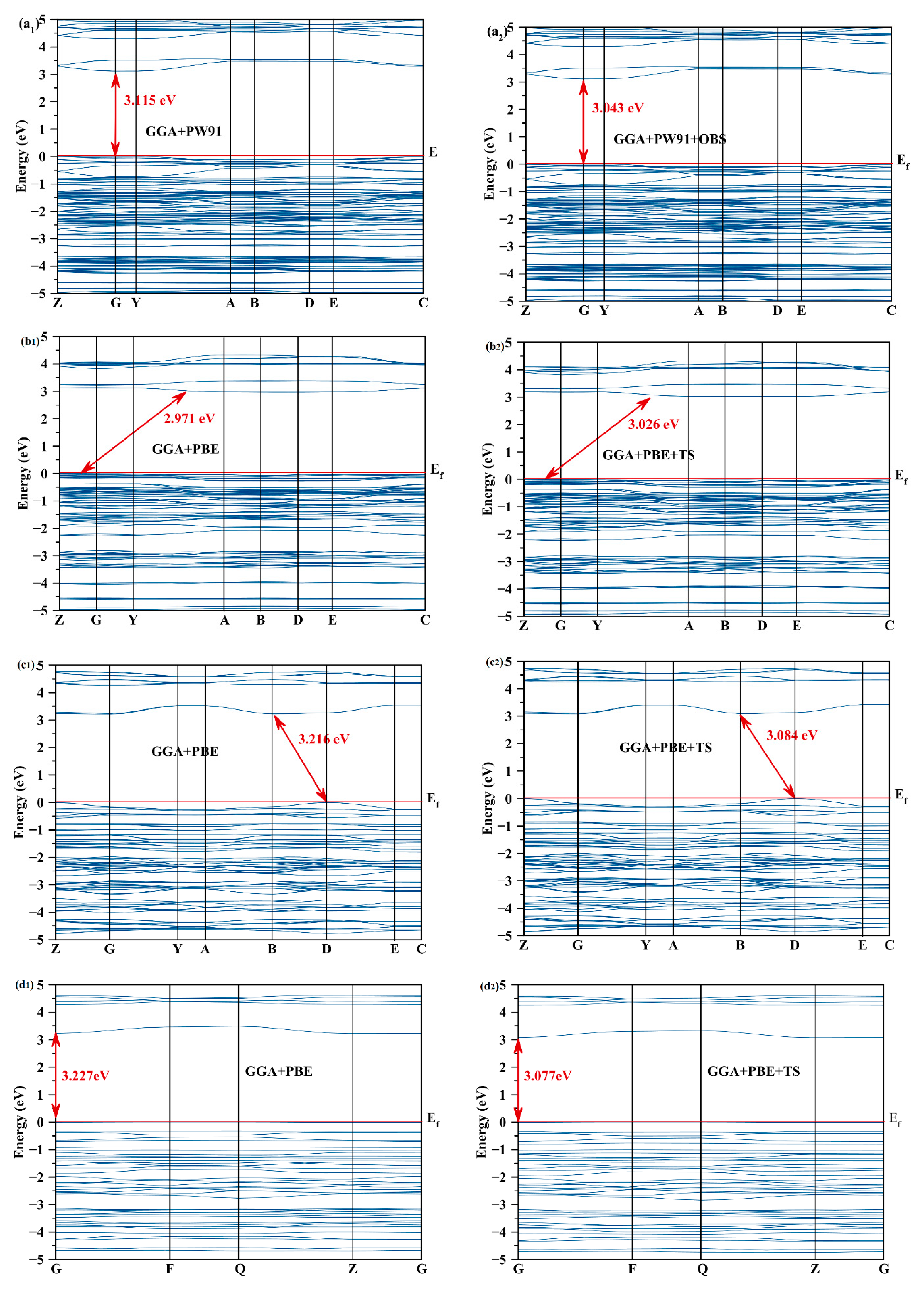
2.4. Effect of the Van der Waals Interactions on the Crystallographic and Electronic Structures
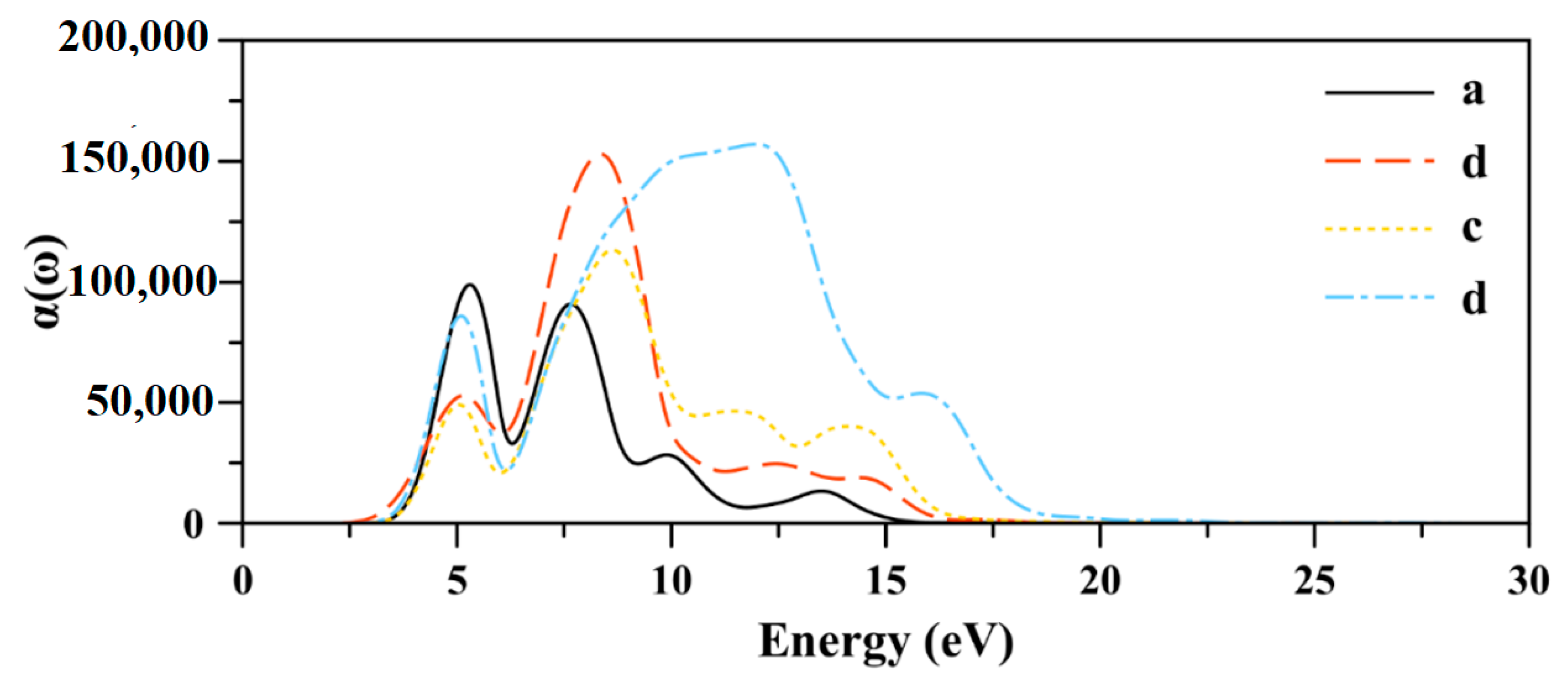
2.5. Absorption
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saidaminov, M.I.; Almutlaq, J.; Sarmah, S.; Dursun, I.; Zhumekenov, A.A.; Begum, R.; Pan, J.; Cho, N.; Mohammed, O.F.; Bakr, O.M. Pure Cs4PbBr6: Highly luminescent zero-dimensional perovskite solids. ACS Energy Lett. 2016, 1, 840–845. [Google Scholar] [CrossRef]
- Chen, L.J.; Dai, J.H.; Lin, J.D.; Mo, T.S.; Lin, H.P.; Yeh, H.C.; Chuang, Y.C.; Jiang, S.A.; Lee, C.R. Wavelength-tunable and highly stable perovskite-quantum-dot-doped lasers with liquid crystal lasing cavities. ACS Appl. Mater. Interfaces 2018, 10, 33307–33315. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.; Kumar, M.H.; Chong, W.K.; Liu, X.; Cai, Y.; Ding, H.; Asta, M.; Grätzel, M.; Mhaisalkar, S.; Mathews, N. Solution-processed tin-based perovskite for near-infrared lasing. Adv. Mater. 2016, 28, 8191–8196. [Google Scholar] [CrossRef]
- Yu, W.; Li, F.; Yu, L.; Niazi, M.R.; Zou, Y.; Corzo, D.; Basu, A.; Ma, C.; Dey, S.; Tietze, M.L. Single crystal hybrid perovskite field-effect transistors. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wei, Z.; Yoo, P.; Shi, E.; Zeller, M.; Zhu, C.; Liao, P.; Dou, L. Highly stable lead-free perovskite field-effect transistors incorporating linear π-conjugated organic ligands. J. Am. Chem. Soc. 2019, 141, 15577–15585. [Google Scholar] [CrossRef] [PubMed]
- Kasel, T.W.; Deng, Z.; Mroz, A.M.; Hendon, C.H.; Butler, K.T.; Canepa, P. Metal-free perovskites for non linear optical materials. Chem. Sci. 2019, 10, 8187–8194. [Google Scholar] [CrossRef]
- Xu, J.; Li, X.; Xiong, J.; Yuan, C.; Semin, S.; Rasing, T.; Bu, X.H. Nonlinear Optical Perovskites: Halide Perovskites for Nonlinear Optics (Adv. Mater. 3/2020). Adv. Mater. 2020, 32, 2070017. [Google Scholar] [CrossRef]
- Liu, Q.; Yin, J.; Zhang, B.B.; Chen, J.K.; Zhou, Y.; Zhang, L.M.; Wang, L.M.; Zhao, Q.; Hou, J.; Shu, J. Theory-Guided Synthesis of Highly Luminescent Colloidal Cesium Tin Halide Perovskite Nanocrystals. J. Am. Chem. Soc. 2021, 143, 5470–5480. [Google Scholar] [CrossRef]
- Ozório, M.S.; Srikanth, M.; Besse, R.; Da Silva, J.L. The role of the A-cations in the polymorphic stability and optoelectronic properties of lead-free ASnI 3 perovskites. Phys. Chem. Chem. Phys. 2021, 23, 2286–2297. [Google Scholar] [CrossRef]
