
Synthesis, Structure and Mg2+ Ionic Conductivity of Isopropylamine Magnesium Borohydride


1
Interdisciplinary Nanoscience Center (iNANO), Department of Chemistry, University of Aarhus, Langelandsgade 140, DK-8000 Aarhus C, Denmark
2
Interdisciplinary Nanoscience Center (iNANO), Aarhus Institute of Advanced Studies (AIAS), Department of Physics and Astronomy, Center for Interstellar Catalysis, University of Aarhus, DK-8000 Aarhus C, Denmark
*
Author to whom correspondence should be addressed.
Inorganics 2023, 11(1), 17; https://doi.org/10.3390/inorganics11010017
Submission received: 18 November 2022
/
Revised: 6 December 2022
/
Accepted: 15 December 2022
/
Published: 30 December 2022
(This article belongs to the Special Issue State-of-the-Art and Progress in Metal-Hydrogen Systems)
Abstract
:The discovery of new inorganic magnesium electrolytes may act as a foundation for the rational design of novel types of solid-state batteries. Here we investigated a new type of organic-inorganic metal hydride, isopropylamine magnesium borohydride, Mg(BH4)2∙(CH3)2CHNH2, with hydrophobic domains in the solid state, which appear to promote fast Mg2+ ionic conductivity. A new synthetic strategy was designed by combination of solvent-based methods and mechanochemistry. The orthorhombic structure of Mg(BH4)2∙(CH3)2CHNH2 was solved ab initio by the Rietveld refinement of synchrotron X-ray powder diffraction data and density functional theory (DFT) structural optimization in space group I212121 (unit cell, a = 9.8019(1) Å, b = 12.1799(2) Å and c = 17.3386(2) Å). The DFT calculations reveal that the three-dimensional structure may be stabilized by weak dispersive interactions between apolar moieties and that these may be disordered. Nanoparticles and heat treatment (at T > 56 °C) produce a highly conductive composite, σ(Mg2+) = 2.86 × 10−7, and 2.85 × 10−5 S cm−1 at −10 and 40 °C, respectively, with a low activation energy, Ea = 0.65 eV. Nanoparticles stabilize the partially eutectic molten state and prevent recrystallization even at low temperatures and provide a high mechanical stability of the composite.
1. Introduction
Due to the increased electrification of society, new ways of storing energy need to be developed, e.g., with significant improvement of the gravimetric and volumetric capacity as well as the safety of batteries, while simultaneously keeping the costs down. In recent years, the increase in capacity in Li-ion batteries using carbon anodes has stagnated and is approaching the theoretical limit [1]. The development of “post-Li-ion” batteries is therefore of critical importance [2]. There are increasing efforts towards sustainability, and the use of more abundant elements, such as sodium and magnesium, could cut the cost of the raw materials both economically and environmentally [3]. The all-solid-state battery is a new promising technology, which is expected to provide several advantages, such as easier assembling and production, as well as an improved safety profile and lifetime. This is partly related to the solid electrolyte, which is expected to be made thinner, provide faster charging and discharging rates, help to avoid short-circuits in the battery, and increase thermal stability during cooling, as the compounds are already solid when compared to traditional liquid electrolytes [4]. The Achilles heel appears to be in creating fast ionic conductivity in the solid state, in particular for divalent cations, such as Mg2+.
Magnesium appears to be less prone to forming dendrites when compared to lithium, however, obtaining fast ionic conductivity in the solid state of divalent cations is very challenging. This is particularly true at relevant ambient temperatures where very few solid-state magnesium-based electrolyte materials have been reported, as compared to monovalent cations such as Li+ and Na+. In recent years, novel magnesium borohydrides have provided high Mg2+ conductivities even at low temperatures (≈10−4 S cm−1 at 40 °C), making this class of materials very interesting [5,6,7,8,9]. These new compounds are built using ionic and covalent bonds and therefore have very low electronic conductivity. Furthermore, metal borohydrides have the benefit of being compatible with metal anodes, which increase both the gravimetric and volumetric densities in a final cell [10,11,12]. However, cationic conductivity still needs to be further increased for battery applications and challenges, and contact and electrochemical stability issues must be addressed [13]. Replacing lithium is challenging, since lithium is light, mobile in the solid state and since compatible electrodes often operate with a large electrochemical stability window [2].
Detailed knowledge of the phenomena that are responsible for fast cation conductivity in the solid state may lead to rational design of novel materials. Recently, borohydrides have shown greatly increased ionic conductivities when coordinated to a neutral ligand [7,8,9,14]. Furthermore, thermal and mechanical properties are also altered, allowing for malleable compounds or even liquid-like electrolytes [8,15]. Here we present the synthesis, structure, and ionic conductivity of a new type of monoisopropylamine magnesium borohydride, Mg(BH4)2∙(CH3)2CHNH2, and incorporation of this complex into a nanocomposite.
2. Results
Initial Characterization. A new synthesis method combining solvent-based techniques to prepare the reactants, and mechanochemistry to form the product has been developed [16,17]. Magnesium borohydride, α-Mg(BH4)2 (denoted, s1), was synthesized by a solvothermal method and then dissolved in isopropylamine (IPA), (CH3)2CHNH2. A white solid was filtered off and dried in vacuum (25 °C, 40 min) to form diisopropylamine magnesium borohydride, Mg(BH4)2∙2(CH3)2CHNH2 (s4). A new crystalline compound was formed by mechanochemical treatment of α-Mg(BH4)2 and Mg(BH4)2∙2(CH3)2CHNH2 in the molar ratio 1:1 at 350 rpm with a total milling time of 120 min. Mechanochemical treatment at a shorter time or with lower ball-to-powder ratio provides partly reacted products (sample s2). This is illustrated in Figure 1, which provides diffraction patterns of the reactants α-Mg(BH4)2 (s1) and Mg(BH4)2∙2(CH3)2CHNH2 (s4) and the new compound (s3). The diffraction pattern of the sample s3 is assigned to a single crystalline phase and all observed Bragg reflections can be accounted for by an orthorhombic unit cell, a = 9.8019(1) Å, b = 12.1799(2) Å and c = 17.3386(2) Å, which is similar to previously proposed data for the suggested composition, Mg(BH4)2·(CH3)2CHNH2 [6].
Structural analysis. The successful synthesis of single-phase monoisopropylamine magnesium borohydride, Mg(BH4)2∙(CH3)2CHNH2, allowed for the measurement of high-quality synchrotron radiation powder X-ray diffraction (SR-PXD) data (see Figure 1). The structure was solved ab initio, in the orthorhombic space group I212121, using direct space methods (implemented in the program FOX). Several other structural models were investigated but rejected due to an unsatisfactory fit to the experimental diffraction data, unrealistic coordination and/or instability when optimised by density functional theory (DFT), see supporting information. Several cycles of DFT structural optimization and Rietveld refinement were conducted to develop the final structural model. The structure is composed of magnesium in two different coordination environments. In the first environment, Mg2+ is coordinated to four BH4−, similar to the structure of α-Mg(BH4)2, however the coordination is planar, whereas it is tetrahedral in α-Mg(BH4)2. In the second environment, magnesium is tetrahedrally coordinated to two BH4− and two (CH3)2CHNH2 molecules. These structural units are bridged by BH4− to form a zigzag 1D chain propagating along the a-axis, forming a ‘flat helix’ as shown in Figure 2. From DFT it was found that bridging tetrahydridoborohydride (BH4−) complexes, Mg–BH4−–Mg, have a bidentate κ2 coordination to Mg in [Mg(BH4)4] and a tridentate κ3 coordination to Mg in [Mg(BH4)2(NH2CH(CH3)2)2]. Terminal BH4− was found to have a bidentate κ2 coordination to Mg. Mg–N (2.08 Å) and terminal Mg–B (2.20 Å) distances are similar to what have been reported previously [6,7,18,19,20].
