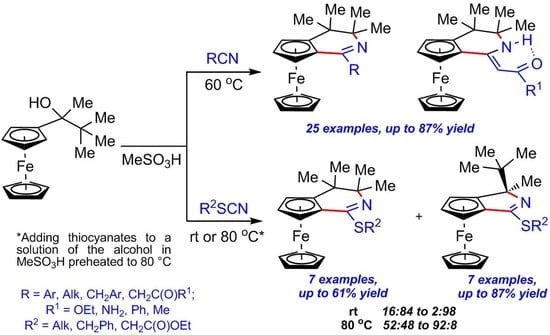
3.2.3. Synthesis of Ferroceno[c]pyridines 3a–m, o–s, u–x (Table 2)
General Procedure (GP). Nitrile 2a–m, o–s, u–x (0.42 mmol) was added to a stirred solution of alcohol 1 (100 mg, 0.35 mmol) in MeSO3H (0.18 mL) at room temperature, and the resulting mixture was heated at 60 °C in an oil bath with vigorous stirring for the indicated time (monitored by TLC). The reaction mixture was then cooled to room temperature, neutralized with 10% aq. Na2CO3 solution (3 mL) and extracted with EtOAc (10 mL × 4). The combined organic phases were washed with water, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel column chromatography.
rac-3,3,4,4-Tetramethyl-1-(4-methylphenyl)-3,4-dihydroferroceno[c]pyridine (3a). The title compound was prepared according to GP using 4-methylbenzonitrile (2a) (0.05 mL, 0.42 mmol); reaction time = 15 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3a (98 mg, 73%) as an orange solid; mp 123.5–125 °C (hexane); Rf 0.08 (petroleum ether/EtOAc 25:1); Rf 0.30 (petroleum ether/TEA 100:1). IR (thin film): 3094, 2973, 2928, 2868, 1588, 1560, 1453, 1372, 1361, 1314, 1301, 1175, 1156, 1108, 1141, 1108, 1002, 933, 899, 822, 755, 745, 495, 467 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 7.96 (br d, 2H, J = 8.2 Hz, H2′, H6′), 7.31 (br d, 2H, J = 7.9 Hz, H3′, H5′), 4.45 (d, 2H, J = 1.9 Hz, H5, H6), 4.37 (t, 1H, J = 1.8 Hz, H7), 4.27 (s, 5H, H Cp), 2.41 (s, 3H, 4′-CH3), 1.45 (s, 3H, 4-CH3), 1.44 (s, 3H, 3-CH3), 1.11 (s, 3H, 4-CH3), 0.74 (s, 3H, 3-CH3). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 162.3 (C1), 138.5 (C4′), 135.7 (C1′), 128.3 (C3′, C5′), 127.1 (C2′, C6′), 98.3 (C4a), 71.4 (C7a), 69.4 (5 CH Cp), 66.9 (C5 or C6), 65.7 (C7), 65.4 (C5 or C6), 61.7 (C3), 33.9 (C4), 28.0 (4-CH3), 24.4 (4-CH3), 23.7 (3-CH3), 23.1 (3-CH3), 20.6 (4′-CH3). MS (EI) m/z (% relative intensity): 385 [M]+ (35), 370 [M–CH3]+ (100), 342 [M–i-C3H7]+ (5), 329 [M–C4H8]+ (18), 314 [M–C4H8–CH3]+ (11), 121 [C5H5Fe]+ (15), 56 [Fe]+ (5). Analysis calculated for C24H27FeN: C, 74.81; H, 7.06; N, 3.64. Found: C, 75.01; H, 7.19; N, 3.56.
rac-3,3,4,4-Tetramethyl-1-phenyl-3,4-dihydroferroceno[c]pyridine (3b). The title compound was prepared according to GP using benzonitrile (2b) (0.043 mL, 0.42 mmol); reaction time = 30 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3b (97 mg, 75%) as a red solid; mp 103–105 °C (hexane); Rf 0.10 (petroleum ether/EtOAc 25:1); Rf 0.35 (petroleum ether/TEA 100:1). IR (thin film): 3093, 2973, 2930, 2868, 1592, 1565, 1452, 1372, 1361, 1317, 1302, 1174, 1156, 1108, 1002, 935, 897, 823, 780, 754, 725, 697, 473, 453 cm−1. 1H NMR (400 MHz, DMSO-d6, 30 °C): δ 8.06–8.03 (m, 2H), 7.55–7.49 (m, 3H), 4.49 (d, 2H, J = 1.8 Hz), 4.40 (t, 1H, J = 1.8 Hz), 4.30 (s, 5H), 1.46 (s, 3H), 1.45 (s, 3H), 1.11 (s, 3H), 0.78 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 162.7 (C), 138.4 (C), 129.2 (CH), 127.9 (2 CH), 127.3 (2 CH), 98.3 (C), 71.3 (C), 69.6 (5 CH), 67.1 (CH), 65.8 (CH), 65.6 (CH), 61.9 (C), 33.9 (C), 28.1 (CH3), 24.7 (CH3), 23.8 (CH3), 23.2 (CH3). MS (EI) m/z (% relative intensity): 371 [M]+ (47), 356 [M–CH3]+ (100), 328 [M–i-C3H7]+ (6), 315 [M–C4H8]+ (22), 300 [M–C4H8–CH3]+ (13), 121 [C5H5Fe]+ (9), 56 [Fe]+ (3). Analysis calculated for C23H25FeN: C, 74.40; H, 6.79; N, 3.77. Found: C, 74.70; H, 7.06; N, 3.77.
rac-1-(4-Methoxyphenyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3c). The title compound was prepared according to GP using 4-methoxybenzonitrile (2c) (0.056 mg, 0.42 mmol); reaction time = 40 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3c (108 mg, 77%) as an orange solid; mp 140.5–142 °C (hexane); Rf 0.02 (petroleum ether/EtOAc 25:1); Rf 0.15 (petroleum ether/TEA 100:1). IR (thin film): 3091, 2973, 2933, 2868, 2836, 1608, 1589, 1562, 1513, 1453, 1441, 1372, 1361, 1309, 1251, 1168, 1155, 1108, 1033, 1002, 933, 899, 836, 825, 751, 660, 544, 518, 478, 461 cm−1. 1H NMR (400 MHz, DMSO-d6, 60 °C): δ 8.07–8.03 (m, 2H), 7.07–7.03 (m, 2H), 4.45 (d, 2H, J = 1.9 Hz), 4.38 (t, 1H, J = 1.8 Hz), 4.27 (s, 5H), 3.86 (s, 3H), 1.45 (s, 3H), 1.43 (s, 3H), 1.12 (s, 3H), 0.71 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 161.9 (C), 160.3 (C), 130.8 (C), 128.8 (2 CH), 113.3 (2 CH), 98.3 (C), 71.4 (C), 69.5 (5 CH), 67.0 (CH), 66.0 (CH), 65.7 (CH), 61.7 (C), 55.1 (CH3), 33.9 (C), 28.5 (CH3), 24.2 (CH3), 24.1 (CH3), 23.0 (CH3). MS (EI) m/z (% relative intensity): 401 [M]+ (38), 386 [M–CH3]+ (100), 371 [M–CH2O]+ (7), 358 [M–i-C3H7 and/or M–CH3–CO]+ (6), 345 [M–C4H8]+ (23), 330 [M–C4H8–CH3]+ (15), 165 (11), 134 (11), 121 [C5H5Fe]+ (28), 56 [Fe]+ (8). Analysis calculated for C24H27FeNO: C, 71.83; H, 6.78; N, 3.49. Found: C, 71.64; H, 7.13; N, 3.44.
rac-1-(3-Methoxyphenyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3d). The title compound was prepared according to GP using 3-methoxybenzonitrile (2d) (0.051 mL, 0.42 mmol); reaction time = 40 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3d (110 mg, 78%) as an orange solid; mp 100–102 °C (hexane); Rf 0.03 (petroleum ether/EtOAc 25:1); Rf 0.20 (petroleum ether/TEA 100:1). IR (thin film): 3094, 2973, 2937, 2869, 2834, 1599, 1568, 1487, 1462, 1435, 1372, 1361, 1305, 1285, 1274, 1238, 1168, 1152, 1108, 1049, 1039, 1002, 948, 882, 824, 802, 791, 755, 742, 697, 663, 534, 516, 473, 449 cm−1. 1H NMR (400 MHz, DMSO-d6, 70 °C): δ 7.61 (dt, 1H, J = 7.6, 1.3 Hz), 7.56 (dd, 1H, J = 2.7, 1.5 Hz), 7.41 (t, 1H, J = 7.9 Hz), 7.06 (ddd, 1H, J = 8.2, 2.7, 1.0 Hz), 4.47 (d, 2H, J = 1.8 Hz), 4.39 (t, 1H, J = 1.8 Hz), 4.27 (s, 5H), 3.88 (s, 3H), 1.47 (s, 3H), 1.46 (s, 3H), 1.11 (s, 3H), 0.78 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 162.5 (C), 158.9 (C), 139.8 (C), 129.0 (CH), 119.9 (CH), 114.9 (CH), 112.7 (CH), 98.3 (C), 71.3 (C), 69.6 (5 CH), 67.1 (CH), 65.8 (CH), 65.6 (CH), 62.0 (C), 55.1 (CH3), 33.9 (C), 28.1 (CH3), 24.6 (CH3), 23.8 (CH3), 23.2 (CH3). MS (EI) m/z (% relative intensity): 401 [M]+ (51), 386 [M–CH3]+ (100), 371 [M–CH2O]+ (6), 358 [M–i-C3H7 and/or M–CH3–CO]+ (6), 345 [M–C4H8]+ (22), 330 [M–C4H8–CH3]+ (12), 121 [C5H5Fe]+ (10), 56 [Fe]+ (3). Analysis calculated for C24H27FeNO: C, 71.83; H, 6.78; N, 3.49. Found: C, 72.05; H, H 7.20; N, 3.49.
rac-1-(2-Methoxyphenyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3e). The title compound was prepared according to GP using 2-methoxybenzonitrile (2e) (0.051 mL, 0.42 mmol); reaction time = 40 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3e (88 mg, 63%) as a brown solid; mp 100–101 °C (hexane); Rf 0.05 (petroleum ether/EtOAc 25:1); Rf 0.25 (petroleum ether/TEA 100:1). IR (thin film): 3096, 2972, 2933, 2869, 2834, 1597, 1494, 1463, 1435, 1372, 1361, 1312, 1267, 1243, 1153, 1117, 1108, 1049, 1027, 1003, 939, 922, 900, 821, 751, 659, 512, 476, 455 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 7.42–7.38 (m, 1H), 7.28 (dd, 1H, J = 7.4, 1.7 Hz), 7.11 (br d, 1H, J = 8.3 Hz), 7.05 (td, 1H, J = 7.4, 1.0 Hz), 4.32 (dd, 1H, J = 2.2, 1.2 Hz), 4.29 (t, 1H, J = 2.4 Hz), 4.12 (s, 5H), 4.02 (dd, 1H, J = 2.4, 1.2 Hz), 3.79 (s, 3H), 1.50 (s, 3H), 1.47 (s, 3H), 1.01 (s, 3H), 1.00 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 162.9 (C), 156.3 (C), 129.6 (C), 129.0 (CH), 128.9 (CH), 119.9 (CH), 111.6 (CH), 97.7 (C), 73.3 (C), 69.2 (5 CH), 66.6 (CH), 64.3 (CH), 64.0 (CH), 61.4 (C), 55.5 (CH3), 34.1 (C), 26.2 (CH3), 26.2 (CH3), 24.4 (CH3), 22.7 (CH3). MS (EI) m/z (% relative intensity): 401 [M]+ (100), 386 [M–CH3]+ (61), 371 [M–CH2O]+ (11), 358 [M–i-C3H7 and/or M–CH3–CO]+ (15), 345 [M–C4H8]+ (50), 330 [M–C4H8–CH3]+ (11), 263 (10), 121 [C5H5Fe]+ (9), 56 [Fe]+ (3). Analysis calculated for C24H27FeNO∙0.15C6H14: C, 72.20; H, 7.08; N, 3.38. Found: C, 72.43; H, 7.10; N, 3.05.