- Duan, X.; Liu, F.; Kwon, H.; Byun, Y.; Minn, I.; Cai, X.; Zhang, J.; Pomper, M.G.; Yang, Z.; Xi, Z. (S)-3-(Carboxyformamido)-2-(3-(carboxymethyl) ureido) propanoic acid as a novel PSMA targeting scaffold for prostate cancer imaging. J. Med. Chem. 2020, 63, 3563–3576. [Google Scholar] [CrossRef]
- Zhou, L.; Liao, J.F.; Huang, Z.G.; Wei, J.H.; Wang, X.D.; Chen, H.Y.; Kuang, D.B. Intrinsic self-trapped emission in 0D lead-free (C4H14N2) 2In2Br10 single crystal. Angew. Chem. 2019, 131, 15581–15586. [Google Scholar] [CrossRef]
- Pious, J.K.; Katre, A.; Muthu, C.; Chakraborty, S.; Krishna, S.; Vijayakumar, C. Zero-dimensional lead-free hybrid perovskite-like material with a quantum-well structure. Chem. Mater. 2019, 31, 1941–1945. [Google Scholar] [CrossRef]
- Yavari, M.; Ebadi, F.; Meloni, S.; Wang, Z.S.; Yang, T.C.J.; Sun, S.; Schwartz, H.; Wang, Z.; Niesen, B.; Durantini, J. How far does the defect tolerance of lead-halide perovskites range? The example of Bi impurities introducing efficient recombination centers. J. Mater. Chem. A 2019, 7, 23838–23853. [Google Scholar] [CrossRef]
- Ke, W.; Kanatzidis, M.G. Prospects for low-toxicity lead-free perovskite solar cells. Nat. Commun. 2019, 10, 965. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Hou, G.J.; Zhu, Z.G.; Yan, Q.B.; Zheng, Q.R.; Su, G. Predicted lead-free perovskites for solar cells. Chem. Mater. 2018, 30, 718–728. [Google Scholar] [CrossRef]
- Kamat, P.V.; Bisquert, J.; Buriak, J. Lead-free perovskite solar cells. ACS Energy Lett. 2017, 2, 904–905. [Google Scholar] [CrossRef]
- Giustino, F.; Snaith, H.J. Toward lead-free perovskite solar cells. ACS Energy Lett. 2016, 1, 1233–1240. [Google Scholar] [CrossRef]
- Gómez, A.; Wang, Q.; Goñi, A.R.; Campoy-Quiles, M.; Abate, A. Ferroelectricity-free lead halide perovskites. Energy Environ. Sci. 2019, 12, 2537–2547. [Google Scholar] [CrossRef]
- Ozório, M.S.; Oliveira, W.X.; Silveira, J.F.; Nogueira, A.F.; Da Silva, J.L. Novel zero-dimensional lead-free bismuth-based perovskites: From synthesis to structural and optoelectronic characterization. Mater. Adv. 2020, 1, 3439–3448. [Google Scholar] [CrossRef]
- Chu, K.B.; Xie, J.L.; Chen, W.J.; Lu, W.X.; Song, J.L.; Zhang, C. A novel bismuth-based hybrid material with highly activity for fast removal of rhodamine B under dark conditions. Polyhedron 2018, 151, 146–151. [Google Scholar] [CrossRef]
- Zhou, C.; Lin, H.; Tian, Y.; Yuan, Z.; Clark, R.; Chen, B.; van de Burgt, L.J.; Wang, J.C.; Zhou, Y.; Hanson, K. Luminescent zero-dimensional organic metal halide hybrids with near-unity quantum efficiency. Chem. Sci. 2018, 9, 586–593. [Google Scholar] [CrossRef]