The bridging BH4− group is not placed in the center between Mg(1) (Mg in [Mg(BH4)4]) and Mg(2) (Mg in [Mg(BH4)2(NH2CH(CH3)2)2]). The distances are Mg(1)–B = 3.14 Å and Mg(2)–B = 2.57 Å for the experimental structure and Mg(1)–B = 2.60 Å and Mg(2)–B = 2.33 Å for the DFT-optimized structure, respectively. This suggests that the bridging BH4− group is more tightly bound to Mg2, as compared to Mg1. The apolar regions in the structure formed by CH3 groups in neighboring IPA molecules are close (C–C, 2.5–2.7 Å). As further discussed below, weak dispersive interactions between the IPA molecules may play a role in stabilizing the 3D structure in the b,c-plane, as seen in Figure 2 (right). Furthermore, the DFT results suggest a certain degree of disorder of the (CH3)2CH– moiety, which could explain the discrepancy between the observed and calculated diffraction patterns as seen in Figure 3.
The DFT structural optimization was done with two different exchange–correlation functionals with and without accounting for van-der-Waals (vdW) interactions, beginning from the experimental structure shown in Figure 2. Independently of the used functions, it was found that the experimental structure is metastable and that it becomes increasingly disordered upon further refinement. This indicates a ‘flat energy landscape’ between the experimental structure and various disordered structural models (see Figures S1–S4 and Table S1 in the supporting information). In all DFT optimizations (with and without vdW), the distance between CH3 groups in neighboring IPA molecules increases to about 3.5–3.9 Å. This indicates that the interactions between the CH3 groups are weak. Calculations for the butane dimer reveal typical vdW C–C distances of around 4 Å [21]. However, the chemical bonding scheme in all the different structural models, both experimental and theoretical, is the same but with different degrees of structural distortion. The distortion primarily occurs for the (CH3)2CH– moieties, which indicates that more than one conformation of these could exist, resulting in static or dynamic disorder to the structure. The DFT results also reveal that some H atoms in the neighboring borohydride and amine groups are close (dH–H = 2.5–2.70 Å), which suggests that di-hydrogen bonds, B−Hδ−···+δH−N, bind the 1-D chains together.
Thermal analysis. Figure 4 shows in situ temperature-resolved synchrotron radiation powder X-ray diffraction of Mg(BH4)2∙1.20IPA (s3) in the temperature range from 20 to 75 °C. Initially, the sample contained one crystalline compound, Mg(BH4)2∙(CH3)2CHNH2. However, during heating, a new set of diffraction peaks appeared at 31 °C, which are assigned to the crystallization of Mg(BH4)2∙2(CH3)2CHNH2. Sample analysis using liquid-state 1H NMR reveals that s3 contains 1.20 IPA per Mg(BH4)2 (see Table 1). Therefore amorphous Mg(BH4)2∙2(CH3)2CHNH2 may be recrystallized upon heating or surface-adsorbed IPA may react with monoisopropylamine magnesium borohydride through a gas–solid reaction. Notice that the boiling point of IPA is 32 °C. At 58 °C, the diffraction from Mg(BH4)2∙2(CH3)2CHNH2 disappears, which is at a significantly lower temperature than previously reported (87 °C) [6]. This may indicate eutectic melting of the composite Mg(BH4)2∙(CH3)2CHNH2-Mg(BH4)2∙2(CH3)2CHNH2 since the diffracted intensity from both compounds decreases above 51 °C. After the disappearance of diffraction from Mg(BH4)2∙2(CH3)2CHNH2 at 58 °C, the remaining diffracted intensity from Mg(BH4)2∙(CH3)2CHNH2 further decreases in intensity and disappears at 73 °C. After storage for 4 months at RT, the diffraction data of s3 measured at RT revealed the crystallization of minor amounts of Mg(BH4)2∙2(CH3)2CHNH2.
Figure 5 displays the thermal analysis of Mg(BH4)2∙1.20(CH3)2CHNH2 (s3), i.e., thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and mass spectrometry (MS) measured simultaneously for the same sample. A thermal DSC event was observed at 56 °C, in accordance with the disappearance of diffraction from Mg(BH4)2∙2(CH3)2CHNH2, and assigned to eutectic melting of a fraction of the sample. This event is accompanied by a weak indication of the release of hydrogen observed by MS. In the temperature range of 108 to 138 °C, a mass loss of Δm/m = 6.59 wt% corresponding to the release of 0.14 molecules of IPA per formula unit was observed, and the remaining sample composition is ~1 IPA per formula unit. Release of IPA in this temperature range, 108 to 138 °C, was also detected by MS, accompanied by a minor release of hydrogen. In the temperature range of 138 to 225 °C a major mass loss of Δm/m = 42.61 wt% was observed, corresponding to a loss of 0.9 IPA. Mass spectroscopy reveals an increasing release of IPA and H2 with a maximum release rate around 200 °C. The release of hydrogen suggests a chemical reaction between the organic moiety (IPA) and the borohydride complex. A total mass loss of 49.20 wt% was observed in the temperature range of RT to 225 °C.
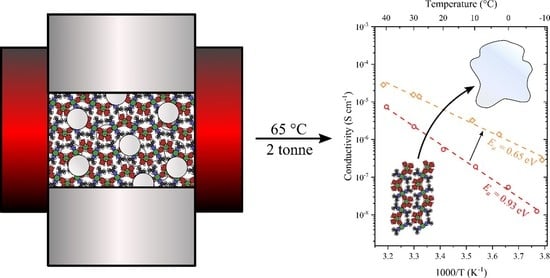
Magnesium ionic conductivity. The Mg2+ ionic conductivity of Mg(BH4)2∙1.20(CH3)2-CHNH2 (s3) was measured by electrochemical impedance spectroscopy (EIS) during heating from −7.9 to 40 °C, see Figure 6. The same sample (s3) was then heated to 40 °C and cooled to 30 °C three times in order to convert amorphous material to Mg(BH4)2-2(CH3)2CHNH2 and to further stabilize the tablet prior to the second EIS measurement. Activation energies, EA, were extracted from a plot of log(σT) versus 1/T as described in the experimental section. The electronic conductivity of this class of compounds is negligible [5,8,9].
The Mg2+ ionic conductivity of Mg(BH4)2∙1.20IPA (s3) is high and increases exponentially in the temperature range of −10 to 40 °C, see Figure 6. The conductivity is slightly higher after thermal treatment, i.e., of the composite (~0.8)Mg(BH4)2∙(CH3)2CHNH2− (~0.2)Mg(BH4)2∙2(CH3)2CHNH2. Noteworthy, a low activation energy was observed for the first measurement, EA = 0.93 eV of s3, which further decreased to EA = 0.86 eV after heat treatment, i.e., of the composite (~0.8)Mg(BH4)2∙(CH3)2CHNH2−(~0.2)Mg(BH4)2∙2(CH3)2CHNH2.