rac-4-(3,3,4,4-Tetramethyl-3,4-dihydroferroceno[c]pyridin-1-yl)aniline (3f). The title compound was prepared according to GP using 4-aminobenzonitrile (2f) (49 mg, 0.42 mmol); reaction time = 50 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 5:1–petroleum ether/TEA 100:1) gave pure compound 3f (89 mg, 66%) as an orange solid; mp 167.5–169 °C (hexane); Rf 0.02 (petroleum ether/EtOAc 5:1); Rf 0.30 (petroleum ether/TEA 100:1). IR (thin film): 3464, 3433, 3378, 3321, 3204, 3094, 2973, 2931, 2868, 1621, 1609, 1584, 1556, 1518, 1454, 1372, 1361, 1315, 1296, 1171, 1155, 1107, 1002, 834, 754, 660, 504, 470 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 7.88–7.85 (m, 2H), 6.68–6.65 (m, 2H), 5.31 (br s, 2H), 4.44–4.42 (m, 2H), 4.37 (t, 1H, J = 1.8 Hz), 4.26 (s, 5H), 1.44 (s, 3H), 1.39 (s, 3H), 1.12 (s, 3H), 0.65 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 161.8 (C), 150.1 (C), 128.6 (2 CH), 126.0 (C), 112.8 (2 CH), 98.4 (C), 71.9 (C), 69.4 (5 CH), 66.6 (CH), 66.3 (CH), 65.4 (CH), 61.4 (C), 33.9 (C), 28.7 (CH3), 24.4 (CH3), 24.0 (CH3), 23.0 (CH3). MS (EI) m/z (% relative intensity): 386 [M]+ (44), 371 [M–CH3]+ (100), 330 [M–C4H8]+ (19), 315 [M–C4H8–CH3]+ (12), 121 [C5H5Fe]+ (10), 56 [Fe]+ (4). HRMS-ESI (m/z): [M + H]+ calculated for C23H27FeN2, 387.1518; found, 387.1523.
rac-2-(3,3,4,4-Tetramethyl-3,4-dihydroferroceno[c]pyridin-1-yl)aniline (3g). The title compound was prepared according to GP using 2-aminobenzonitrile (2g) (49 mg, 0.42 mmol); reaction time = 180 min. Purification by silica gel column chromatography (petroleum ether/acetone 15:1) gave pure compound 3g (40 mg, 30%) as an orange oil; Rf 0.20 (petroleum ether/acetone 15:1). IR (thin film): 3452, 3202, 3093, 2973, 2929, 2868, 1613, 1580, 1540, 1450, 1372, 1361, 1308, 1264, 1159, 1108, 1002, 936, 818, 750, 659, 493, 472, 453 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 8.25 (dd, 1H, J = 7.9, 1.6 Hz), 7.14–7.10 (m, 1H), 6.87 (br s, 2H), 6.75 (dd, 1H, J = 8.2, 1.3 Hz), 6.70–6.65 (m, 1H), 4.47 (t, 1H, J = 2.4 Hz), 4.44 (dd, 1H, J = 2.4, 1.2 Hz), 4.33 (dd, 1H, J = 2.5, 1.2 Hz), 4.29 (s, 5H), 1.46 (s, 3H), 1.43 (s, 3H), 1.13 (s, 3H), 0.75 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 165.3 (C), 148.5 (C), 130.1 (CH), 129.6 (CH), 118.0 (C), 116.0 (CH), 114.0 (CH), 98.1 (C), 71.9 (C), 69.6 (5 CH), 67.3 (CH), 67.1 (CH), 65.5 (CH), 61.6 (C), 33.2 (C), 28.2 (CH3), 24.2 (CH3), 23.8 (CH3), 23.75 (CH3). MS (EI) m/z (% relative intensity): 386 [M]+ (100), 371 [M–CH3]+ (96), 330 [M–C4H8]+ (27), 303 (10), 265 [M–C5H5Fe]+ (11), 121 [C5H5Fe]+ (16), 56 [Fe]+ (7). Analysis calculated for C23H26FeN2: C, 71.51; H, 6.78; N, 7.25. Found: C, 71.56; H, 7.17; N, 7.04.
rac-1-(4-Bromphenyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3h). The title compound was prepared according to GP using 4-bromobenzonitrile (2h) (76 mg, 0.42 mmol); reaction time = 30 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3h (129 mg, 82%) as a brown solid; mp 85–86.5 °C (hexane); Rf 0.12 (petroleum ether/EtOAc 25:1); Rf 0.40 (petroleum ether/TEA 100:1). IR (thin film): 3094, 2974, 2931, 2869, 1588, 1556, 1488, 1452, 1391, 1372, 1361, 1310, 1155, 1108, 1070, 1011, 934, 898, 824, 754, 697, 518, 506, 475, 440 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 8.00–7.99 (m, 2H), 7.68–7.65 (m, 2H), 4.43 (d, 2H, J = 1.8 Hz), 4.34 (t, 1H, J = 1.9 Hz), 4.24 (s, 5H), 1.40 (s, 6H), 1.07 (s, 3H), 0.70 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 161.6 (C), 137.4 (C), 130.8 (2 CH), 129.2 (2 CH), 122.6 (C), 98.2 (C), 70.9 (C), 69.4 (5 CH), 67.1 (CH), 65.6 (CH), 65.5 (CH), 62.0 (C), 33.8 (C), 28.0 (CH3), 24.3 (CH3), 23.6 (CH3), 22.9 (CH3). MS (EI) m/z (% relative intensity): 449 [M]+ (64), 434 [M–CH3]+ (100), 406 [M–i-C3H7]+ (4), 393 [M–C4H8]+ (23), 378 [M–C4H8–CH3]+ (3), 369 [M–HBr]+ (3), 314 (43), 299 (57), 191 (13), 121 [C5H5Fe]+ (14), 56 [Fe]+ (5). Analysis calculated for C23H24BrFeN∙0.1C6H14: C, 61.78; H, 5.58; N, 3.05. Found: C, 61.61; H, 5.92; N, 3.05.
rac-1-(3-Bromphenyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3i). The title compound was prepared according to GP using 3-bromobenzonitrile (2i) (76 mg, 0.42 mmol); reaction time = 40 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3i (125 mg, 80%) as a brown solid; mp 131–132 °C (hexane); Rf 0.03 (petroleum ether/EtOAc 25:1); Rf 0.35 (petroleum ether/TEA 100:1). IR (thin film): 3094, 2974, 2930, 2869, 1591, 1555, 1451, 1372, 1361, 1302, 1186, 1155, 1108, 1070, 999, 937, 902, 870, 824, 791, 756, 738, 709, 692, 518, 474, 453 cm−1. 1H NMR (400 MHz, CDCl3, 30 °C): δ 8.23 (t, 1H, J = 1.8 Hz), 7.92 (dt, 1H, J = 7.8, 1.2 Hz), 7.53 (ddd, 1H, J = 7.9, 2.1, 1.1 Hz), 7.29 (t, 1H, J = 7.8 Hz), 4.35–4.33 (m, 2H), 4.27 (dd, 1H, J = 2.3, 1.4 Hz), 4.23 (s, 5H), 1.49 (s, 3H), 1.46 (s, 3H), 1.10 (s, 3H), 0.81 (s, 3H). 13C NMR (100 MHz, CDCl3, 30 °C): 163.5 (C), 141.4 (C), 132.3 (CH), 131.1 (CH), 129.9 (CH), 126.3 (CH), 122.4 (C), 99.2 (C), 71.9 (C), 70.0 (5 CH), 67.4 (CH), 66.2 (CH), 66.0 (CH), 62.9 (C), 34.7 (C), 28.7 (CH3), 25.2 (CH3), 24.0 (CH3), 23.6 (CH3). MS (EI) m/z (% relative intensity): 449 [M]+ (90), 434 [M–CH3]+ (100), 406 [M–i-C3H7]+ (10), 393 [M–C4H8]+ (48), 378 [M–C4H8–CH3]+ (7), 314 (32), 299 (16), 249 (12), 234 (12), 218 (14), 191 (22), 121 [C5H5Fe]+ (25), 56 [Fe]+ (10). Analysis calculated for C23H24BrFeN: C, 61.36; H, 5.37; N, 3.11. Found: C, 61.60; H, 5.39; N, 3.04.
rac-1-(2-Bromphenyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3j). The title compound was prepared according to GP using 2-bromobenzonitrile (2j) (76 mg, 0.42 mmol); reaction time = 40 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3j (120 mg, 76%) as an orange solid; mp 110–111 °C (hexane); Rf 0.02 (petroleum ether/EtOAc 25:1); Rf 0.20 (petroleum ether/TEA 100:1). IR (thin film): 3095, 2973, 2930, 2868, 1604, 1476, 1452, 1432, 1342, 1372, 1361, 1312, 1193, 1154, 1108, 1086, 1049, 1029, 1021, 1003, 923, 861, 822, 767, 751, 742, 693, 546, 486, 468, 447 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 7.65 (dd, 1H, J = 8.0, 1.1 Hz), 7.51–7.43 (m, 2H), 7.36–7.31 (m, 1H), 4.36 (dd, 2H, J = 2.4, 1.2Hz), 4.32 (t, 1H, J = 2.4 Hz), 4.17 (s, 5H), 3.90 (dd, 1H, J = 2.4, 1.1 Hz), 1.47 (s, 3H), 1.46 (s, 3H), 1.03 (s, 3H), 0.99 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): 163.4 (C), 140.1 (C), 132.3 (CH), 129.51 (C), 129.46 (C), 127.1 (CH), 120.2 (C), 97.8 (C), 71.8 (C), 69.4 (5 CH), 67.0 (CH), 64.4 (CH), 64.1 (CH), 61.8 (C), 34.1 (C), 26.6 (CH3), 25.8 (CH3), 24.4 (CH3), 22.2 (CH3). MS (EI) m/z (% relative intensity): 449 [M]+ (100), 406 [M–i-C3H7]+ (5), 393 [M–C4H8]+ (28), 384 (13), 370 [M–Br]+ (20), 355 [M–CH3–Br]+ (22), 354 [M–CH3–HBr]+ (22), 326 (19), 312 (18), 262 (11), 248 (59), 234 (62), 218 (22), 192 (29), 178 (31), 165 (15), 152 (10), 121 [C5H5Fe]+ (18), 56 [Fe]+ (8). Analysis calculated for C23H24BrFeN∙0.2C6H14: C, 62.18; H, 5.78; N, 3.00. Found: C, 62.44; H, 5.63; N, 3.02.