- Pandech, N.; Kongnok, T.; Palakawong, N.; Limpijumnong, S.; Lambrecht, W.R.; Jungthawan, S. Effects of the van der Waals Interactions on Structural and Electronic Properties of CH3NH3 (Pb, Sn)(I, Br, Cl) 3 Halide Perovskites. ACS Omega 2020, 5, 25723–25732. [Google Scholar] [CrossRef] [PubMed]
- Ozório, M.S.; Dias, A.C.; Silveira, J.F.; Da Silva, J.L. Theoretical Investigation of the Role of Anion and Trivalent Cation Substitution in the Physical Properties of Lead-Free Zero-Dimensional Perovskites. J. Phys. Chem. C 2022, 126, 7245–7255. [Google Scholar] [CrossRef]
- Knutson, J.L.; Martin, J.D.; Mitzi, D.B. Tuning the band gap in hybrid tin iodide perovskite semiconductors using structural templating. Inorg. Chem. 2005, 44, 4699–4705. [Google Scholar] [CrossRef] [PubMed]
- Saparov, B.; Mitzi, D.B. Organic–inorganic perovskites: Structural versatility for functional materials design. Chem. Rev. 2016, 116, 4558–4596. [Google Scholar] [CrossRef]
- Moussa, O.B.; Chebbi, H.; Arfaoui, Y.; Falvello, L.R.; Tomas, M.; Zid, M.F. Structural study, vibrational and optical properties, Hirshfeld surface analysis and DFT investigation of a novel organic cation hexachloridostannate (IV), (C5H8N3) 2 [SnCl6]. J. Mol. Struct. 2019, 1195, 344–354. [Google Scholar] [CrossRef]
- Sayer, I.; Dege, N.; Ghalla, H.; Moliterni, A.; Naïli, H. Crystal structure, DFT studies and thermal characterization of new luminescent stannate (IV) based inorganic-organic hybrid compound. J. Mol. Struct. 2021, 1224, 129266. [Google Scholar] [CrossRef]
- Feddaoui, I.; Abdelbaky, M.S.; García-Granda, S.; Essalah, K.; Nasr, C.B.; Mrad, M. Synthesis, crystal structure, vibrational spectroscopy, DFT, optical study and thermal analysis of a new stannate (IV) complex based on 2-ethyl-6-methylanilinium (C9H14N) 2 [SnCl6]. J. Mol. Struct. 2019, 1186, 31–38. [Google Scholar] [CrossRef]
- Mathlouthi, M.; Valkonen, A.; Rzaigui, M.; Smirani, W. Structural characterization, spectroscopic, thermal, AC conductivity and dielectric properties and antimicrobial studies of (C8H12N) 2 [SnCl6]. Phase Transit. 2017, 90, 399–414. [Google Scholar] [CrossRef]
- Ferjani, H. Crystal structure, optical property and Hirshfeld surface analysis of bis [1-(prop-2-en-1-yl)-1H-imidazol-3-ium] hexachloridostannate (IV). Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1624–1628. [Google Scholar] [CrossRef]
- El-Mellouhi, F.; Marzouk, A.; Bentria, E.T.; Rashkeev, S.N.; Kais, S.; Alharbi, F.H. Hydrogen bonding and stability of hybrid organic–inorganic perovskites. ChemSusChem 2016, 9, 2648–2655. [Google Scholar] [CrossRef]