Previous investigations reveal a significant increase in the Mg2+ conductivity of composites containing different crystalline isopropylamine magnesium borohydride compounds, which was also observed here. Furthermore, the compounds were able to strip and plate magnesium with an oxidative stability of 1.2 V vs. Mg/Mg2+ [6]. Adding 50 wt% Al2O3 (13 nm) to the sample (s3) resulted in sample s5. Initially, the conductivity was similar to the original sample; however, as the sample was heated to above the melting point of the Mg(BH4)2∙2(CH3)2CHNH2 (at 56 °C) (see Figure 4), the conductivity significantly increased (see Figure S4). The sample deformed during this transition and became thinner and wider. This was accounted for during calculation of the conductivity. During cooling, this highly conductive state with low activation energy (0.60 eV) was maintained to 30 °C. The highly conductive state shows a similar (but lower) conductivity to that of Mg(BH4)2∙1.5(CH3)2CH–NH2 containing 50 nm MgO particles [6]. This difference is due to the lower content of the eutectic molten state in Mg(BH4)2∙1.20IPA-Al2O3 (s5) as compared to Mg(BH4)2∙1.5-MgO. As the sample deformed during heating, a new pellet was made by applying a pressure of 2.5 Gpa for 30 s, releasing the pressure and then heating to 65 °C while in the press. Finally, a pressure of 2.5 Gpa was applied to the sample for 30 s at RT. This resulted in a highly viscous sample, likely due to the melt stabilization effect mentioned in refs. [5,8]. Due to the soft nature of the sample, the sample was relaxed onto the electrodes at RT for 16 h before measurements to ensure optimal contact. The conductivity of Mg(BH4)2∙1.20IPA−Al2O3 (50 wt%, s5) is shown in Figure 6.
The “paste-like” nature of sample s5 was maintained even after storage at −18 °C for 24 h, indicating that recrystallization was inhibited by the presence of nanoparticles. The thermal stability of Mg(BH4)2∙1.20(CH3)2CHNH2 (s3) was limited by the presence of ~20% Mg(BH4)2∙2(CH3)2CHNH2, which represents eutectic melting with a fraction of the sample at T = 56 °C. Thus, this sample (s3) has low thermal stability as compared to other similar compounds, i.e., Mg(BH4)2∙2NH3BH3 (47 °C) [9], Mg(BH4)2∙NH2(CH2)2NH2 (75 °C) [22] and Mg(BH4)2∙NH3 (90 °C) [7]. However, the activation energy for Mg2+ cationic conductivity is significantly lower for Mg(BH4)2∙(CH3)2CHNH2 (s3), EA = 0.86 eV, as compared to similar compounds presented in Figure 6 This advantage is even more pronounced for the nanocomposite Mg(BH4)2∙(CH3)2CHNH2−Al2O3 (s5), with an activation energy of 0.65 eV. Thus, the advantage of this new material is a moderate temperature dependence of the cationic conductivity, which has an exceptionally high value at low temperatures. Furthermore, the soft nature of the sample may improve contact with the electrodes, which is a major challenge in these all-solid-state systems.
3. Discussion
Recently, the Mg2+ conductivity mechanism of Mg(BH4)2∙NH3 was investigated using diffraction, structure refinements and DFT. The structure of Mg(BH4)2∙NH3 was found to be very flexible owing to a three-dimensional network of di-hydrogen bonds, B−Hδ−···+δH−N, but also to the BH4−−Mg2+ coordination, which varies from in edge to corner coordination (κ1 to κ3). Furthermore, the migration of Mg2+ cations is also assisted by a neutral molecule, NH3, which is exchanged between the framework and interstitial magnesium [7,23]. The IPA analogue, Mg(BH4)2∙(CH3)2CHNH2, investigated here, resembles the above-mentioned compound by having a one-dimensional chain-like structure and that the organic and inorganic moieties are interconnected by weak interactions.
In the following section we discuss and compare the experimental and DFT-optimized structural models, as well as the role of weak dispersive interactions. In the experimental structure obtained from the Rietveld refinement, see Figure 2, the isopropyl groups point towards each other: C−H···H−C (dH–H = 1.9–3.4 Å). From DFT we find that this ordered structure is metastable compared to more disordered structures. The metastable DFT structure and the experimental structure are very similar, except for the terminal BH4− which is closer to the isopropyl groups in the experimental structure, as seen in Figure S2. This could hint at a B−Hδ−···H−C interaction, which is absent in the DFT structure. In the more disordered (and more stable) DFT structures, we found a distortion of the helixes; see Figures S1 and S3. While the helixes remain, terminal BH4− moves from a planar coordination to a tetrahedral coordination. Terminal Mg(1)–B distances increased from 2.2 Å to 2.4 Å and BH4− has a bidentate (κ2) coordination compared to the tridentate (κ3) coordination found from the Rietveld refinement. For bridging Mg(1)–B–Mg(2), the BH4− group is still not placed in the center between Mg(1) and Mg(2) (B–((IPA)2Mg(2))–B = 2.57 Å and B–((BH4−)2Mg(1))–B = 2.31 Å) with a bidentate (κ2) coordination to Mg(1) and a tridentate (κ3) coordination to Mg(2). Both the experimental and theoretical models found a coordination number of eight for Mg2+. However, experimental hydrogen positions obtained by the Rietveld refinement are a result of anti-bump restraints in the ab initio structural solution process and are not refined.
In the more disordered DFT models, the C−H···H−C distances significantly increased (dH–H = 2.5–3.7 Å) compared to the experimental structure. However, in all structures, the neighbouring IPA molecules are rather close and within distances that are typical for weak dispersive interactions [21]. It therefore seems plausible that such interactions are important for stabilizing the 3D structure. We did not observe any significant difference between DFT structures optimized with and without account for van der-Waals interactions, but this may be because the unit cell is kept fixed at the experimentally measured size during the relaxation, which limits how far the different groups are able to move with respect to each other. In Figure S3, several DFT structures that lie between the metastable and most stable structure in energy can be seen. The existence of these shallow local minima on the potential energy surface (and likely many more) supports the claim that the sample is highly disordered due to the flat energy landscape. The disordered DFT-optimized structural models often have a slightly poorer fit to the diffraction data, which relates to the very different structural descriptions. DFT provides an atomic scale ‘momentary’ view of the unit cell, whereas diffraction provides an average structure over time and space. Furthermore, DFT describes the 0 K potential energy surface, whereas the powder pattern was measured at room temperature. Failure to converge to a refined model has also been observed for ammine rare earth borohydrides RE(BH4)3∙4NH3 (RE3+ = La, Ce, Pr, Nd) [24,25].
Thus, both DFT and diffraction reveal a new composition and structure of the compound investigated here, which has some degree of structural disorder and is held together by weak interactions. The high Mg2+ ionic conductivity and moderate activation energy for cation migration are assigned to these structural properties. Composites of crystalline materials, Mg(BH4)2∙x(CH3)2CHNH2, x = 1 or 2, have higher conductivity and eutectic melting. The molten state can be stabilized by nanoparticles to form a mechanically and thermally more stable nanocomposite. This nanocomposite has higher Mg2+ conductivity and lower activation energy, which has previously been assigned to surface effects, e.g., the wetting of nanoparticles by thin layers of eutectic molten liquid. These new phenomena for the rational design of functional battery materials are demonstrated in this work.
4. Materials and Methods
4.1. Synthesis
Magnesium borohydride, Mg(BH4)2, was synthesized as described in refs. [26,27]. Anhydrous toluene (purity 99.8%, 40 mL) and anhydrous dimethylsulphide borane, (CH3)2SBH3 (90% in toluene, 10 mL) were added to a round-bottomed flask. While stirring, di-n-butylmagnesium Mg(C4H9)2 (1.0 M in heptane with up to 1 wt% triethylaluminum, 28 mL) was slowly added within 2 min, ensuring an excess of (CH3)2SBH3 at all times [26]. A white precipitate immediately formed upon addition of Mg(C4H9)2 and the reaction continued for 20 h with stirring at room temperature. The product, Mg(BH4)2·½S(CH3)2, was washed with toluene and heated to 143 °C for 4.5 h in a evacuated Schlenk tube to form α-Mg(BH4)2, denoted sample s1.