rac-3,3,4,4-Tetramethyl-1-(4-(trifluoromethyl)phenyl)-3,4-dihydroferroceno[c]pyridine (3k). The title compound was prepared according to GP using 4-(trifluoromethyl)benzonitrile (2k) (71 mg, 0.42 mmol); reaction time = 15 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave pure compound 3k (117 mg, 76%) as an orange solid; mp 121.5–123 °C (hexane); Rf 0.16 (petroleum ether/EtOAc 50:1); Rf 0.30 (petroleum ether/TEA 100:1). IR (thin film): 3095, 2976, 2934, 2871, 1620, 1594, 1564, 1453, 1373, 1362, 1327, 1313, 1165, 1127, 1107, 1081, 1067, 1018, 1003, 849, 824, 758, 474, 444 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 8.24 (br d, 2H, J = 7.9 Hz), 7.87 (br d, 2H, J = 7.9 Hz), 4.50 (d, 2H, J = 1.8 Hz), 4.41 (t, 1H, J = 1.8 Hz), 4.31 (s, 5H), 1.464 (s, 3H), 1.456 (s, 3H), 1.11 (s, 3H), 0.78 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): 161.8 (C), 141.9 (C), 129.3 (q, 2JC,F = 31.8 Hz, C), 127.9 (s, 2 CH), 124.8 (q, 3JC,F = 3.8 Hz, 2 CH), 124.1 (q, 1JC,F = 271.0 Hz, CF3), 98.2 (C), 70.8 (C), 69.5 (5 CH), 67.2 (CH), 65.6 (CH), 65.4 (CH), 62.2 (C), 33.9 (C), 27.9 (CH3), 24.5 (CH3), 23.5 (CH3), 23.0 (CH3). 19F NMR (377 MHz, DMSO-d6, 50 °C): δ 101.5 (s, CF3). MS (EI) m/z (% relative intensity): 439 [M]+ (88), 424 [M–CH3]+ (60), 396 [M–i-C3H7]+ (8), 383 [M–C4H8]+ (55), 368 (16), 284 (100), 243 (11), 227 (13), 121 [C5H5Fe]+ (13), 56 [Fe]+ (3). Analysis calculated for C24H24F3FeN: C, 65.62; H, 5.51; N, 3.19. Found: C, 65.92; H, 5.78; N, 3.11.
rac-3,3,4,4-Tetramethyl-1-(3-(trifluoromethyl)phenyl)-3,4-dihydroferroceno[c]pyridine (3l). The title compound was prepared according to GP using 3-(trifluoromethyl)benzonitrile (2l) (0.056 mL, 0.42 mmol); reaction time = 30 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave pure compound 3l (129 mg, 84%) as a brown oil; Rf 0.15 (petroleum ether/EtOAc 50:1); Rf 0.30 (petroleum ether/TEA 100:1). IR (thin film): 3093, 2976, 2931, 2870, 1599, 1574, 1454, 1433, 1373, 1362, 1336, 1296, 1266, 1185, 1166, 1127, 1109, 1095, 1072, 1002, 825, 808, 702, 690, 473, 448 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 8.40–8.39 (m, 1H), 8.33–8.30 (m, 1H), 7.84–7.81 (m, 1H), 7.75–7.71 (m, 1H), 4.49–4.47 (m, 2H), 4.34 (dd, 1H, J = 2.4, 1.3 Hz), 4.27 (s, 5H), 1.419 (s, 3H), 1.415 (s, 3H), 1.10 (s, 3H), 0.70 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 161.5 (C), 138.9 (C), 131.0 (CH), 129.1 (CH), 128.8 (q, 2JC,F = 31.6 Hz, C), 125.6 (q, 3JC,F = 3.7 Hz, CH), 124.1 (q, 1JC,F = 272.3 Hz, CF3), 123.5 (q, 3JC,F = 4.2, CH), 98.3 (C), 70.8 (C), 69.4 (5 CH), 67.2 (CH), 65.8 (CH), 65.4 (CH), 62.2 (C), 33.9 (C), 28.2 (CH3), 24.1 (CH3), 23.6 (CH3), 22.7 (CH3). 19F NMR (377 MHz, DMSO-d6, 50 °C): δ 101.6 (s, CF3). MS (EI) m/z (% relative intensity): 439 [M]+ (100), 424 [M–CH3]+ (44), 396 [M–i-C3H7]+ (17), 383 [M–C4H8]+ (74), 368 [M–C4H8–CH3]+ (11), 284 (59), 121 [C5H5Fe]+ (10), 56 [Fe]+ (2). Analysis calculated for C24H24F3FeN: C, 65.62; H, 5.51; N, 3.19. Found: C, 65.99; H, 5.63; N, 3.21.
rac-3,3,4,4-Tetramethyl-1-(2-(trifluoromethyl)phenyl)-3,4-dihydroferroceno[c]pyridine (3m). The title compound was prepared according to GP using 2-(trifluoromethyl)benzonitrile (2m) (0.056 mL, 0.42 mmol); reaction time = 40 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave pure compound 3m (100 mg, 65%) as brown oil; Rf 0.04 (petroleum ether/EtOAc 50:1); Rf 0.20 (petroleum ether/TEA 100:1). IR (thin film): 3095, 2977, 2932, 2870, 1603, 1580, 1456, 1447, 1373, 1362, 1315, 1264, 1163, 1136, 1109, 1059, 1034, 1003, 823, 768, 683, 545, 449 cm−1. 1H NMR (400 MHz, DMSO-d6, 30 °C): δ 7.80–7.76 (m, 2H), 7.65–7.61 (m, 2H), 4.36 (dd, 1H, J = 2.4, 1.2 Hz), 4.33 (t, 1H, J = 2.5 Hz), 4.22 (s, 5H), 3.81 (dd, 1H, J = 2.5, 1.2 Hz), 1.46 (s, 3H), 1.45 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 162.1 (C), 138.1 (C, broad), 131.7 (CH), 129.8 (CH), 128.0 (CH), 126.3 (q, 2JC,F = 30.6 Hz, C), 126.0 (q, 3JC,F = 4.9 Hz, CH), 123.8 (q, 1JC,F = 274.3 Hz, CF3), 97.8 (C), 72.2 (C), 69.5 (5 CH), 67.2 (CH), 64.1 (CH), 63.7 (CH), 61.7 (C), 34.2 (C), 26.8 (CH3), 25.3 (CH3), 24.5 (CH3), 21.9 (CH3). 19F NMR (376 MHz, DMSO-d6, 50 °C): δ 106.7 (s, CF3). MS (EI) m/z (% relative intensity): 439 [M]+ (100), 424 [M–CH3]+ (10), 396 [M–i-C3H7]+ (19), 383 [M–C4H8]+ (19), 368 [M–C4H8–CH3]+ (2), 284 (22), 121 [C5H5Fe]+ (5), 56 [Fe]+ (1). HRMS-ESI (m/z): [M + H]+ calculated for C24H25F3FeN, 440.1283; found, 440.1291.
rac-1,3,3,4,4-Pentamethyl-3,4-dihydroferroceno[c]pyridine (3o). The title compound was prepared according to GP using acetonitrile (2o) (0.02 mL, 0.42 mmol); reaction time = 15 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3o (82 mg, 76%) as a red oil; Rf 0.07 (petroleum ether/EtOAc 25:1); Rf 0.25 (petroleum ether/TEA 100:1). IR (thin film): 3094, 2972, 2868, 1619, 1465, 1438, 1387, 1373, 1361, 1301, 1164, 1135, 1108, 1027, 1001, 854, 821, 658, 511, 487, 475, 458 cm−1. 1H NMR (400 MHz, DMSO-d6, 30 °C): δ 4.46 (t, 1H, J = 1.8 Hz), 4.32 (d, 2H, J = 1.8 Hz), 4.23 (s, 5H), 2.18 (s, 3H), 1.40 (s, 3H), 1.31 (s, 3H), 1.00 (s, 3H), 0.72 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 162.5 (C), 97.8 (C), 73.5 (C), 69.2 (5 CH), 66.3 (CH), 65.1 (CH), 63.5 (CH), 61.2 (C), 33.9 (C), 28.2 (CH3), 25.1 (CH3), 23.8 (CH3), 23.6 (CH3), 22.7 (CH3). MS (EI) m/z (% relative intensity): 309 [M]+ (100), 294 [M–CH3]+ (24), 266 [M–i-C3H7]+ (52), 251 [M–i-C3H7–CH3]+ (11), 226 (12), 186 [C5H5FeC5H5]+ (13), 162 (14), 146 (11), 129 (10), 121 [C5H5Fe]+ (44), 115 (20), 91 (10), 56 [Fe]+ (19), 42 (12). Analysis calculated for C18H23FeN: C, 69.91; H, 7.88; N, 4.53. Found: C, 69.55; H, 7.89; N, 4.49.
rac-1-Ethyl-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3p). The title compound was prepared according to GP using propiononitrile (2p) (0.03 mL, 0.42 mmol); reaction time = 20 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave pure compound 3p (90 mg, 80%) as a red oil; Rf 0.05 (petroleum ether/EtOAc 50:1); Rf 0.10 (petroleum ether/TEA 100:1). IR (thin film): 3095, 2972, 2932, 2870, 1615, 1465, 1442, 1387, 1360, 1287, 1248, 1194, 1158, 1128, 1108, 1028, 1002, 922, 870, 821, 511, 482 cm−1. 1H NMR (400 MHz, CDCl3, 30 °C): δ 4.33 (dd, 1H, J = 2.4, 1.1 Hz), 4.25 (t, 1H, J = 2.4 Hz), 4.20 (dd, 1H, J = 2.4, 1.1 Hz), 4.15 (s, 5H), 2.50 (qt, 2H, J = 7.6, 3.6 Hz), 1.40 (s, 6H), 1.25 (t, 3H, J = 7.6 Hz), 0.97 (s, 3H), 0.89 (s, 3H). 13C NMR (100 MHz, CDCl3, 30 °C): δ 168.0 (C), 99.0 (C), 73.1 (C), 69.7 (5 CH), 66.9 (CH), 65.0 (CH), 63.7 (CH), 61.4 (C), 34.7 (C), 30.6 (CH2), 27.7 (CH3), 26.3 (CH3), 24.5 (CH3), 23.6 (CH3), 13.1 (CH3). MS (EI) m/z (% relative intensity): 323 [M]+ (100), 308 [M–CH3]+ (18), 280 [M–i-C3H7]+ (34), 267 [M–C4H8]+ (18), 265 [M–i-C3H7–CH3]+ (13), 121 [C5H5Fe]+ (20), 56 [Fe]+ (9). HRMS-ESI (m/z): [M + H]+ calculated for C19H26FeN, 324.1409; found, 324.1411.
rac-1-Isobutyl-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3q). The title compound was prepared according to GP using isovaleronitrile (2q) (0.044 mL, 0.42 mmol); reaction time = 15 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave pure compound 3q (92 mg, 75%) as a red oil; Rf 0.07 (petroleum ether/EtOAc 25:1); Rf 0.30 (petroleum ether/TEA 100:1). IR (thin film): 3097, 2969, 2930, 2868, 1613, 1464, 1386, 1372, 1361, 1304, 1259, 1245, 1224, 1194, 1159, 1144, 1135, 1108, 1028, 1002, 862, 821, 754, 510, 447 cm−1. 1H NMR (400 MHz, C6D6, 50 °C): δ 4.06 (dd, 1H, J = 2.5, 1.1 Hz), 4.00 (s, 6H), 3.96 (t, 1H, J = 2.4 Hz), 2.49–2.32 (m, 3H), 1.52 (s, 3H), 1.37 (s, 3H), 1.09 (d, 3H, J = 6.2 Hz), 1.06 (d, 3H, J = 6.3 Hz,), 1.01 (s, 3H), 0.97 (s, 3H). 13C NMR (100 MHz, C6D6, 50 °C): δ 164.3 (C), 99.2 (C), 74.8 (C), 69.8 (5 CH), 66.8 (CH), 65.0 (CH), 63.6 (CH), 61.9 (C), 46.0 (CH2), 34.9 (C), 28.0 (CH3), 26.8 (CH), 26.3 (CH3), 24.8 (CH3), 24.0 (CH3), 23.2 (CH3), 23.0 (CH3). MS (EI) m/z (% relative intensity): 351 [M]+ (100), 336 [M–CH3]+ (45), 308 [M–i-C3H7]+ (40), 251 (19), 121 [C5H5Fe]+ (47), 56 [Fe]+ (12). Analysis calculated for C21H29FeN: C, 71.80; H, 8.32; N, 3.99. Found: C, 71.82; H, 8.73; N, 3.80.