- Yu, C.J.; Jong, U.G.; Ri, M.H.; Ri, G.C.; Pae, Y.H. Electronic structure and photoabsorption property of pseudocubic perovskites CH3NH3PbX3 (X = I, Br) including van der Waals interaction. J. Mater. Sci. 2016, 51, 9849–9854. [Google Scholar] [CrossRef]
- Srikant, V.; Clarke, D.R. On the optical band gap of zinc oxide. J. Appl. Phys. 1998, 83, 5447–5451. [Google Scholar] [CrossRef]
- Zolfaghari, P.; De Wijs, G.; De Groot, R. The electronic structure of organic–inorganic hybrid compounds:(NH4) 2CuCl4, (CH3NH3) 2CuCl4 and (C2H5NH3) 2CuCl4. J. Phys. Condens. Matter 2013, 25, 295502. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, Z.; Ge, C.; Li, H.; Hao, M.; Wan, C.; Song, Y.; Li, B.; Dong, Q. Multiple Hydrogen Bond-Induced Structural Distortion for Broadband White-Light Emission in Two-Dimensional Perovskites. CCS Chem. 2021, 3, 2576–2583. [Google Scholar] [CrossRef]
- Bernasconi, A.; Page, K.; Dai, Z.; Tan, L.Z.; Rappe, A.M.; Malavasi, L. Ubiquitous Short-Range Distortion Hybrid Perovskites Hydrogen-Bonding role: The MAPbCl3 case. J. Phys. Chem. C 2018, 122, 28265–28272. [Google Scholar] [CrossRef]
- Kaiba, A.; Geesi, M.H.; Riadi, Y.; Ibnouf, E.O.; Aljohani, T.A.; Guionneau, P. A new Organic–Inorganic hybrid compound (NH3(CH2)2C6H5)2[SnCl6]: Crystal structure, characterization, Hirshfeld surface analysis, DFT calculation, vibrational properties and biological evaluation. J. Solid State Chem. 2021, 304, 122587. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Gao, W.; Gao, X.; Abtew, T.A.; Sun, Y.Y.; Zhang, S.; Zhang, P. Quasiparticle band gap of organic-inorganic hybrid perovskites: Crystal structure, spin-orbit coupling, and self-energy effects. Phys. Rev. B 2016, 93, 085202. [Google Scholar] [CrossRef]
- Laref, A.; Al-Enazi, M.; Al-Qahtani, H.; Laref, S.; Wu, X. Impact of fluorine on organic cation for determining the electronic and optical properties of CH3− xFxNH3PbI3 (x = 0, 1, 2, 3) hybrid perovskites-based photovoltaic devices. Sol. Energy 2019, 177, 517–530. [Google Scholar] [CrossRef]
- Gebhardt, J.; Kim, Y.; Rappe, A.M. Influence of the dimensionality and organic cation on crystal and electronic structure of organometallic halide perovskites. J. Phys. Chem. C 2017, 121, 6569–6574. [Google Scholar] [CrossRef]
- Maheshwari, S.; Patwardhan, S.; Schatz, G.C.; Renaud, N.; Grozema, F.C. The effect of the magnitude and direction of the dipoles of organic cations on the electronic structure of hybrid halide perovskites. Phys. Chem. Chem. Phys. 2019, 21, 16564–16572. [Google Scholar] [CrossRef] [PubMed]