Diisopropylamine magnesium borohydride, Mg(BH4)2∙2(CH3)2CHNH2 was synthesized as described in ref. [6], by dissolving finely ground α-Mg(BH4)2 (300 mg) in 1.2 mL isopropylamine, (CH3)2CHNH2, IPA) while stirring. The reaction was allowed to continue for 30 min in an ice-bath, after which the product was dried for 40 min in vacuum at 25 °C and denoted s4.
Monoisopropylamine magnesium borohydride, Mg(BH4)2∙(CH3)2CHNH2 was mechanochemically synthesized using Mg(BH4)2∙2(CH3)2CHNH2 (s4, 277.9 mg, 1.5 mmol) and α-Mg(BH4)2 (s1, 101.9 mg, 1.9 mmol). The reactants were ball-milled in a WC vial using three 10 mm WC balls for 60 repetitions of 2 min, with the milling intervened by 2 min of pause, i.e., total milling time 120 min and a ball-to-sample ratio of ~54/1. This sample is denoted s3.
A nanocomposite was prepared by adding nano particulate, 50 wt% Al2O3 (13 nm), to sample s3 mechanochemically. Al2O3 (100.1 mg) and s3 (100.5 mg) were milled at 350 rpm for 2 min intervened by 2 min of pause, i.e., total milling time 120 min and a ball-to-sample ratio of ~68/1.
4.2. Characterization
Powder X-ray diffraction (PXD) data were obtained using a Rigaku Smartlab diffractometer equipped with a monochromatic rotating Cu source (λ = 1.54056 Å). Sample preparation was done in an argon environment where samples were packed in 0.5 mm (outer diameter) borosilicate capillaries and sealed with grease to avoid air exposure.
In situ synchrotron radiation powder X-ray diffraction (SR-PXD) data were acquired at the I11 beamline at the Diamond light source, Oxford, UK (λ = 0.826366 Å) [28]. The samples were measured in the temperature range from 20 to 75 °C at a heating rate of 2 °C per min.
The software FOX was used for indexing of the unit cell whereafter the structure was solved by ab initio structure determination [29,30]. Subsequently, atomic positions were refined by Rietveld refinements with the software Fullprof, treating BH4− and (CH3)2CHNH2 as rigid bodies [30,31].
Density functional theory (DFT) structural optimization was carried out using the GPAW code v. 21.1.0 with a plane wave basis set [32,33], the Atomic Simulation Environment (ASE) software package [34] and the exchange–correlation functionals PBE [35] and BEEF-vdW [36]. Several structure optimization runs were carried out using different plane-wave cutoff energies between 340 eV and 550 eV and either (1∙1∙1) or (2∙2∙2) k point sampling. The optimizations were initiated from the experimental structure as shown in Figure 2 and terminated when the maximum force on any atom fell below 0.01 eV/Å. The small changes to the numerical settings caused the optimization runs to find different local minima on the potential energy surface, corresponding to a structure that resembled the experimental one, as well as several more disordered structures. During the optimizations, all atoms were allowed to relax, and the unit cell was kept fixed to the experimentally determined size. The structures and energies presented in the supporting information were obtained from a final structural optimization run of the obtained minimum energy structures using a cutoff energy of 500 eV and (1∙1∙1) k point sampling.
Nuclear magnetic resonance (NMR) measurements were performed on a Bruker Ascend 400 MHz spectrometer equipped with a 1H–13C–15N 5 mm TXI liquid state probe. Samples were dissolved in deuterated dimethylsulfoxide in NMR tubes before measuring. The integrated intensities of 1H on BH4− and isopropylamine were used to calculate the sample composition. The integral of these peaks was normalized to their abundance in the sample as described in ref. [6].
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were measured using a PerkinElmer STA 6000 coupled with a mass spectrometer (MS) (Hiden Analytical HPR-20 QMS sampling system). Approximately 2 mg was placed in a closed Al2O3 crucible and was heated from 30 to 300 °C (ΔT/Δt = 2 °C/min) with an argon purge rate of 30 mL/min. The lid had a small hole for outlet gas, which was examined using mass spectrometry for hydrogen (m/z = 2) and isopropylamine (m/z = 44).
Electrochemical impedance spectroscopy (EIS) measurements were performed from 7∙107 Hz to 1 Hz using a Biologic MTZ-35 impedance analyzer with a symmetrical molybdenum sample holder equipped with a 4 probe setup. Samples were pressed in a hydraulic press at 1 tonne at RT for 1 min. Samples were heated using a custom-made furnace at 2 °C/min and kept at a constant temperature for one minute when the set temperature was reached. Data were fitted using an Q1/(R1+Q2) equivalent circuit, where R1 represents the charge transfer resistance and Q1 and Q2 are constant phase elements. Q1 accounts for the depressed semicircles of real systems and Q2 is used to represent the mass transfer, as the observed mass transfer cannot be described by standard capacitors or Warburg elements. When fitted, Q1 will act as a capacitor (α~0.97), essentially creating an RC circuit with R1 and Q2 acting as mass transfer elements (α~0.7). From the charge transfer resistance, ionic conductivity is calculated as d/A/R1, where d is the thickness of the pellet and A is the area. Activation energies (Ea) were extracted from linear fits to log(σi) versus 1/T, where σi is the ionic conductivity. Because of the 4 probe setup, we assumed the resistance of the setup to be negligible. First, the sample was cooled to −7.9 °C using an ethanol and dry ice bath where it was held for 15 min to ensure the temperature had stabilized. Using a heat blower, the mixture was brought up to 0 °C where dry ice was added to stabilize the temperature. Above RT, the custom-made oven was used instead. The sample was subjected to temperature increases between 30 and 40 °C three times before being cooled to −2.6 °C. Due to sample s2 becoming soft, EIS measurements were only conducted up to 40 °C.
5. Conclusions
The compound Mg(BH4)2∙(CH3)2CHNH2 was synthesized using a mechanochemical method. From SR-PXD it was found to have a polar helical 1D structure consisting of alternating magnesium environments ([Mg(BH4)4] and [Mg(BH4)2(NH2CH(CH3)2)2]). These are surrounded by weakly interacting apolar layers to form a 3D structure. The structure was solved in I212121 with the unit cell parameters a = 9.8019(1) Å, b = 12.1799(2) Å and c = 17.3386(2) Å. Discrepancies between the refined model and collected data were attributed to disorder, especially from the weakly interacting isopropyl groups (CH3)2CH–. From DFT optimization it was found that the structure is metastable and that more stable distorted structures could exist. This could also explain the discrepancies between the refined model and the data. From SR-PXD, the compound was found to have a melting point of 73 °C while Mg(BH4)2∙2(CH3)2CHNH2 formed during heating and melted at 56 °C, which is lower than reported previously. This is assigned to eutectic melting. From TGA-DSC-MS investigations, IPA release first occurs at 108 °C however this might be due to surface-coordinated IPA being released. The compound was found to have a very low activation energy of 0.85 eV, which is likely caused by weak hydrophobic interactions in interstitial sites. The composite (~0.8)Mg(BH4)2∙(CH3)2CHNH2−(~0.2)Mg(BH4)2∙2(CH3)2 CHNH2 was melted onto 13 nm Al2O3 nanoparticles at 60 °C while in a compressed state after applying a pressure of 2.5 GPa. This formed a paste-like material which lowered the activation energy to 0.65 eV and increased conductivity at 40 °C by a factor of three and at −10 °C by a factor of ten. This paste-like state remains stable for more than 24 h and at temperatures lower than −10 °C.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11010017/s1, Figure S1: Most stable structure of Mg(BH4)2∙(CH3)2CHNH2, Figure S2: Comparison between the metastable structures, Figure S3: Structures gained from DFT, Figure S4: Rejected model found from ab inito structural solution, Figure S5: Ionic conductivity of Mg(BH4)2∙IPA@Al2O3 (s5) without preheating, Figure S6: Nyquist plot of Mg(BH4)2∙1.2IPA-Al2O3; Table S1: DFT relative potential energies.