rac-1-(Adamantan-1-yl)methyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3r). The title compound was prepared according to GP using 1-adamantaneacetonitrile (2r) (61 mg, 0.42 mmol); reaction time = 25 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave pure compound 3r (113 mg, 73%) as a red solid; mp 137–139.5 °C (MeOH); Rf 0.10 (petroleum ether/EtOAc 50:1); Rf 0.40 (petroleum ether/TEA 100:1). IR (thin film): 3096, 2970, 2902, 2846, 1460, 1387, 1372, 1360, 1314, 1293, 1262, 1208, 1150, 1121, 1108, 1029, 1002, 821, 753, 660, 508, 448 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.11 (dd, 1H, J = 2.5, 1.2 Hz), 4.01 (s, 5H), 3.99 (dd, 1H, J = 2.4, 1.1 Hz), 3.96 (t, 1H, J = 2.4 Hz), 2.41 (d, 1H, J = 13.0 Hz), 2.35 (d, 1H, J = 13.0 Hz), 2.00–1.90 (m, 6H), 1.76–1.70 (m, 9H), 1.55 (s, 3H), 1.36 (s, 3H), 1.11 (s, 3H), 0.97 (s, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 163.5 (C), 99.3 (C), 76.0 (C), 69.9 (5 CH), 66.9 (CH), 64.70 (CH), 64.68 (CH), 61.9 (C), 50.5 (CH2), 43.8 (3 CH2), 37.6 (3 CH2), 34.7 (C), 34.4 (C), 29.5 (3 CH), 27.6 (CH3), 26.8 (CH3), 25.5 (CH3), 23.8 (CH3). MS (EI) m/z (% relative intensity): 443 [M]+ (100), 428 [M–CH3]+ (11), 400 [M–i-C3H7]+ (33), 385 [M–i-C3H7–CH3]+ (12), 359 (44), 308 [M–Ad]+ (12), 251 (19), 135 [Ad]+ (14), 121 [C5H5Fe]+ (33), 56 [Fe]+ (6). Analysis calculated for C28H37FeN∙0.55MeOH: C, 74.37; H, 8.57; N, 3.04. Found: C, 74.73; H, 8.95; N, 3.00.
rac-1-(Adamantan-1-yl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (3s). The title compound was prepared according to GP using adamantane-1-carbonitrile (2s) (68 mg, 0.42 mmol); reaction time = 20 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 75:1–petroleum ether/TEA 100:1) gave pure compound 3s (107 mg, 71%) as an orange oil; Rf 0.10 (petroleum ether/EtOAc 75:1); Rf 0.40 (petroleum ether/TEA 100:1). IR (thin film): 3096, 2970, 2927, 2903, 2848, 1597, 1450, 1370, 1359, 1292, 1250, 1162, 1108, 1002, 820, 758, 475, 456 cm−1. 1H NMR (400 MHz, C6D6, 50 °C): δ 4.34 (dd, 1H, J = 2.5, 1.1 Hz), 4.04 (t, 1H, J = 2.5 Hz), 4.02 (s, 5H), 4.00 (dd, 1H, J = 2.5, 1.1 Hz), 2.22–2.11 (m, 6H), 2.09–2.06 (m, 3H), 1.79 (br t, 6H, J = 3.2 Hz), 1.51 (s, 3H), 1.34 (s, 3H), 1.05 (s, 3H), 0.94 (s, 3H). 13C NMR (100 MHz, C6D6, 50 °C): δ 169.3 (C), 100.2 (C), 70.8 (C), 70.1 (5 CH), 67.2 (CH), 66.4 (CH), 63.9 (CH), 60.9 (C), 42.2 (C), 41.8 (3 CH2), 37.7 (3 CH2), 34.6 (C), 29.6 (3 CH), 26.7 (CH3), 26.4 (CH3), 25.1 (CH3), 23.6 (CH3). MS (EI) m/z (% relative intensity): 429 [M]+ (100), 414 [M–CH3]+ (14), 386 [M–C3H7]+ (17), 373 [M–C4H8]+ (37), 294 [M–Ad]+ (11), 121 [C5H5Fe]+ (11), 56 [Fe]+ (2). Analysis calculated for C27H35FeN: C, 75.52; H, 8.22; N, 3.26. Found: C, 74.98; H, 8.49; N, 3.04.
rac-Ethyl (Z)-2-(3,3,4,4-Tetramethyl-3,4-dihydroferroceno[c]pyridin-1(2H)-ylidene)acetate (3u). The title compound was prepared according to GP using ethyl cyanoacetate 2u (0.045 mL, 0.42 mmol); reaction time = 20 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1) gave pure product 3u (79 mg, 59%) as an orange oil; Rf 0.30 (petroleum ether/EtOAc 25:1). IR (thin film): 3288, 3096, 2975, 2930, 2870, 1646, 1602, 1499, 1465, 1442, 1366, 1295, 1187, 1154, 1108, 1072, 1044, 1002, 822, 784, 756, 629, 470 cm−1. 1H NMR (400 MHz, CDCl3, 30 °C): δ 8.23 (br d, 1H, NH), 4.84 (s, 1H, H2), 4.53 (dd, 1H, J = 2.5, 1.2 Hz, H7′), 4.27 (t, 1H, J = 2.5 Hz, H6′), 4.21 (dd, 1H, J = 2.5, 1.2 Hz, H5′), 4.18–4.09 (m, 2H, OCH2CH3, partially overlapped), 4.14 (s, 5H, H Cp), 1.55 (s, 3H, 4′-CH3), 1.37 (s, 3H, 3′-CH3), 1.30 (t, 3H, J = 7.1 Hz, OCH2CH3), 1.06 (s, 3H, 4′-CH3), 0.95 (s, 3H, 3′-CH3). 13C NMR (100 MHz, CDCl3, 30 °C): δ 170.9 (C1), 160.7 (C1′), 98.3 (C4a’), 78.6 (C2), 74.1 (C7a’), 70.5 (5 CH, Cp), 67.3 (C6′), 66.0 (C5′), 64.2 (C7′), 58.4 (OCH2CH3), 57.5 (C3′), 36.4 (C4′), 28.9 (4′-CH3), 25.8 (3′-CH3), 25.1 (4′-CH3), 23.8 (3′-CH3), 14.9 (OCH2CH3). GC–MS analyses of product 3u indicated the presence of a peak in the chromatogram that, based on its MS spectra, belonged to compound 3o. The latter was likely produced by the thermolysis of the parent compound in the GC instrument injector. HRMS-ESI (m/z): [M + H]+ calculated for C21H28FeNO2, 382.1464; found, 382.1458.
rac-(Z)-2-(3,3,4,4-Tetramethyl-3,4-dihydroferroceno[c]pyridin-1(2H)-ylidene)acetamide (3v). The title compound was prepared according to GP using cyanoacetamide 2v (35 mg, 0.42 mmol); reaction time = 30 min. Purification by silica gel column chromatography (petroleum ether/acetone 3:1) followed by recrystallization from hexane gave pure product 3v (42 mg, 34%) as an orange solid, m.p. 147–149 °C (hexane); Rf 0.30 (petroleum ether/acetone 3:1). IR (thin film): 3440, 3326, 3213, 3098, 2972, 2927, 2870, 2856, 1684, 1621, 1572, 1457, 1364, 1337, 1151, 1108, 1002, 823, 756, 666, 507, 471 cm−1. 1H NMR (400 MHz, CDCl3,30 °C): δ 8.80 (br s, 1H, NH), 4.74 (s, 1H, H2), 4.67 (br s, 2H, NH2), 4.47 (dd, 1H, J = 2.5, 1.2 Hz, H7′), 4.24 (t, 1H, J = 2.5 Hz, H6′), 4.20 (dd, 1H, J = 2.5, 1.2 Hz, H5′), 4.14 (s, 5H, H Cp), 1.55 (s, 3H, 4′-CH3), 1.36 (s, 3H, 3′-CH3), 1.05 (s, 3H, 4′-CH3), 0.94 (s, 3H, 3′-CH3). 13C NMR (100 MHz, CDCl3, 30 °C): δ 172.9 (C1), 158.9 (C1′), 98.5 (C4a’), 80.0 (C2), 74.4 (C7a’), 70.4 (5 CH, Cp), 66.9 (C6′), 65.9 (C5′), 63.6 (C7′), 57.1 (C3′), 36.3 (C4′), 28.8 (4′-CH3), 25.8 (3′-CH3), 25.1 (4′-CH3), 23.8 (3′-CH3). GC–MS analyses of product 3v indicated the presence of a peak in the chromatogram that, based on its MS spectra, belonged to compound 3o. The latter was likely produced by the thermolysis of the parent compound in the GC instrument injector. HRMS-ESI (m/z): [M + H]+ calculated for C19H25FeN2O, 353.1311; found, 353.1307.
rac-(Z)-1-Phenyl-2-(3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridin-1(2H)-ylidene)ethan-1-one (3w). The title compound was prepared according to GP using 3-oxo-3-phenylpropanenitrile 2w (61 mg, 0.42 mmol); reaction time = 20 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 10:1) gave pure compound 3w (126 mg, 87%) as a red oil; Rf 0.30 (petroleum ether/EtOAc 10:1). IR (thin film): 2975, 2929, 1594, 1580, 1543, 1451, 1438, 1366, 1324, 1304, 1265, 1229, 1153, 1108, 1055, 1026, 1002, 824, 755, 732, 711, 620, 512, 487, 461 cm−1. 1H NMR (400 MHz, CDCl3, 30 °C): δ 10.99 (br d, 1H, NH), 7.95–7.91 (m, 2H, H2′’, H6′’), 7.44–7.41 (m, 3H, H3′’, H4′’, H5′’), 5.98 (s, 1H, H2), 4.69 (dd, 1H, J = 2.6, 1.2 Hz, H7′), 4.38 (t, 1H, J = 2.5 Hz, H6′), 4.31 (dd, 1H, J = 2.5, 1.2 Hz, H5′), 4.18 (s, 5H, H Cp), 1.57 (s, 3H, 4′-CH3), 1.46 (s, 3H, 3′-CH3), 1.10 (s, 3H, 4′-CH3), 1.03 (s, 3H, 3′-CH3). 13C NMR (100 MHz, CDCl3, 30 °C): δ 187.2 (C1), 163.2 (C1′), 141.2 (C1′’), 130.4 (C4′’), 128.3 (C3′’, C5′’), 127.0 (C2′’, C6′’), 98.3 (C4a’), 88.0 (C2), 73.5 (C7a’), 70.7 (5 CH, Cp), 67.9 (C6′), 66.6 (C5′), 64.8 (C7′), 57.9 (C3′), 36.3 (C4′), 28.8 (4′-CH3), 25.7 (3′-CH3), 25.0 (4′-CH3), 23.5 (3′-CH3). MS (EI) m/z (% relative intensity): 413 [M]+ (100), 370 [M–i-C3H7]+ (19), 348 (42), 308 [M–C(O)C6H5]+ (12), 121 [C5H5Fe]+ (9), 56 [Fe]+ (2). HRMS-ESI (m/z): [M + H]+ calculated for C25H28FeNO, 414.1420; found, 414.1515.