- Ferjani, H.; Ben Smida, Y.; Onwudiwe, D.C.; Elamin, N.Y.; Ezzine, S.; Almotlaq, N.S. An Experimental and Theoretical Study of the Optical Properties of (C2H7N4O) 2BiCl5 for an Optoelectronic Application. Inorganics 2022, 10, 48. [Google Scholar] [CrossRef]
- Adala, N.; Marzougui, B.; Smida, Y.B.; Marzouki, R.; Ferhi, M.; Onwudiwe, D.C.; Hamzaoui, A.H. An experimental and theoretical study of the structural, optical, electrical, and dielectric properties of PrAsO4. J. Alloys Compd. 2022, 910, 164894. [Google Scholar] [CrossRef]
- Lakhdar, M.H.; Smida, Y.B.; Amlouk, M. Synthesis, optical characterization and DFT calculations of electronic structure of Sb2O3 films obtained by thermal oxidation of Sb2S3. J. Alloys Compd. 2016, 681, 197–204. [Google Scholar] [CrossRef]
- Lee, I.H.; Martin, R.M. Applications of the generalized-gradient approximation to atoms, clusters, and solids. Phys. Rev. B 1997, 56, 7197. [Google Scholar] [CrossRef]
- Juran, T.R.; Smeu, M. Hybrid density functional theory modeling of Ca, Zn, and Al ion batteries using the Chevrel phase Mo 6 S 8 cathode. Phys. Chem. Chem. Phys. 2017, 19, 20684–20690. [Google Scholar] [CrossRef]
- Umari, P.; Mosconi, E.; Angelis, F.D. Relativistic GW calculations on CH3NH3PbI3 and CH3NH3SnI3 Perovskites for Solar Cell Applications. Sci. Rep. 2014, 4, 4467. [Google Scholar] [CrossRef]
- Chang, L.; Besteiro, L.V.; Sun, J.; Santiago, E.Y.; Gray, S.K.; Wang, Z.; Govorov, A.O. Electronic structure of the plasmons in metal nanocrystals: Fundamental limitations for the energy efficiency of hot electron generation. ACS Energy Lett. 2019, 4, 2552–2568. [Google Scholar] [CrossRef]
- Toll, J.S. Causality and the dispersion relation: Logical foundations. Phys. Rev. 1956, 104, 1760. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hummer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef]
- Peter, Y.Y.; Cardona, M. Electronic Band Structures, Fundamentals of Semiconductors; Springer: Berlin/Heidelberg, Germany, 1996; pp. 17–105. [Google Scholar]
- Cardona, M.; Peter, Y.Y. Fundamentals of Semiconductors; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Smida, Y.B.; Ferjani, H.; Boukhachem, A.; Onwudiwe, D.C.; Elamin, N.Y.; Hamzaoui, A.H. Ab initio study of the optoelectronic properties of α-Ba2SnS4. Mater. Sci. Semicond. Process. 2022, 150, 106917. [Google Scholar] [CrossRef]
- Flor, G.; Orobengoa, D.; Tasci, E.; Perez-Mato, J.M.; Aroyo, M.I. Comparison of structures applying the tools available at the Bilbao Crystallographic Server. J. Appl. Crystallogr. 2016, 49, 653–664. [Google Scholar] [CrossRef]