Author Contributions
Formal analysis, investigation, data curation, visualization, L.G.K.; Formal analysis, investigation, data curation, M.B.A.; methodology, software, validation, resources, M.A.; Conceptualization, supervision, project administration, funding acquisition T.R.J.; All authors contributed to writing—original draft preparation and to review and editing; All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the Danish Council for Independent Research, Technology and Production Solid-State Magnesium Batteries—SOS-MagBat (9041-00226B) and the Danish Natural Science Research Councils (DanScatt). Funding from the Danish Ministry of Higher Education and Science through the SMART Lighthouse is gratefully acknowledged.
Data Availability Statement
SR-PXD data as well as EIS data will be made available on DOI: 10.6084/m9.figshare.20579484.
Acknowledgments
Affiliation with the Center for Integrated Materials Research (iMAT) at Aarhus University is gratefully acknowledged. The synchrotron data used in this study were collected at the I11 beamline at the Diamond light source. We thank Stephen Thompson and Eamonn Connolly for setting up the experiment.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wu, F.; Maier, J.; Yu, Y. Guidelines and Trends for Next-Generation Rechargeable Lithium and Lithium-Ion Batteries. Chem. Soc. Rev. 2020, 49, 1569–1614. [Google Scholar] [CrossRef] [PubMed]
- Kulova, T.L.; Fateev, V.N.; Seregina, E.A.; Grigoriev, A.S. A Brief Review of Post-Lithium-Ion Batteries. Int. J. Electrochem. Sci. 2020, 15, 7242–7259. [Google Scholar] [CrossRef]
- U.S. Geological Survey. Mineral Commodity Summaries 2022; Mineral Commodity Summaries: Reston, VA, USA, 2022; Volume 2022, p. 202. [Google Scholar] [CrossRef]
- Famprikis, T.; Canepa, P.; Dawson, J.A.; Islam, M.S.; Masquelier, C. Fundamentals of Inorganic Solid-State Electrolytes for Batteries. Nat. Mater. 2019, 18, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Skov, L.N.; Grinderslev, J.B.; Rosenkranz, A.; Lee, Y.-S.; Jensen, T.R. Towards Solid-State Magnesium Batteries: Ligand-Assisted Superionic Conductivity. Batter. Supercaps 2022, 5, e202200163. [Google Scholar] [CrossRef]
- Kristensen, L.G.; Amdisen, M.B.; Skov, L.N.; Jensen, T.R. Fast Magnesium Ion Conducting Isopropylamine Magnesium Borohydride Enhanced by Hydrophobic Interactions. Phys. Chem. Chem. Phys. 2022, 24, 18185–18197. [Google Scholar] [CrossRef]
- Yan, Y.; Dononelli, W.; Jørgensen, M.; Grinderslev, J.B.; Lee, Y.-S.; Cho, Y.W.; Černý, R.; Hammer, B.; Jensen, T.R. The Mechanism of Mg2+ Conduction in Ammine Magnesium Borohydride Promoted by a Neutral Molecule. Phys. Chem. Chem. Phys. 2020, 22, 9204–9209. [Google Scholar] [CrossRef]
- Yan, Y.; Grinderslev, J.B.; Jørgensen, M.; Skov, L.N.; Skibsted, J.; Jensen, T.R. Ammine Magnesium Borohydride Nanocomposites for All-Solid-State Magnesium Batteries. ACS Appl. Energy Mater. 2020, 3, 9264–9270. [Google Scholar] [CrossRef]
- Kisu, K.; Kim, S.; Inukai, M.; Oguchi, H.; Takagi, S.; Orimo, S. Magnesium Borohydride Ammonia Borane as a Magnesium Ionic Conductor. ACS Appl. Energy Mater. 2020, 3, 3174–3179. [Google Scholar] [CrossRef]
- Xiao, N.; McCulloch, W.D.; Wu, Y. Reversible Dendrite-Free Potassium Plating and Stripping Electrochemistry for Potassium Secondary Batteries. J. Am. Chem. Soc. 2017, 139, 9475–9478. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, J.; Han, L.; Wang, Z.; Wang, H.; Zhao, Q.; Liu, J.; Pan, F. A MOF-Based Single-Ion Zn2+ Solid Electrolyte Leading to Dendrite-Free Rechargeable Zn Batteries. Nano Energy 2019, 56, 92–99. [Google Scholar] [CrossRef]
- Cao, C.; Li, Y.; Feng, Y.; Peng, C.; Li, Z.; Feng, W. A Solid-State Single-Ion Polymer Electrolyte with Ultrahigh Ionic Conductivity for Dendrite-Free Lithium Metal Batteries. Energy Storage Mater. 2019, 19, 401–407. [Google Scholar] [CrossRef]
- Xia, S.; Wu, X.; Zhang, Z.; Cui, Y.; Liu, W. Practical Challenges and Future Perspectives of All-Solid-State Lithium-Metal Batteries. Chem 2019, 5, 753–785. [Google Scholar] [CrossRef]
- Yan, Y.; Grinderslev, J.B.; Lee, Y.-S.; Jørgensen, M.; Cho, Y.W.; Černý, R.; Jensen, T.R. Ammonia-Assisted Fast Li-Ion Conductivity in a New Hemiammine Lithium Borohydride, LiBH4·1/2NH3. Chem. Commun. 2020, 56, 3971–3974. [Google Scholar] [CrossRef]
- Blanchard, D.; Nale, A.; Sveinbjörnsson, D.; Eggenhuisen, T.M.; Verkuijlen, M.H.W.; Suwarno; Vegge, T.; Kentgens, A.P.M.; Jongh, P.E. de Nanoconfined LiBH4 as a Fast Lithium Ion Conductor. Adv. Funct. Mater. 2015, 25, 184–192. [Google Scholar] [CrossRef]
- Huot, J.; Cuevas, F.; Deledda, S.; Edalati, K.; Filinchuk, Y.; Grosdidier, T.; Hauback, B.C.; Heere, M.; Jensen, T.R.; Latroche, M.; et al. Mechanochemistry of Metal Hydrides: Recent Advances. Materials 2019, 12, 2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, B.; Grinderslev, J.B.; Møller, K.T.; Paskevicius, M.; Jensen, T.R. From Metal Hydrides to Metal Borohydrides. Inorg. Chem. 2018, 57, 10768–10780. [Google Scholar] [CrossRef] [PubMed]