rac-(Z)-1-(3,3,4,4-Tetramethyl-3,4-dihydroferroceno[c]pyridin-1(2H)-ylidene)propan-2-one (3x). The title compound was prepared according to GP using 3-oxobutyronitrile 2x (0.036 mL, 0.42 mmol); reaction time = 20 min. Purification by silica gel column chromatography (petroleum ether/EtOAc 25:1) gave pure compound 3x (99 mg, 81%) as a red oil, which solidified on long-term standing; mp 158.5–162 °C; Rf 0.20 (petroleum ether/EtOAc 25:1). IR (thin film): 3094, 2974, 2927, 2870, 1603, 1566, 1509, 1449, 1365, 1354, 1317, 1262, 1201, 1153, 1108, 1005, 961, 823, 757, 715, 508, 469 cm−1. 1H NMR (400 MHz, CDCl3, 30 °C): δ 10.43 (br s, 1H, NH), 5.28 (s, 1H, H2), 4.55 (dd, 1H, J = 2.5, 1.2 Hz, H7′), 4.32 (t, 1H, J = 2.5 Hz, H6′), 4.25 (dd, 1H, J = 2.4, 1.1 Hz, H5′), 4.15 (s, 5H, H Cp), 2.07 (s, 3H, 1-CH3), 1.53 (s, 3H, 4′-CH3), 1.39 (s, 3H, 3′-CH3), 1.06 (s, 3H, 4′-CH3), 0.97 (s, 3H, 3′-CH3). 13C NMR (100 MHz, CDCl3, 30 °C): δ 194.3 (C1), 161.2 (C1′), 98.4 (C4a’), 91.1(C2), 73.2 (C7a’), 70.6 (5 CH, Cp), 67.6 (C6′), 66.4 (C5′), 64.5 (C7′), 57.5 (C3′), 36.2 (C4′), 29.2 (1-CH3), 28.7 (4′-CH3), 25.6 (3′-CH3), 25.1 (4′-CH3), 23.6 (3′-CH3). MS (EI) m/z (% relative intensity): 351 [M]+ (100), 308 [M–C(O)CH3) or/and i-C3H7]+ (27), 286 (61), 242 (10), 228 (10), 121 [C5H5Fe]+ (9), 56 [Fe]+ (2). HRMS-ESI (m/z): [M + H]+ calculated for C20H26FeNO, 352.1358; found, 352.1354.
3.2.4. Synthesis of Ferroceno[c]pyridines 8a–c and 9a–c (Table 3)
General Procedure 1 (GP1). Phenylacetonitrile 7a–c (0.42 mmol) was added to the stirred solution of alcohol 1 (100 mg, 0.35 mmol) in MeSO3H (0.18 mL) at room temperature, and the resulting mixture was heated at 60 °C in an oil bath with vigorous stirring for the indicated time (monitored by TLC). The reaction mixture was then cooled to room temperature, neutralized with 10% aq. Na2CO3 solution (3 mL), and extracted with EtOAc (10 mL × 4). The combined organic phases were washed with water, dried over anhydrous Na2SO4, and filtered. Method A: Dried organic extract was concentrated in vacuo, and the crude residue was purified by silica gel column chromatography to obtain a mixture of compounds 8a–c and 9a–c; the ratios of 8a–c and 9a–c were determined by NMR 1H immediately after the isolation of the mixed fraction. Storage of this mixture at room temperature exposed to air until full conversion of 8a–c to 9a–c (monitored by TLC, reaction time 1), followed by purification by silica gel column chromatography, afforded pure compound 9a–c. Method B: Dried organic extract was stored at room temperature exposed to air until full conversion of 8a–c to 9a–c (monitored by TLC, reaction time 1) and then concentrated in vacuo. The crude residue was purified by silica gel column chromatography to afford pure compound 9a–c.
rac-1-(3,4-Dimethoxybenzyl)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (8a) and rac-(3,4-Dimethoxyphenyl)(3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine)methanone (9a). The title compounds were prepared according to GP1 using 3,4-dimethoxyphenylacetonitrile (7a) (74 mg, 0.42 mmol); reaction time = 10 min; reaction time 1 = 1 day. Method A: Purification by silica gel column chromatography (petroleum ether/CH2Cl2 3:1–petroleum ether/EtOAc/Et3N 100:5:3) gave an inseparable mixture of 7a, 8a and 9a (108 mg, 7a/8a/9a = 31:54:15; 8a/9a = 78:22). Storage of the mixture of 7a, 8a and 9a at room temperature exposed to air until full conversion of 8a to 9a, followed by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 25:1), afforded pure compound 9a (39 mg, 24%). Method B: Purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 25:1) gave pure 9a (33 mg, 21%). Data for 8a [spectroscopic data for 8a were obtained only from the mixture of 7a, 8a and 9a]: Rf 0.15 (petroleum ether/EtOAc 5:1). 1H NMR (DMSO-d6, 400 MHz, 30 °C): δ 7.04 (d, 1H, J = 1.8 Hz), 7.01–6.94 (m, 2H, overlapped), 4.32–4.24 (m, 3H), 4.00 (s, 5H), 3.78 (s, 3H, overlapped), 3.76 (s, 3H), 3.71 (d, 1H, J = 13.5 Hz), 3.66 (d, 1H, J = 13.5 Hz), 1.42 (s, 3H), 1.40 (s, 3H), 0.96 (s, 3H), 0.93 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): 148.5 (C), 147.4 (C), 131.2 (C), 120.7 (CH), 112.2 (CH), 111.9 (CH), 98.0 (C), 72.0 (C, broad), 69.3 (5 CH), 66.8 (CH, broad), 64.2 (CH, broad), 63.8 (CH, broad), 61.2 (C), 55.5 (CH3O), 55.3 (CH3O), 42.5 (CH2), 34.0 (C), 26.5 (CH3), 26.3 (CH3), 24.6 (CH3), 22.9 (CH3); one quaternary carbon peak is missing, probably due to the broadening of the signal. MS (EI) m/z (% relative intensity): 445 [M]+ (100), 402 [M–i-C3H7]+ (13), 364 (10), 294 [M–(CH3O)2C6H3CH2]+ (17), 266 (15), 121 [C5H5Fe]+ (9), 56 (2). Data for 9a: red oil; Rf 0.27 (petroleum ether/EtOAc 5:1). IR (thin film): 3085, 2975, 2936, 1658, 1593, 1514, 1463, 1417, 1321, 1274, 1260, 1242, 1209, 1175, 1138, 1117, 1025, 1003, 823, 757, 666, 448 cm−1. 1H NMR(DMSO-d6, 400 MHz, 30 °C): δ 7.82 (dd, 1H, J = 8.4, 2.0 Hz), 7.70 (d, 1H, J = 1.9 Hz), 7.22 (d, 1H, J = 8.5 Hz), 4.51–4.50 (m, 2H), 4.45 (t, 1H, J = 2.4 Hz), 4.17 (s, 5H), 3.92 (s, 3H), 3.88 (s, 3H), 1.54 (s, 3H), 1.47 (s, 3H), 1.10 (s, 3H), 0.89 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 30 °C): δ 189.9 (C), 163.4 (C), 153.7 (C), 148.5 (C), 127.5 (C), 125.5 (CH), 111.8 (CH), 111.0 (CH), 98.0 (C), 69.5 (5 CH), 67.8 (CH), 65.9 (CH), 64.6 (CH), 62.3 (C), 55.8 (CH3O), 55.4 (CH3O), 34.3 (C), 28.3 (CH3), 25.4 (CH3), 23.2 (CH3). MS (EI) m/z (% relative intensity): 459 [M]+ (100), 444 [M–CH3]+ (13), 403 [M–C4H8]+ (40), 321 [M–(CH3O)2C6H4]+ (13), 306 [M–(CH3O)2C6H4–CH3]+ (19), 294 [M–(CH3O)2C6H3C(O)]+ (21), 237 (17), 165 [(CH3O)2C6H3C(O)]+ (22), 121 [C5H5Fe]+ (16), 56 [Fe]+ (3). Analysis calculated for C26H29FeNO3: C, 67.98; H, 6.36; N, 3.05. Found: C, 68.20; H, 6.80; N, 3.15.
rac-1-Benzyl-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (8b) and rac-Phenyl(3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine)methanone (9b). The title compounds were prepared according to GP1 using phenylacetonitrile (7b) (0.48 mL, 0.42 mmol); reaction time = 15 min; reaction time 1 = 2 days. Method A: Purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave, in order of elution, 9b (20 mg, 14%) and an inseparable mixture of 8b and 9b (56 mg, 8b/9b = 89:11); calculated 8b/9b = 66:34. Storage of the mixture of 8b and 9b at room temperature exposed to air until full conversion of 8b to 9b, followed by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 25:1), afforded pure compound 9b (29 mg, 21%). Overall yield of 9b: 49 mg, 35%. Method B: Purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 25:1) gave pure 9b (33 mg, 24%). Data for 8b [spectroscopic data for 8b were obtained only from the mixture of 8b and 9b]: Rf 0.10 (petroleum ether/EtOAc 25:1). 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 7.47–7.43 (m, 2H), 7.40–7.36 (m, 2H), 7.27–7.24 (m, 1H), 4.27–4.25 (m, 3H), 3.97 (s, 5H), 3.79 (d, 1H, J = 13.8 Hz), 3.75 (d, 1H, J = 13.8 Hz), 1.42 (s, 3H), 1.41 (s, 3H), 0.93 (s, 3H), 0.91 (s, 3H). The 13C spectrum of mixture 8b and 9b was complex, including broadening and overlapping of some signals, and it was impossible to clearly assign the peaks corresponding to 8b. MS (EI) m/z (% relative intensity): 385 [M]+ (100), 342 [M–i-C3H7]+ (14), 329 [M–C4H8]+ (10), 304 (11), 121 [C5H5Fe]+ (16), 56 [Fe]+ (5). Data for 9b: red solid; mp 120.5–122 °C (hexane); Rf 0.20 (petroleum ether/EtOAc 25:1). IR (thin film): 3086, 2975, 2930, 2868, 1669, 1595, 1458, 1448, 1374, 1362, 1324, 1274, 1225, 1154, 1122, 1108, 1002, 876, 830, 737, 692, 493, 451 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 8.14–8.12 (m, 2H), 7.75–7.71 (m, 1H), 7.65–7.61 (m, 2H), 4.55 (dd, 1H, J = 2.4, 1.2 Hz), 4.49 (dd, 1H, J = 2.4, 1.2 Hz), 4.46 (t, 1H, J = 2.4 Hz), 4.18 (s, 5H), 1.55 (s, 3H), 1.48 (s, 3H), 1.09 (s, 3H), 0.91 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 191.2 (C), 163.1 (C), 135.0 (C), 133.3 (CH), 129.8 (2 CH), 128.3 (2 CH), 97.9 (C), 69.3 (5 CH), 67.7 (CH), 65.7 (CH), 64.5 (CH), 62.4 (C), 34.2 (C), 28.0 (CH3), 25.3 (CH3), 23.1 (CH3), 22.9 (CH3). MS (EI) m/z (% relative intensity): 399 [M]+ (100), 384 [M–CH3]+ (13), 343 [M–C4H8]+ (33), 294 [M–CH2C6H5]+ (11), 246 (18), 237 (13), 121 [C5H5Fe]+ (19), 56 [Fe]+ (6). HRMS-ESI (m/z): [M + H]+ calculated for C24H26FeNO, 400.1358; found, 400.1363.