- Papi, H.; Jalali-Asadabadi, S.; Nourmohammadi, A.; Ahmad, I.; Nematollahia, J.; Yazdanmehr, M. Optical properties of ideal γ-Al2O3 and with oxygen point defects: An ab initio study. RSC Adv. 2015, 5, 55088–55099. [Google Scholar] [CrossRef]
- Bergerhoff, G.; Berndt, M.; Brandenburg, K.; Degen, T. Concerning inorganic crystal structure types. Acta Crystallogr. Sect. B Struct. Sci. 1999, 55, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Ortmann, F.; Bechstedt, F.; Schmidt, W. Semiempirical van der Waals correction to the density functional description of solids and molecular structures. Phys. Rev. B 2006, 73, 205101. [Google Scholar] [CrossRef] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Benghia, A.; Dahame, T.; Bentria, B. First principle calculation of physical properties of barium based chalcogenides BaM4S7 (M = Ga, Al); a DFT, DFT-D and hybrid functional HSE06 study. Opt. Mater. 2016, 54, 269–275. [Google Scholar] [CrossRef]
- Wang, W.; Fan, L.; Wang, G.; Li, Y. CO2 and SO2 sorption on the alkali metals doped CaO (100) surface: A DFT-D study. Appl. Surf. Sci. 2017, 425, 972–977. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, W.; Xiao, H. Comparative DFT-and DFT-D-based molecular dynamics studies of pressure effects in crystalline 1, 3, 5-triamino-2, 4, 6-trinitrobenzene at room temperature. RSC Adv. 2014, 4, 53149–53156. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, G.; Gong, X. 2, 4-Diazido-5-iodo-pyrimidine crystal under high pressure: A comparison of DFT and DFT-D studies. Comput. Theor. Chem. 2012, 1000, 60–69. [Google Scholar] [CrossRef]
- Gao, H.; Wei, W.; Li, L.; Tan, Y.; Tang, Y. Mechanical properties of a 2D lead-halide perovskite, (C6H5CH2NH3) 2PbCl4, by nanoindentation and first-principles calculations. J. Phys. Chem. C 2020, 124, 19204–19211. [Google Scholar] [CrossRef]
- Xu, Z.W.; Zhang, C.R.; Wu, Y.Z.; Gong, J.J.; Wang, W.; Liu, Z.J.; Chen, H.S. Density functional theory study on the electronic structures and related properties of Ag-doped CH3NH3PbI3 perovskite. Results Phys. 2019, 15, 102709. [Google Scholar] [CrossRef]
- Goerigk, L. A comprehensive overview of the DFT-D3 London-dispersion correction. In Non-Covalent Interactions in Quantum Chemistry and Physics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 195–219. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Rao, E.N.; Vaitheeswaran, G.; Reshak, A.H.; Auluck, S. Role of spin–orbit interaction on the nonlinear optical response of CsPbCO 3 F using DFT. Phys. Chem. Chem. Phys. 2017, 19, 31255–31266. [Google Scholar]
- Kormányos, A.; Zólyomi, V.; Drummond, N.D.; Rakyta, P.; Burkard, G.; Fal’Ko, V.I. Monolayer MoS 2: Trigonal warping, the Γ valley, and spin-orbit coupling effects. Phys. Rev. B 2013, 88, 045416. [Google Scholar] [CrossRef]