- Černý, R.; Filinchuk, Y.; Hagemann, H.; Yvon, K. Magnesium Borohydride: Synthesis and Crystal Structure. Angew. Chem. Int. Ed. 2007, 46, 5765–5767. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, L.H.; Ban, V.; Møller, K.T.; Lee, Y.-S.; Cho, Y.W.; Besenbacher, F.; Filinchuk, Y.; Skibsted, J.; Jensen, T.R. Synthesis, Crystal Structure, Thermal Decomposition, and 11B MAS NMR Characterization of Mg(BH4)2(NH3BH3)2. J. Phys. Chem. C 2014, 118, 12141–12153. [Google Scholar] [CrossRef]
- Jepsen, L.H.; Ley, M.B.; Filinchuk, Y.; Besenbacher, F.; Jensen, T.R. Tailoring the Properties of Ammine Metal Borohydrides for Solid-State Hydrogen Storage. ChemSusChem 2015, 8, 1452–1463. [Google Scholar] [CrossRef]
- Goursot, A.; Mineva, T.; Kevorkyants, R.; Talbi, D. Interaction between N-Alkane Chains: Applicability of the Empirically Corrected Density Functional Theory for Van Der Waals Complexes. J. Chem. Theory Comput. 2007, 3, 755–763. [Google Scholar] [CrossRef]
- Roedern, E.; Kühnel, R.-S.; Remhof, A.; Battaglia, C. Magnesium Ethylenediamine Borohydride as Solid-State Electrolyte for Magnesium Batteries. Sci. Rep. 2017, 7, 46189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinderslev, J.B.; Skov, L.N.; Andreasen, J.G.; Ghorwal, S.; Skibsted, J.; Jensen, T.R. Methylamine Lithium Borohydride as Electrolyte for All-Solid-State Batteries. Angew. Chem. Int. Ed. 2022, 61, e202203484. [Google Scholar] [CrossRef] [PubMed]
- Grinderslev, J.B.; Ley, M.B.; Lee, Y.-S.; Jepsen, L.H.; Jørgensen, M.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. Ammine Lanthanum and Cerium Borohydrides, M(BH4)3·nNH3; Trends in Synthesis, Structures, and Thermal Properties. Inorg. Chem. 2020, 59, 7768–7778. [Google Scholar] [CrossRef]
- Grinderslev, J.B.; Jensen, T.R. Trends in the Series of Ammine Rare-Earth-Metal Borohydrides: Relating Structural and Thermal Properties. Inorg. Chem. 2021, 60, 2573–2589. [Google Scholar] [CrossRef] [PubMed]
- Zanella, P.; Crociani, L.; Masciocchi, N.; Giunchi, G. Facile High-Yield Synthesis of Pure, Crystalline Mg(BH4)2. Inorg. Chem. 2007, 46, 9039–9041. [Google Scholar] [CrossRef]
- Filinchuk, Y.; Richter, B.; Jensen, T.R.; Dmitriev, V.; Chernyshov, D.; Hagemann, H. Porous and Dense Magnesium Borohydride Frameworks: Synthesis, Stability, and Reversible Absorption of Guest Species. Angew. Chem. Int. Ed. Engl. 2011, 50, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.P.; Parker, J.E.; Potter, J.; Hill, T.P.; Birt, A.; Cobb, T.M.; Yuan, F.; Tang, C.C. Beamline I11 at Diamond: A New Instrument for High Resolution Powder Diffraction. Rev. Sci. Instrum. 2009, 80, 075107. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Sørensen, L.H.; Filinchuk, Y.; Besenbacher, F.; Jensen, T.R. Screening of Metal Borohydrides by Mechanochemistry and Diffraction. Angew. Chem., Int. Ed. 2012, 51, 3582–3586. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Černý, R. FOX, ‘free Objects for Crystallography’: A Modular Approach Toab Initiostructure Determination from Powder Diffraction. J. Appl. Crystallogr. 2002, 35, 734–743. [Google Scholar] [CrossRef]
- Favre-Nicolin, V. Free Objects for Crystallography. Available online: https://sourceforge.net/projects/objcryst/ (accessed on 18 November 2022).
- Mortensen, J.J.; Hansen, L.B.; Jacobsen, K.W. Real-Space Grid Implementation of the Projector Augmented Wave Method. Phys. Rev. B 2005, 71, 035109. [Google Scholar] [CrossRef] [Green Version]
- Enkovaara, J.; Rostgaard, C.; Mortensen, J.J.; Chen, J.; Dułak, M.; Ferrighi, L.; Gavnholt, J.; Glinsvad, C.; Haikola, V.; Hansen, H.A.; et al. Electronic Structure Calculations with GPAW: A Real-Space Implementation of the Projector Augmented-Wave Method. J. Phys. Condens. Matter 2010, 22, 253202. [Google Scholar] [CrossRef]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The Atomic Simulation Environment—a Python Library for Working with Atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellendorff, J.; Lundgaard, K.T.; Møgelhøj, A.; Petzold, V.; Landis, D.D.; Nørskov, J.K.; Bligaard, T.; Jacobsen, K.W. Density Functionals for Surface Science: Exchange-Correlation Model Development with Bayesian Error Estimation. Phys. Rev. B 2012, 85, 235149. [Google Scholar] [CrossRef]
Figure 1.
Powder X-ray diffraction patterns of samples s1 to s4 measured at the I11 beamline at the Diamond light source, Oxford (UK) (s3, λ = 0.826366(3) Å) and in-house (s1, s2, s4, λ = 1.5406 Å). The pattern of sample s3 is distinct to that of the reactants, α-Mg(BH4)2 (s1) and Mg(BH4)2∙2IPA (s4), and is assigned to a new compound, Mg(BH4)2∙(CH3)2CHNH2. Sample s2 was treated in a less intense manner by mechanochemistry, which resulted in an incomplete reaction.
Figure 1.
Powder X-ray diffraction patterns of samples s1 to s4 measured at the I11 beamline at the Diamond light source, Oxford (UK) (s3, λ = 0.826366(3) Å) and in-house (s1, s2, s4, λ = 1.5406 Å). The pattern of sample s3 is distinct to that of the reactants, α-Mg(BH4)2 (s1) and Mg(BH4)2∙2IPA (s4), and is assigned to a new compound, Mg(BH4)2∙(CH3)2CHNH2. Sample s2 was treated in a less intense manner by mechanochemistry, which resulted in an incomplete reaction.
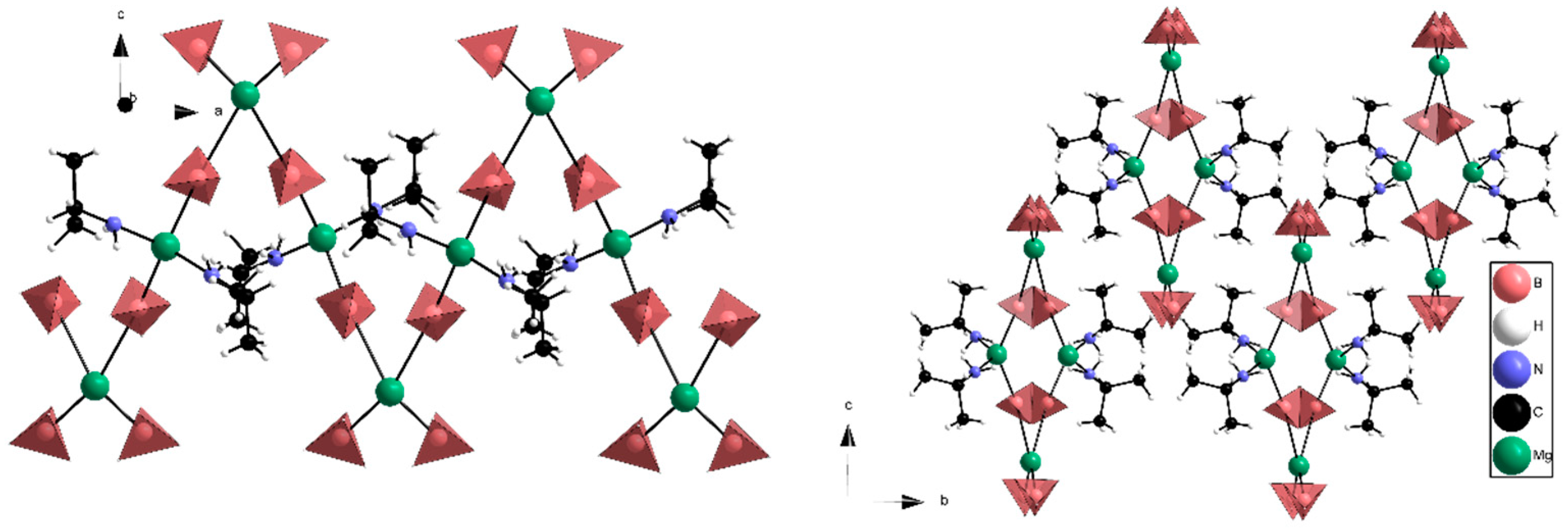
Figure 2.