rac-3,3,4,4-Tetramethyl-1-(4-nitrobenzyl)-3,4-dihydroferroceno[c]pyridine (8c) and rac-(4-Nitrophenyl)(3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine)methanone (9c). The title compounds were prepared according to GP1 using 2-(4-nitrophenyl)acetonitrile (7c) (68 mg, 0.42 mmol); reaction time = 15 min; reaction time 1 = 6 days. Method A: Purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave an inseparable mixture of 8c and 9c (41 mg, 8c/9c = 65:35; contaminated with unidentified compounds). Storage of the mixture of 8c and 9c at room temperature exposed to air until full conversion of 8c to 9c, followed by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1), afforded pure compound 9c (25 mg, 16%). Method B: Purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1) gave pure compound 9c (21 mg, 14%). Data for 8c [spectroscopic data for 8c were obtained only from the mixture of 8c and 9c, contaminated with identified compounds]: Rf 0.13 (petroleum ether/EtOAc 25:1). 1H NMR (400 MHz, C6D6, 30 °C): 7.90–7.88 (m, 2H), 7.14–7.11 (m, 2H), 3.93 (dd, 1H, J = 2.5, 1.1 Hz), 3.89 (t, 1H, J = 2.4 Hz), 3.79 (dd, 1H, J = 2.5, 1.1 Hz), 3.77 (s, 5H), 3.63 (d, 1H, J = 14 Hz), 3.57 (d, 1H, J = 14.1 Hz). A great signal overlapping was observed in the region of 0.73–1.16 ppm, and it was impossible to clearly assign the peaks of methyl groups corresponding to 8c. 13C spectrum of mixture 8c and 9c was complex, including overlapping of the some signals, and it was impossible to clearly assign the peaks corresponding to 8c. MS (EI) m/z (% relative intensity): 430 [M]+ (100), 400 [M–C2H6]+ (13), 387 [M–i-C3H7]+ (11), 374 [M–C4H8]+ (10), 294 (14), 263 (10), 237 (10), 121 [C5H5Fe]+ (10), 56 [Fe]+ (3). Data for 9c: brown oil; Rf 0.25 (petroleum ether/EtOAc 25:1). IR (thin film): 3105, 3047, 2978, 2926, 2867, 2850, 1677, 1601, 1588, 1527, 1457, 1374, 1362, 1347, 1318, 1273, 1217, 1155, 1122, 1108, 1002, 989, 882, 857, 824, 737, 711, 697, 507, 483, 447 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 8.04–8.01 (m, 2H), 7.77–7.74 (m, 2H), 4.92 (dd, 1H, J = 2.4, 1.2 Hz), 4.08 (t, 1H, J = 2.4 Hz), 4.05 (dd, 1H, J = 2.5, 1.2 Hz), 4.00 (s, 5H), 1.44 (s, 3H), 1.35 (s, 3H), 0.95 (s, 3H), 0.89 (s, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 189.9 (C), 163.7 (C), 150.4 (C), 141.1 (C), 131.6 (2 CH), 123.2 (2 CH), 98.7 (C), 70.2 (5 CH), 68.6 (CH), 66.6 (CH), 66.2 (CH), 63.9 (C), 35.0 (C), 29.0 (CH3), 25.7 (CH3), 23.4 (CH3), 23.4 (CH3). MS (EI) m/z (% relative intensity): 444 [M]+ (100), 429 [M–CH3]+ (14), 414 [M–C2H6]+ (10), 388 [M–C4H8]+ (41), 306 (10), 294 (15), 277 (13), 237 (17), 121 [C5H5Fe]+ (19), 56 [Fe]+ (4). HRMS-ESI (m/z): [M + H]+ calculated for C24H25FeN2O3, 445.1209; found, 445.1208.
3.2.5. Synthesis of Ferroceno[c]pyridines 11a–g and Ferroceno[c]pyrroles 12a–g (Table 5 and Table 6)
General Procedure 2 (GP2). Thiocyanate 10a–g (0.42 mmol) was added to the stirred solution of alcohol 1 (100 mg, 0.35 mmol) in MeSO3H (0.18 mL) at room temperature, and the resulting mixture was stirred at this temperature for the indicated time (monitored by TLC). The reaction mixture was then worked up as described in GP.
General Procedure 3 (GP3). Thiocyanate 10a–g (0.42 mmol) was added to the solution of 1 in MeSO3H, and preheated to 80 °C with stirring. The resulting mixture was then stirred at this temperature for 3 min, cooled to room temperature, and worked up as described in GP.
rac-1-(Ethylthio)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (11a) and (1R*,Sp*)-1-(tert-Butyl)-3-(ethylthio)-1-methyl-1H-ferroceno[c]pyrrole (12a). The title compounds were prepared according to GP2 (reaction time = 25 min) and GP3 using ethyl thiocyanate (10a) (0.037 mL, 0.42 mmol). GP2: purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave, in order of elution, a mixture of 11a and 12a (6 mg, 5%, 11a/12a = 19:81) and 12a (108 mg, 87%). GP3: purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 100:1–petroleum ether/TEA 100:1) gave, in order of elution, 11a (70 mg, 57%) and a mixture of 11a and 12a (36 mg, 29%, 11a/12a = 22:78). Data for 11a: brown solid; mp 81–83 °C (hexane); Rf 0.61 (petroleum ether/EtOAc 25:1). IR (thin film): 3096, 2971, 2927, 2868, 1580, 1448, 1372, 1361, 1287, 1239, 1151, 1141, 1108, 1097, 1002, 932, 899, 822, 512, 478 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.41 (dd, 1H, J = 2.4, 1.1 Hz, H7), 4.10 (s, 5H, H Cp), 3.99 (dd, 1H, J = 2.4, 1.1 Hz, H5), 3.92 (t, 1H, J = 2.4 Hz, H6), 3.22–3.12 (m, 1H, SCH2CH3), 3.11–3.02 (m, 1H, SCH2CH3), 1.50 (s, 3H, 3-CH3), 1.40 (s, 3H, 4-CH3), 1.28 (t, 3H, J = 7.3 Hz, SCH2CH3), 0.98 (s, 3H, 4-CH3), 0.88 (s, 3H, 3-CH3). 13C NMR (100 MHz, C6D6, 30 °C): δ 161.3 (C1), 99.1 (C4a), 75.0 (C7a), 70.5 (5 CH Cp), 66.5 (C6), 65.7 (C5), 64.3 (C3), 63.9 (C7), 35.7 (C4), 29.2 (4-CH3), 25.2 (4-CH3), 24.8 (3-CH3), 24.2 (3-CH3), 22.9 (SCH2CH3), 15.1 (SCH2CH3). MS (EI) m/z (% relative intensity): 355 [M]+ (100), 340 [M–CH3]+ (1), 326 [M–C2H5]+ (34), 293 (17), 269 (75), 172 (11), 121 [C5H5Fe]+ (12), 117 (22), 56 [Fe]+ (4). Analysis calculated for C19H25FeNS: C, 64.23; H, 7.09; N, 3.94; S, 9.02. Found: C, 64.04; H, 7.36; N, 3.83; S, 8.74. Data for 12a: brown solid; mp 79.5–81.5 °C (hexane); Rf 0.47 (petroleum ether/EtOAc 25:1). IR (thin film): 3096, 2969, 2936, 2908, 2871, 1522, 1451, 1391, 1366, 1299, 1149, 1141, 1106, 1003, 887, 821, 648, 508, 488 cm−1. 1H NMR (400 MHz, C6D6, 50 °C): δ 4.08 (dd, 1H, J = 2.3, 0.6 Hz, H4), 4.05 (dd, 1H, J = 2.1, 0.6 Hz, H6), 4.04 (s, 5H, H Cp), 3.87 (t, 1H, J = 2.3 Hz, H5), 3.25–3.16 (m, 1H, SCH2CH3), 3.07–2.98 (m, 1H, SCH2CH3), 1.76 (s, 3H, 1-CH3), 1.30 (t, 3H, J = 7.3 Hz, SCH2CH3), 0.90 (s, 9H, C(CH3)3). 13C NMR (100 MHz, C6D6, 50 °C): δ 165.8 (C3), 110.1 (C6a), 90.8 (C3a), 78.5 (C1), 71.0 (C5), 69.8 (5 CH Cp), 62.8 (C6), 57.0 (C4), 37.8 (C(CH3)3), 26.1 (C(CH3)3), 24.5 (SCH2CH3), 23.1 (1-CH3), 15.2 (SCH2CH3). MS (EI) m/z (% relative intensity): 355 [M]+ (23), 298 [M–t-C4H9]+ (100), 270 (18), 121 [C5H5Fe]+ (6), 56 [Fe]+ (2). Analysis calculated for C19H25FeNS: C, 64.23; H, 7.09; N, 3.94; S, 9.02. Found: C, 63.94; H, 7.32; N, 3.86; S, 8.80.
rac-3,3,4,4-Tetramethyl-1-(methylthio)-3,4-dihydroferroceno[c]pyridine (11b) and (1R*,Sp*)-1-(tert-Butyl)-1-methyl-3-(methylthio)-1H-ferroceno[c]pyrrole (12b). The title compounds were prepared according to GP2 (reaction time = 25 min) and GP3 using methyl thiocyanate (10b) (0.028 mL, 0.42 mmol). GP2: purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave, in order of elution, a mixture of 11b and 12b (33 mg, 28%, 11b/12b = 9:91) and 12b (76 mg, 64%). GP3: purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 100:1–petroleum ether/TEA 100:1) gave, in order of elution, 11b (41 mg, 35%) and a mixture of 11b and 12b (54 mg, 45%, 11b/12b = 14:86). Data for 11b: brown solid; mp 70–74 °C (hexane); Rf 0.61 (petroleum ether/EtOAc 25:1). IR (thin film): 2973, 2924, 1581, 1449, 1372, 1361, 1286, 1241, 1151, 1108, 1097, 1002, 899, 822, 626, 512, 478 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.41 (dd, 1H, J = 2.5, 1.1 Hz), 4.09 (s, 5H), 3.99 (dd, 1H, J = 2.4, 1.2 Hz), 3.92 (t, 1H, J = 2.4 Hz), 2.40 (s, 3H), 1.52 (s, 3H), 1.40 (s, 3H), 0.98 (s, 3H, CH3), 0.88 (s, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 161.9 (C), 99.1 (C), 74.9 (C), 70.5 (5 CH), 66.6 (CH), 65.7 (CH), 64.4 (C), 63.8 (CH), 35.8 (C), 29.2 (CH3), 27.3 (C), 25.2 (CH3), 24.7 (CH3), 24.3 (CH3), 11.4 (CH3). MS (EI) m/z (% relative intensity): 341 [M]+ (100), 326 [M–CH3]+ (16), 293 (22), 269 (32), 252 (15), 237 (11), 173 (11), 121 [C5H5Fe]+ (10), 117 (21), 89 (10), 56 [Fe]+ (4). Analysis calculated for C18H23FeNS: C, 63.35; H, 6.79; N, 4.10; S, 9.39. Found: C, 63.34; H, 6.66; N, 4.04; S, 9.60. Data for 12b: brown solid; mp 97–106.5 °C (hexane); Rf 0.38 (petroleum ether/EtOAc 25:1). IR (thin film): 3079, 2981, 2970, 2953, 2939, 2907, 2871, 1525, 1453, 1364, 1297, 1286, 1144, 1103, 1004, 926, 812, 653, 508, 482 cm−1. 1H NMR (400 MHz, C6D6, 50 °C): δ 4.07 (dd, 1H, J = 2.3, 0.7 Hz), 4.05 (dd, 1H, J = 2.2, 0.7 Hz), 4.03 (s, 5H), 3.87 (t, 1H, J = 2.3 Hz), 2.42 (s, 3H), 1.76 (s, 3H), 0.89 (s, 9H). 13C NMR (100 MHz, C6D6, 50 °C): δ 166.5 (C), 110.4 (C), 90.5 (C), 78.5 (C), 71.0 (CH), 69.8 (5 CH), 62.9 (CH), 56.9 (CH), 37.7 (C), 26.0 (3 CH3), 23.2 (CH3), 12.7 (CH3). MS (EI) m/z (% relative intensity): 341 [M]+ (23), 284 [M–t-C4H9]+ (100), 269 (10), 218 (14), 121 [C5H5Fe]+ (12), 56 [Fe]+ (3). Analysis calculated for C18H23FeNS: C, 63.35; H, 6.79; N, 4.10; S, 9.39. Found: C, 63.13; H, 6.99; N, 4.06; S, 9.05.