- Kormányos, A.; Zólyomi, V.; Drummond, N.D.; Burkard, G. Spin-orbit coupling, quantum dots, and qubits in monolayer transition metal dichalcogenides. Phys. Rev. X 2014, 4, 011034. [Google Scholar] [CrossRef]
- Pan, J.; Metzger, W.K.; Lany, S. Spin-orbit coupling effects on predicting defect properties with hybrid functionals: A case study in CdTe. Phys. Rev. B 2018, 98, 054108. [Google Scholar] [CrossRef]
- Aroyo, M.I.; Perez-Mato, J.M.; Capillas, C.; Kroumova, E.; Ivantchev, S.; Madariaga, G.; Kirov, A.; Wondratschek, H. Bilbao Crystallographic Server: I. Databases and crystallographic computing programs. Z. Krist.-Cryst. Mater. 2006, 221, 15–27. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Compound Name | Crystal System/ Space Group | Number of Donor Atoms | N-H⋯Cl Hydrogen Bonds | π-π Interactions | (eV) | Ref |
|---|---|---|---|---|---|---|---|
| a | (NH3(CH2)2C6H5)2SnCl6 | Monoclinic P21/c | 1 Nitrogen atoms NH3+ | 4 Strong | 4 Weak | - | [1] |
| b | (C6H10N2) SnCl6 | Monoclinic P21/c | 2 Nitrogen atoms NH+/NH3+ | 5 Strong | 2 Weak | 3.56 Thin film | [2] |
| c | (C9H14N)2 SnCl6 | Monoclinic C2/m | 1 Nitrogen atom NH3+ | 4 Strong | 1 Weak | 5.2 Solid | [3] |
| d | (C8H12N)2 SnCl6 | Triclinic P-1 | 1 Nitrogen atom NH3+ | 5 Strong | 2 Weak | 4.11 In water | [4] |
| Compound | Space Group | (Å) | Δ (10−5) | Inorganic Interlayer Distance (Å) | dCl⋯Cl (Å) |
|---|---|---|---|---|---|
| a | P21/c | 2.423994 | 1 | 13.326 | 9.760 |
| b | P21/c | 2.422345 | 5.5 | 7.668 | 3.456 |
| c | C2/m | 2.424254 | 1.4 | 10.517 | 7.175 |
| d | P-1 | 2.427381 | 5.4 | 10.684 | 6.858 |
| Energy | Ev | Ec | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| States | ||||||||||
| Compound | a | −1.45 | −0.20 | −1.52 | −0.18 | 4.71 | 3.43 | 3.43 | 4.72 | ------ |
| b | −0.67 | −1.08 | −0.73 | −0.80 | 3.34 | 4.18 | 4.18 | 3.31 | 3.31 | |
| c | −1.50 | −0.35 | −0.47 | −0.31 | 3.34 | 3.37 | 3.38 | 4.50 | 6.36 | |
| d | −1.51 | −0.50 | −1.58 | −0.45 | 3.27 | 3.27 | 3.28 | 4.41 | 7.70 | |
| Label | Transition | ||
|---|---|---|---|
| Ea | 4.91 | H|s C|p Sn|s Cl|p | 4.91 4.88 |
| Eb | 4.79 | Sn|s Cl|p | 4.85 |
| Ec | 4.80 | Sn|s Cl|p H|s Cl|p | 4.87 4.84 |
| Ed | 4.78 | Sn|s Cl|p H|s Cl|p | 4.78 4.78 |
| Methods | a | b | c | d |
|---|---|---|---|---|
| GGA-PBE | 3.042 | 3.049 | 3.152 | 3.147 |
| GGA-PBE+TS | 2.988 | 3.056 | 3.063 | 3.072 |
| GGA-PBE-Grimme | 2.985 | 3.058 | 3.078 | 3.092 |
| GGA-PW91 | 3.081 | 3.057 | 3.158 | 3.167 |
| GGA-PW91+OBS | 3.341 | 3.057 | 3.172 | 3.151 |