Experimental structure of Mg(BH4)2∙(CH3)2CHNH2 viewed in the a-c plane (left) and b-c plane (right). In the a-c plane the chain-like structure with alternating Mg2+ environments is visible. The b-c plane shows how the chains arrange to form flat polar helixes, which are surrounded by apolar regions. Atoms: magnesium (green), carbon (black), nitrogen (blue), BH4− (red tetrahedron) and hydrogen (white).
Figure 2.
Experimental structure of Mg(BH4)2∙(CH3)2CHNH2 viewed in the a-c plane (left) and b-c plane (right). In the a-c plane the chain-like structure with alternating Mg2+ environments is visible. The b-c plane shows how the chains arrange to form flat polar helixes, which are surrounded by apolar regions. Atoms: magnesium (green), carbon (black), nitrogen (blue), BH4− (red tetrahedron) and hydrogen (white).

Figure 3.
Rietveld refinement of the structural model of Mg(BH4)2∙(CH3)2CHNH2 in the space group I212121, using the powder X-ray diffraction pattern of Mg(BH4)2∙1.20IPA (s3). R-factors: Rp = 1.26, Rwp = 2.47, conventional R-factors: Rp = 37.6 and Rwp = 23.2 and RBragg = 20.3.
Figure 3.
Rietveld refinement of the structural model of Mg(BH4)2∙(CH3)2CHNH2 in the space group I212121, using the powder X-ray diffraction pattern of Mg(BH4)2∙1.20IPA (s3). R-factors: Rp = 1.26, Rwp = 2.47, conventional R-factors: Rp = 37.6 and Rwp = 23.2 and RBragg = 20.3.
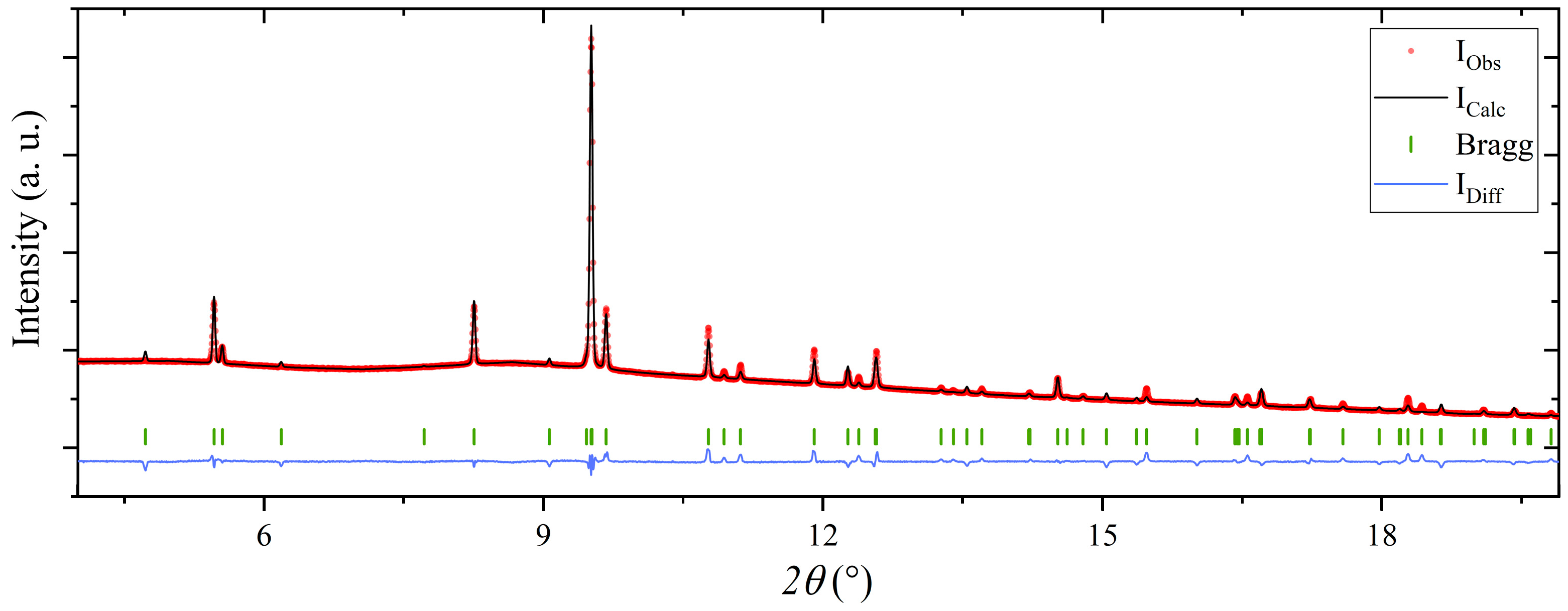
Figure 4.
(a) Temperature-resolved in situ synchrotron radiation powder X-ray diffraction of Mg(BH4)2∙1.20IPA (s3) from 20 to 75 °C (heating rate of 2 °C/min). (b,c) Selected diffraction patterns at 46 and 22 °C, respectively. The data were acquired at the I11 beamline of the Diamond light source (λ = 0.826366 Å).
Figure 4.
(a) Temperature-resolved in situ synchrotron radiation powder X-ray diffraction of Mg(BH4)2∙1.20IPA (s3) from 20 to 75 °C (heating rate of 2 °C/min). (b,c) Selected diffraction patterns at 46 and 22 °C, respectively. The data were acquired at the I11 beamline of the Diamond light source (λ = 0.826366 Å).

Figure 5.
Simultaneous measurement of thermogravimetry (TGA), differential scanning calorimetry (DSC) and mass spectroscopy (MS) of Mg(BH4)2·IPA (s3). (Top) TGA data and the gradient of the differential scanning calorimetry data. (Bottom) MS analysis of hydrogen and isopropylamine release.
Figure 5.
Simultaneous measurement of thermogravimetry (TGA), differential scanning calorimetry (DSC) and mass spectroscopy (MS) of Mg(BH4)2·IPA (s3). (Top) TGA data and the gradient of the differential scanning calorimetry data. (Bottom) MS analysis of hydrogen and isopropylamine release.