rac-3,3,4,4-Tetramethyl-1-(propylthio)-3,4-dihydroferroceno[c]pyridine (11c) and (1R*,Sp*)-1-(tert-Butyl)-1-methyl-3-(propylthio)-1H-ferroceno[c]pyrrole (12c). The title compounds were prepared according to GP2 (reaction time = 25 min) and GP3 using propyl thiocyanate (10c) (0.043 mL, 0.42 mmol). GP2: purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave, in order of elution, a mixture of 11c and 12c (29 mg, 23%, 11c/12c = 24:76) and 12c (97 mg, 75%). GP3: purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 100:1–petroleum ether/TEA 100:1) gave, in order of elution, 11c (68 mg, 53%) and a mixture of 11c and 12c (37 mg, 29%, 11c/12c = 14:86). Data for 11c: brown oil; Rf 0.67 (petroleum ether/EtOAc 25:1). IR (thin film): 3097, 2969, 2930, 2870, 1580, 1449, 1372, 1361, 1286, 1238, 1151, 1141, 1108, 1097, 1002, 932, 899, 822, 512, 478 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.43 (dd, 1H, J = 2.4, 1.1 Hz), 4.11 (s, 5H), 3.99 (dd, 1H, J = 2.4, 1.1 Hz), 3.93 (t, 1H, J = 2.4 Hz), 3.24–3.09 (m, 2H), 1.70 (h, 2H, J = 7.3 Hz), 1.49 (s, 3H), 1.40 (s, 3H), 0.98 (s, 3H), 0.96 (t, 3H, J = 7.3 Hz), 0.88 (s, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 161.4 (C), 99.1 (C), 75.0 (C), 70.5 (5 CH), 66.6 (CH), 65.7 (CH), 64.4 (C), 63.9 (CH), 35.7 (C), 30.4 (CH2), 29.2 (CH3), 25.2 (CH3), 24.7 (CH3), 24.2 (CH3), 23.5 (CH2), 13.7 (CH3). MS (EI) m/z (% relative intensity): 369 [M]+ (100), 326 [M–C3H7]+ (38), 293 (20), 269 (70), 252 (7), 172 (11), 121 [C5H5Fe]+ (12), 117 (27), 56 [Fe]+ (3). HRMS-ESI (m/z): [M + H]+ calculated for C20H28FeNS, 370.1286; found, 370.1288. Data for 12c: brown oil; Rf 0.51 (petroleum ether/EtOAc 25:1). IR (thin film): 3096, 2966, 2935, 2871, 1521, 1453, 1366, 1288, 1149, 1108, 1003, 933, 887, 820, 648, 508, 488 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.09 (dd, 1H, J = 2.3, 0.7 Hz), 4.04 (s, 5H), 4.03 (dd, 1H, J = 2.3, 0.7 Hz), 3.85 (t, 1H, J = 2.3 Hz), 3.32–3.26 (m, 1H), 3.07–3.00 (m, 1H), 1.76 (s, 3H), 1.79–1.65 (m, 2H), 0.94–0.90 (m, 12H). 13C NMR (100 MHz, C6D6, 30 °C): δ 165.9 (C), 110.0 (C), 90.7 (C), 78.4 (C), 71.1 (CH), 69.8 (5 CH), 62.8 (CH), 57.0 (CH), 37.7 (C), 32.0 (CH2), 26.0 (3 CH3), 23.5 (CH2), 23.2 (CH3), 13.5 (CH3). MS (EI) m/z (% relative intensity): 369 [M]+ (19), 312 [M–t-C4H9]+ (100), 270 (24), 204 (5), 121 [C5H5Fe]+ (7), 56 [Fe]+ (2). HRMS-ESI (m/z): [M + H]+ calculated for C20H28FeNS, 370.1286; found, 370.1287.
rac-1-(Isopropylthio)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (11d) and (1R*,Sp*)-1-(tert-Butyl)-3-(isopropylthio)-1-methyl-1H-ferroceno[c]pyrrole (12d). The title compounds were prepared according to GP2 (reaction time = 40 min) and GP3 using isopropyl thiocyanate (10d) (0.044 mL, 0.42 mmol). GP2: purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 25:1–petroleum ether/TEA 100:1) gave, in order of elution, 11d (9 mg, 7%), a mixture of 11d and 12d (30 mg, 23%, 11d/12d = 18:82) and 12d (68 mg, 53%). GP3: purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 100:1–petroleum ether/TEA 100:1) gave, in order of elution, 11d (79 mg, 61%) and a mixture of 11d and 12d (25 mg, 19%, 11d/12d = 70:30). Data for 11d: brown oil; Rf 0.47 (petroleum ether/EtOAc 100:1). IR (thin film): 3097, 2971, 2927, 2866, 1578, 1463, 1449, 1415, 1388, 1372, 1286, 1234, 1151, 1141, 1116, 1108, 1095, 1057, 1035, 1023, 1002, 932, 899, 822, 651, 512, 478 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.40 (dd, 1H, J = 2.4, 1.1 Hz), 4.19 (hept, 1H, J = 6.8 Hz), 4.10 (s, 5H), 3.99 (dd, 1H, J = 2.5, 1.2 Hz), 3.91 (t, 1H, J = 2.4 Hz), 1.51 (s, 3H), 1.41 (s, 3H), 1.40 (d, 3H, J = 6.6 Hz), 1.35 (d, 3H, J = 6.8 Hz), 0.98 (s, 3H), 0.90 (s, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 161.8 (C), 99.0 (C), 75.0 (C), 70.5 (5 CH), 66.5 (CH), 65.7 (CH), 64.3 (C), 63.9 (CH), 35.6 (C), 33.5 (CH), 29.3 (CH3), 25.1 (CH3), 24.8 (CH3), 24.2 (CH3), 23.4 (CH3), 23.2 (CH3). MS (EI) m/z (% relative intensity): 369 [M]+ (52), 326 [M–i-C3H7]+ (38), 269 (100), 172 (10), 121 [C5H5Fe]+ (17), 117 (25), 56 [Fe]+ (5). Analysis calculated for C20H27FeNS: C, 65.04; H, 7.37; N, 3.79; S, 8.68. Found: C, 65.44; H, 7.45; N, 3.57; S, 8.66. HRMS-ESI (m/z): [M + H]+ calculated for C20H28FeNS, 370.1286; found, 370.1284. Data for 12d: brown oil; Rf 0.50 (petroleum ether/EtOAc 25:1). IR (thin film): 3097, 2966, 2934, 2911, 2868, 1519, 1478, 1451, 1424, 1392, 1382, 1366, 1294, 1241, 1226, 1148, 1108, 1102, 1004, 934, 886, 820, 660, 513, 487 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.14 (hept, 1H, J = 6.8 Hz), 4.07 (dd, 1H, J = 2.3, 0.7 Hz), 4.04 (s, 5H), 4.03 (dd, 1H, J = 2.2, 0.8 Hz), 3.85 (t, 1H, J = 2.2 Hz), 1.76 (s, 3H), 1.39 (d, 3H, J = 6.8 Hz), 1.35 (d, 3H, J = 6.9 Hz), 0.91 (s, 9H). 13C NMR (100 MHz, C6D6, 30 °C): δ 165.9 (C), 109.7 (C), 90.9 (C), 78.6 (C), 71.1 (CH), 69.8 (5 CH), 62.8 (CH), 57.0 (CH), 37.8 (C), 35.3 (CH), 26.1 (3 CH3), 23.6 (CH3), 23.2 (CH3), 23.1 (CH3). MS (EI) m/z (% relative intensity): 369 [M]+ (18), 312 [M–t-C4H9]+ (61), 270 (100), 204 (15), 121 [C5H5Fe]+ (14), 56 [Fe]+ (6). HRMS-ESI (m/z): [M + H]+ calculated for C20H28FeNS, 370.1286; found, 370.1288.
rac-1-(Benzylthio)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (11e) and (1R*,Sp*)-3-(Benzylthio)-1-(tert-butyl)-1-methyl-1H-ferroceno[c]pyrrole (12e). The title compounds were prepared according to GP2 (reaction time = 25 min) and GP3 using benzyl thiocyanate (10e) (62.6 mg, 0.42 mmol). GP2: purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave, in order of elution, a mixture of 11e and 12e (80 mg, 55%, 11e/12e = 22:78) and 12e (54 mg, 37%). GP3: purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 100:1–petroleum ether/TEA 100:1) gave, in order of elution, a mixture of starting thiocyanate 10e and 11e (70 mg, 10e/11e = 16:84) and a mixture of 11e and 12e (57 mg, 39%, 11e/12e = 27:73). The mixture of 10e and 11e was additionally chromatographed on silica gel (benzene–benzene/TEA 100:1) to yield pure compound 11e (61 mg, 42%). Data for 11e: brown solid; mp 62.5–67.5 °C; Rf 0.79 (petroleum ether/EtOAc 25:1). IR (thin film): 3086, 3062, 3028, 2972, 2926, 2867, 2855, 1581, 1495, 1450, 1372, 1361, 1287, 1236, 1151, 1108, 1096, 1035, 1002, 932, 898, 823, 759, 700, 512, 477 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 7.42–7.40 (m, 2H), 7.16–7.11 (m, 3H), 7.05–7.01 (m, 1H), 4.44 (dd, 1H, J = 13.6 Hz), 4.40 (d, 1H, J = 13.6 Hz), 4.36 (dd, 1H, J = 2.4, 1.1 Hz), 4.05 (s, 5H), 3.97 (dd, 1H, J = 2.5, 1.1 Hz), 3.90 (t, 1H, J = 2.4 Hz), 1.52 (s, 3H), 1.38 (s, 3H), 0.97 (s, 3H), 0.89 (s, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 161.1 (C), 139.9 (C), 129.6 (2 C), 128.5 (2 C), 127.0 (C), 99.1 (C), 74.6 (C), 70.5 (5 CH), 66.7 (CH), 65.8 (CH), 64.6 (C), 63.8 (CH), 35.7 (C), 32.8 (CH2), 29.2 (CH3), 25.2 (CH3), 24.6 (CH3), 24.3 (CH3). MS (EI) m/z (% relative intensity): 417 [M]+ (69), 402 [M–CH3]+ (2), 326 [M–CH2C6H5]+ (40), 269 (100), 132 (9), 121 [C5H5Fe]+ (9), 56 [Fe]+ (2). Analysis calculated for C24H27FeNS: C, 69.06; H, 6.52; N, 3.36; S, 7.68. Found: C, 69.13; H, 6.75; N, 3.05; S, 7.83. Data for 12e: brown oil, Rf 0.65 (petroleum ether/EtOAc 25:1). IR (thin film): 3088, 3063, 3029, 2969, 2952, 2909, 2870, 1521, 1495, 1478, 1453, 1392, 1366, 1299, 1225, 1194, 1149, 1103, 1003, 929, 886, 821, 767, 699, 649, 508, 488, 472, 439 cm−1. 1H NMR (400 MHz, DMSO-d6, 50 °C): δ 7.50–7.47 (m, 2H), 7.36–7.31 (m, 2H), 7.29–7.24 (m, 1H), 4.46 (d, 1H, J = 13.6 Hz), 4.41 (dd, 1H, J = 2.2, 0.7 Hz), 4.41 (d, 1H, J = 13.6 Hz), 4.31 (dd, 1H, J = 2.3, 0.7 Hz), 4.26 (t, 1H, J = 2.2 Hz), 4.14 (s, 5H), 1.71 (s, 3H), 0.74 (s, 3H). 13C NMR (100 MHz, DMSO-d6, 50 °C): δ 164.3 (C), 138.4 (C), 128.6 (2 CH), 127.9 (2 CH), 126.7 (CH), 108.9 (C), 89.0 (C), 77.5 (C), 70.7 (CH), 69.0 (5 CH), 62.4 (CH), 56.2 (CH), 32.8 (CH2), 25.2 (3 CH3), 22.4 (CH3). MS (EI) m/z (% relative intensity): 417 [M]+ (23), 360 [M–t-C4H9]+ (100), 269 (37), 121 [C5H5Fe]+ (7), 56 [Fe]+ (2). HRMS-ESI (m/z): [M + H]+ calculated for C24H28FeNS, 418.1286; found, 418.1294.