| Compound | Scheme | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | V (Å3) | Eg (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a | Exp | 7.345 | 25.667 | 11.971 | - | 90.106 | - | 2256.8 | - |
| GGA-PW91 | 7.4315 | 25.8404 | 12.1654 | - | 90.1286 | - | 2336.15 | 3.115 | |
| GGA-PW91+OBS | 7.2527 | 25.5844 | 11.8856 | - | 90.2596 | - | 2205.42 | 3.043 | |
| b | Exp | 7.0566 | 13.5254 | 14.8999 | - | 94.703 | - | 1417.31 | - |
| GGA-PBE | 7.1286 | 13.6778 | 15.0881 | - | 94.4060 | - | 1466.80 | 2.971 | |
| GGA-PBE+TS | 7.0537 | 13.5895 | 14.9690 | - | 94.5887 | - | 1430.27 | 3.026 | |
| c | Exp | 19.7521 | 7.2293 | 9.2181 | - | 103.046 | - | 1282.31 | - |
| GGA-PBE | 19.8713 | 7.3406 | 9.3495 | - | 103.363 | - | 1326.86 | 3.216 | |
| GGA-PBE+TS | 19.7328 | 7.1942 | 9.3148 | - | 103.4064 | - | 1286.31 | 3.084 | |
| d | Exp | 7.4904 | 7.9864 | 10.6842 | 91.572 | 90.470 | 117.5410 | 566.34 | - |
| GGA-PBE | 7.6320 | 8.0769 | 10.7584 | 91.4226 | 90.5358 | 117.9866 | 585.28 | 3.227 | |
| GGA-PBE+TS | 7.5157 | 7.9947 | 10.6920 | 91.5932 | 90.4239 | 117.6723 | 568.58 | 3.077 |
| Structure | XC | S | dmax. (Å) | dav. (Å) | Δ |
|---|---|---|---|---|---|
| a (NH3(CH2)2C6H5)2[SnCl6] | GGA-PW91 | 0.0069 | 0.5130 | 0.1939 | 0.027 |
| GGA-PW91+OBS | 0.0051 | 0.5240 | 0.2065 | 0.043 | |
| b (C6H10N2)[SnCl6] | GGA-PBE | 0.0067 | 0.2736 | 0.0961 | 0.015 |
| GGA-PBE+TS | 0.0023 | 0.2854 | 0.0993 | 0.022 | |
| c (C9H14N)2[SnCl6] | GGA-PBE | 0.0071 | 0.3841 | 0.1425 | 0.035 |
| GGA-PBE+TS | 0.0040 | 0.3909 | 0.1486 | 0.026 | |
| d (C8H12N)2[SnCl6] | GGA-PBE | 0.0070 | 0.1707 | 0.0824 | 0.035 |
| GGA-PBE+TS | 0.0012 | 0.1873 | 0.0870 | 0.022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferjani, H.; Smida, Y.B.; Al-Douri, Y. First-Principles Calculations to Investigate the Effect of Van der Waals Interactions on the Crystal and Electronic Structures of Tin-Based 0D Hybrid Perovskites. Inorganics 2022, 10, 155. https://doi.org/10.3390/inorganics10100155
Ferjani H, Smida YB, Al-Douri Y. First-Principles Calculations to Investigate the Effect of Van der Waals Interactions on the Crystal and Electronic Structures of Tin-Based 0D Hybrid Perovskites. Inorganics. 2022; 10(10):155. https://doi.org/10.3390/inorganics10100155
Chicago/Turabian StyleFerjani, Hela, Youssef Ben Smida, and Yarub Al-Douri. 2022. "First-Principles Calculations to Investigate the Effect of Van der Waals Interactions on the Crystal and Electronic Structures of Tin-Based 0D Hybrid Perovskites" Inorganics 10, no. 10: 155. https://doi.org/10.3390/inorganics10100155