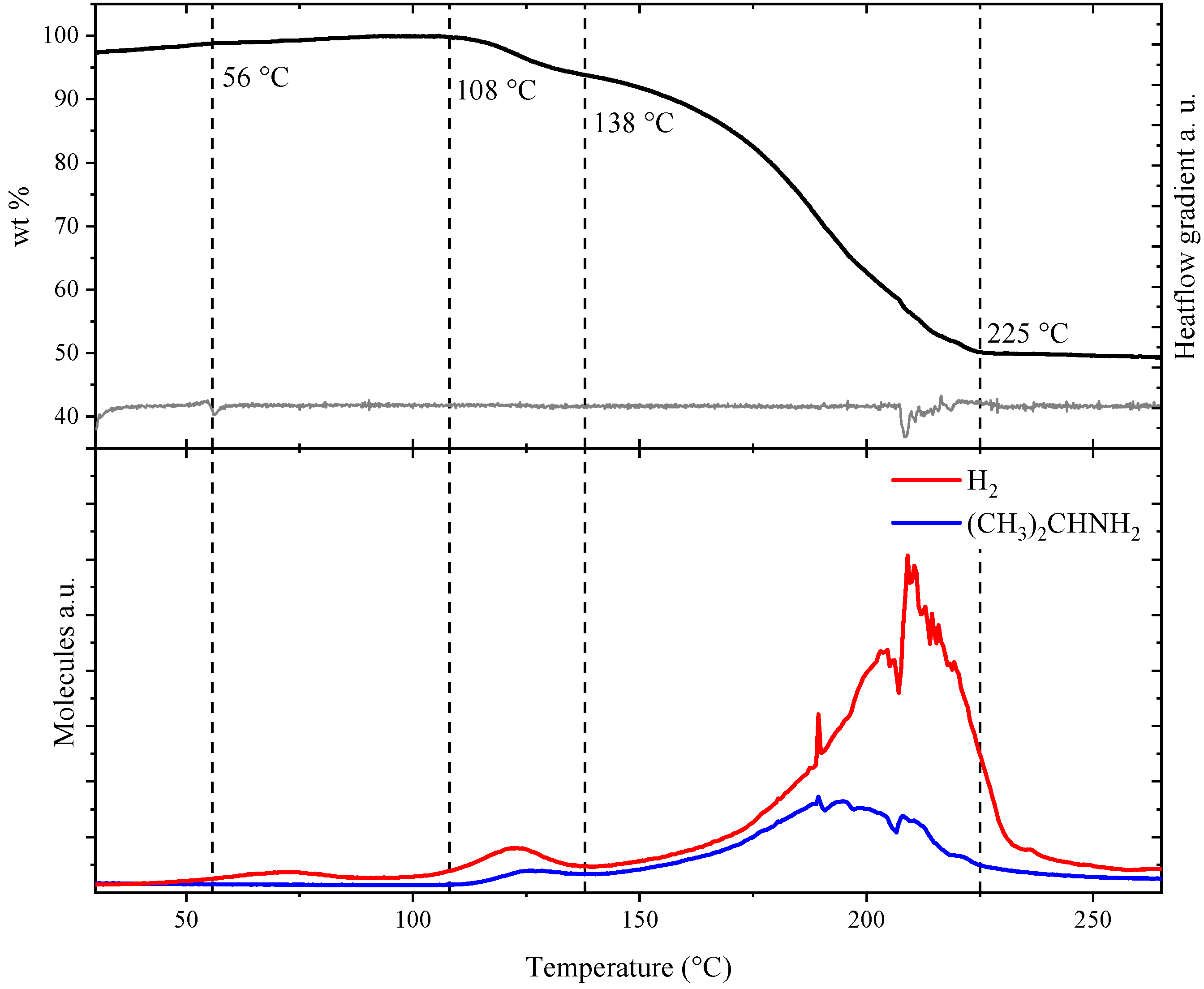
Figure 6.
Magnesium (Mg2+) ionic conductivity as a function of temperature measured by electrochemical impedance spectroscopy (EIS) of Mg(BH4)2∙(CH3)2CHNH2 (s3) during heating from −7.9 to 40 °C (data shown as circles). The same sample (s3) was then heated and cooled three times between 30 and 40 °C before being cooled to −2.6 °C and measured by EIS during heating to 40 °C (open spheres). Nyquist plots of Mg(BH4)2-1.20IPA-Al2O3 (s5) are provided in Figure S6. Magnesium ionic conductivity data and activation energies of Mg(BH4)2∙NH2(CH2)2NH2, Mg(BH4)2∙NH3 and Mg(BH4)2∙2NH3BH3, and of Mg(BH4)2∙1.5IPA−MgO are also included for comparison (data from refs. [6,7,9,22]).
Figure 6.
Magnesium (Mg2+) ionic conductivity as a function of temperature measured by electrochemical impedance spectroscopy (EIS) of Mg(BH4)2∙(CH3)2CHNH2 (s3) during heating from −7.9 to 40 °C (data shown as circles). The same sample (s3) was then heated and cooled three times between 30 and 40 °C before being cooled to −2.6 °C and measured by EIS during heating to 40 °C (open spheres). Nyquist plots of Mg(BH4)2-1.20IPA-Al2O3 (s5) are provided in Figure S6. Magnesium ionic conductivity data and activation energies of Mg(BH4)2∙NH2(CH2)2NH2, Mg(BH4)2∙NH3 and Mg(BH4)2∙2NH3BH3, and of Mg(BH4)2∙1.5IPA−MgO are also included for comparison (data from refs. [6,7,9,22]).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of the investigated samples including synthesis method, molar reactant ratios, crystalline compounds observed in the product using powder X-ray diffraction, and the (CH3)2CHNH2 content as measured by liquid-state 1H nuclear magnetic resonance.
Table 1.
Overview of the investigated samples including synthesis method, molar reactant ratios, crystalline compounds observed in the product using powder X-ray diffraction, and the (CH3)2CHNH2 content as measured by liquid-state 1H nuclear magnetic resonance.
| Sample | Synthesis Method | Reactants | Reactant Ratio (s3/(s1 + s3)) | Crystalline Compounds | IPA Content (1H NMR) |
|---|---|---|---|---|---|
| s1 | Solvent-based | - | - | α-Mg(BH4)2 | - |
| s2 | Mechanochem. | s1 & s4 | 0.44 | Mg(BH4)2∙(CH3)2CHNH2 | 1.20 |
| s3 | Mechanochem. | s1 & s4 | 0.44 | Mg(BH4)2∙(CH3)2CHNH2 | 1.20 |
| s4 | Solvent-based | - | - | Mg(BH4)2∙2(CH3)2CHNH2 | 2.20 |
| s5 | Mechanochem. | s3 & Al2O3 | - | Mg(BH4)2∙(CH3)2CHNH2 + Al2O3 (50 wt%) | 1.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kristensen, L.G.; Amdisen, M.B.; Andersen, M.; Jensen, T.R. Synthesis, Structure and Mg2+ Ionic Conductivity of Isopropylamine Magnesium Borohydride. Inorganics 2023, 11, 17. https://doi.org/10.3390/inorganics11010017
AMA Style
Kristensen LG, Amdisen MB, Andersen M, Jensen TR. Synthesis, Structure and Mg2+ Ionic Conductivity of Isopropylamine Magnesium Borohydride. Inorganics. 2023; 11(1):17. https://doi.org/10.3390/inorganics11010017
Chicago/Turabian StyleKristensen, Lasse G., Mads B. Amdisen, Mie Andersen, and Torben R. Jensen. 2023. "Synthesis, Structure and Mg2+ Ionic Conductivity of Isopropylamine Magnesium Borohydride" Inorganics 11, no. 1: 17. https://doi.org/10.3390/inorganics11010017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.