rac-1-(Hexylthio)-3,3,4,4-tetramethyl-3,4-dihydroferroceno[c]pyridine (11f) and (1R*,Sp*)-1-(tert-Butyl)-3-(hexylthio)-1-methyl-1H-ferroceno[c]pyrrole (12f). The title compounds were prepared according to GP2 (reaction time = 40 min) and GP3 using hexyl thiocyanate (10f) (0.065 mL, 0.42 mmol). GP2: purification by silica gel column chromatography (petroleum ether/CH2Cl2 1:1–petroleum ether/EtOAc 100:1–petroleum ether/TEA 100:1) gave, in order of elution, a mixture of 11f and 12f (38 mg, 26%, 11f/12f = 22:78) and 12f (94 mg, 65%). GP3: purification by silica gel column chromatography (petroleum ether–petroleum ether/EtOAc 200:1–petroleum ether/EtOAc 50:1–petroleum ether/TEA 100:1) gave, in order of elution, 11f (79 mg, 55%) and a mixture of 11f and 12f (45 mg, 31%, 11f/12f = 42:58). Data for 11f: brown oil; Rf 0.83 (petroleum ether/EtOAc 25:1). IR (thin film): 3097, 2970, 2927, 2857, 1580, 1449, 1372, 1361, 1286, 1240, 1151, 1141, 1108, 1097, 1035, 1002, 932, 899, 822, 512, 478, 453 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.44 (dd, 1H, J = 2.4, 1.2 Hz), 4.12 (s, 5H), 3.99 (dd, 1H, J = 2.4, 1.1 Hz), 3.93 (t, 1H, J = 2.4 Hz), 3.30–3.15 (m, 2H), 1.76–1.69 (m, 2H), 1.52 (s, 3H), 1.45–1.36 (m, 2H, partially overlapped), 1.41 (s, 3H), 1.28–1.22 (m, 4H), 0.99 (s, 3H), 0.90 (s, 3H), 0.88–0.85 (m, 3H). 13C NMR (100 MHz, C6D6, 30 °C): δ 161.5 (C), 99.1 (C), 75.1 (C), 70.5 (5 CH), 66.6 (CH), 65.7 (CH), 64.4 (C), 63.9 (CH), 35.7 (C), 31.8 (CH2), 30.1 (CH2), 29.2 (CH3), 29.0 (CH2), 28.4 (CH2), 25.2 (CH3), 24.8 (CH3), 24.3 (CH3), 22.9 (CH2), 14.2 (CH3). MS (EI) m/z (% relative intensity): 411 [M]+ (100), 326 [M–C6H13]+ (58), 293 (28), 269 (84), 172 (13), 132 (11), 121 [C5H5Fe]+ (15), 117 (29), 56 [Fe]+ (3). HRMS-ESI (m/z): [M + H]+ calculated for C23H34FeNS, 412.1756; found, 412.1760. Data for 12f: brown oil; Rf 0.56 (petroleum ether/EtOAc 25:1). IR (thin film): 3096, 2955, 2930, 2871, 2858, 1521, 1478, 1453, 1425, 1391, 1366, 1298, 1286, 1225, 1193, 1149, 1108, 1035, 1003, 933, 887, 820, 648, 508, 487, 472, 439 cm−1. 1H NMR (400 MHz, C6D6, 30 °C): δ 4.11 (dd, 1H, J = 2.4, 0.7 Hz), 4.06 (s, 5H), 4.04 (dd, 1H, J = 2.3, 0.5 Hz), 3.86 (t, 1H, J = 2.2 Hz), 3.37–3.31 (m, 1H), 3.16–3.09 (m, 1H), 1.81–1.69 (m, 2H, partially overlapped), 1.77 (s, 3H), 1.41–1.33 (m, 2H), 1.27–1.18 (m, 4H), 0.92 (s, 9H), 0.87–0.83 (m, 3H). 13C NMR (100 MHz, C6D6, 50 °C): 166.1 (C), 110.1 (C), 90.8 (C), 78.5 (C), 71.1 (CH), 69.9 (5 CH), 62.9 (CH), 57.0 (CH), 37.8 (C), 31.8 (CH2), 30.3 (CH2), 30.2 (CH2), 28.9 (CH2), 26.1 (3 CH3), 23.2 (CH3), 22.9 (CH2), 14.1 (CH3). MS (EI) m/z (% relative intensity): 411 [M]+ (17), 354 [M–t-C4H9]+ (100), 270 (21), 121 [C5H5Fe]+ (3), 56 [Fe]+ (1). HRMS-ESI (m/z): [M + H]+ calculated for C23H34FeNS, 412.1756; found, 412.1762.
rac-Ethyl 2-((3,3,4,4-Tetramethyl-3,4-dihydroferroceno[c]pyridine)thio)acetate (
11g)
and (1R*,Sp*)-Ethyl 2-((1-(tert-Butyl)-1-methyl-1H-ferroceno[c]pyrrole)thio)acetate (
12g). The title compounds were prepared according to GP2 (reaction time = 1 h) and GP3 (reaction time = 15 min) using ethyl 2-thiocyanatoacetate (
10g) (0.064 mL, 0.53 mmol). GP2: purification by silica gel column chromatography (petroleum ether/EtOAc 25:1) gave, in order of elution,
11g (8 mg, 6%; according to
1H NMR analysis contained ~1% of
3u;
11g/
3u = 99:1) and
12g (106 mg, 73%). GP3: purification by silica gel column chromatography (petroleum ether/EtOAc 25:1) gave, in order of elution,
11g (41 mg, according to
1H NMR analysis contained 4% of
3u;
11g/
3u = 96:4; calculated yield of
11g: 27%) and
12g (38 mg, 26%). Data for
11g: brown oil;
Rf 0.25 (petroleum ether/EtOAc 25:1). IR (thin film): 3096, 2975, 2927, 2869, 2856, 1739, 1647, 1587, 1463, 1448, 1389, 1372, 1363, 1289, 1261, 1242, 1151, 1108, 1097, 1034, 1002, 933, 899, 823, 744, 712, 627, 583, 512, 479, 453 cm
−1.
1H NMR (400 MHz, C
6D
6, 30 °C): δ 4.36 (dd, 1H,
J = 2.5, 1.1 Hz), 4.13 (s, 5H), 4.02–3.97 (m, 4H), 3.91 (t, 1H,
J = 2.4 Hz), 3.77 (d, 1H,
J = 15.8 Hz), 1.44 (s, 3H), 1.37 (s, 3H), 0.99 (t, 3H,
J = 7.1 Hz), 0.95 (s, 3H), 0.85 (s, 3H).
13C NMR (100 MHz, C
6D
6, 30 °C): δ 169.2 (C), 160.2 (C), 99.2 (C), 74.2 (C), 70.6 (5 CH), 66.8 (CH), 65.8 (CH), 64.6 (C), 63.6 (CH), 61.0 (CH
2), 35.8 (C), 31.2 (CH
2), 29.1 (CH
3), 25.2 (CH
3), 24.5 (CH
3), 24.1 (CH
3), 14.3 (CH
3). GC–MS analyses of product
11g indicated the presence in the chromatogram of a peak that, based on corresponding MS spectra, belonged to compound
3o. The latter was likely produced by the thermolysis of the parent compound in the GC instrument injector. HRMS-ESI (
m/z): [M + H]
+ calculated for C
21H
28FeNO
2S, 414.1185; found, 414.1186. Data for
12g: orange oil, which solidified on long-term standing; mp 51–52.5 °C;
Rf 0.32 (petroleum ether/EtOAc 25:1). IR (thin film): 3096, 2973, 2954, 2908, 2871, 1740, 1527, 1478, 1452, 1392, 1367, 1294, 1262, 1226, 1153, 1104, 1032, 1004, 934, 887, 822, 650, 488 cm
−1.
1H NMR (400 MHz, C
6D
6, 50 °C): δ 4.07 (s, 5 CH), 4.05 (dd, 1H,
J = 2.4, 0.6 Hz), 4.04 (dd, 1H,
J = 2.2, 0.7 Hz), 4.00–3.95 (m, 2H), 3.93 (d, 1H,
J = 15.7 Hz, partially overlapped), 3.85 (d, 1H,
J = 15.8 Hz), 3.86 (t, 1H,
J = 2.3 Hz), 1.71 (s, 3H), 0.98 (t, 3H,
J = 7.1 Hz), 0.86 (s, 9H).
13C NMR (100 MHz, C
6D
6, 50 °C): 168.6 (C), 164.8 (C), 110.4 (C), 89.6 (C), 78.7 (C), 71.2 (CH), 70.0 (5 CH), 63.1 (CH), 61.2 (CH
2), 57.0 (CH), 37.7 (C), 32.5 (CH
2), 25.9 (3 CH
3), 23.0 (CH
3), 14.2 (CH
3). GC–MS analysis of compound
12g indicated the presence of a peak of substance with a molecular mass of 309. MS (EI)
m/z (% relative intensity): 309 [M]
+ (31), 253 [M–C
4H
8]
+ (60), 252 [M–
t-C
4H
9]
+ (100), 211 (8), 121 [C
5H
5Fe]
+ (11), 56 [Fe]
+ (3). Apparently, this is a 1-(
tert-butyl)-1,3-dimethyl-1
H-ferroceno[
c]pyrrole yielded by the thermolysis of
12g in the GC injector. The proposed mechanism for the thermolysis of ferroceno[
c]pyrrole
12g is performed in
Supplementary Materials (Scheme S2). HRMS-ESI (
m/z): [M + H]
+ calculated for C
21H
28FeNO
2S, 414.1185; found, 414.1191.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





