

Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook

School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459, Singapore
*
Author to whom correspondence should be addressed.
Inorganics 2023, 11(12), 464; https://doi.org/10.3390/inorganics11120464
Submission received: 27 September 2023
/
Revised: 10 November 2023
/
Accepted: 22 November 2023
/
Published: 30 November 2023
(This article belongs to the Special Issue 10th Anniversary of Inorganics: Inorganic Materials)
Abstract
:To mitigate the effect of CO2 on climate change, significant efforts have been made in the past few decades to capture CO2, which can then be further sequestered or converted into value-added compounds, such as methanol and hydrocarbons, by using thermochemical or electrocatalytic processes. However, CO2 capture and conversion have primarily been studied independently, resulting in individual processes that are highly energy-intensive and less economically viable due to high capital and operation costs. To enhance the overall process efficiency, integrating CO2 capture and conversion into a single system offers an opportunity for a more streamlined process that can reduce energy and capital costs. This strategy can be achieved by employing dual function materials (DFMs), which possess the unique capability to simultaneously adsorb and convert CO2. These materials combine basic metal oxides with active metal catalytic sites that enable both sorption and conversion functions. In this review paper, we focus on the recent strategies that utilize mixed metal oxides as DFMs. Their material design and characteristics, reaction mechanisms, as well as performance and limitations will be discussed. We will also address the challenges associated with this integrated system and attempt to provide insights for future research endeavors.
1. Introduction
With the concentration of carbon dioxide (CO2) in the earth’s atmosphere increasing at an unprecedented rate, climate change has become one of the most pressing challenges of our time due to its adverse impact on environmental and ecological welfare, as reported by the Intergovernmental Panel on Climate Change (IPCC) [1]. Addressing this issue requires a comprehensive approach that encompasses CO2 emission reductions, effective CO2 storage, and the sustainable utilization of CO2 as chemical feedstock to close the carbon cycle. Respectively, CO2 capture and CO2 conversion have garnered considerable attention as potential solutions to mitigate its impact and, concurrently, create other value-added products. These approaches are not only necessary to slow down global warming, but also to offer economic opportunities across various industries [2,3]. Naims shows that the economic feasibility for carbon capture and utilization (CCU) is specific to the technology; that it depends on the process efficiency in using CO2 as a cheaper alternative feedstock to more expensive raw materials produced from fossil fuel [4].
Being primarily emitted from combustion processes, the strategies for CO2 capture are determined by several factors, such as its partial pressure, operating conditions, and flue gas compositions. In general, there are typically three types of capture categories, as illustrated in Figure 1, namely (1) pre-combustion, (2) oxyfuel combustion, and (3) post-combustion, depending on the installation location [5]. A general comparison of their advantages and disadvantages in terms of their energy requirement, costs, CO2 recovery, and limitations is presented in Table 1. Pre-combustion capture removes CO2 before combustion is completed. It is typically integrated with a gasifier or reformer where syngas is produced. Through the intermediate removal of CO2, H2-rich gas is obtained, which can be further used for heat/power generation. Oxyfuel combustion employs either nearly-pure oxygen environments for combustion, or chemical looping combustion (CLC), in which solid oxides are used as oxygen carriers, resulting in a high concentration of CO2 that allows for more efficient and inexpensive separation. However, the combustion itself would be relatively costly due to high oxygen purity demands. Lastly, post-combustion capture removes CO2 from the flue gas. It is considered the most established technology [5], where it typically employs liquid amine absorption or solid adsorption. Due to the low CO2 partial pressure in the flue gas (ca. 10–15%), large volumes of gas, as well as large amounts of energy and equipment size are required, making post-combustion capture processes relatively expensive. For comparison, the mean capture cost from high CO2 purity sources via oxyfuel combustion ranges from USD 10 to USD 50 per ton of CO2 mitigated, while that from post-combustion flue gas amine scrubbing ranges from USD 50 to USD 120 [3]. Following its capture, high-purity CO2 can be recovered via temperature or pressure swing regeneration, after which it is compressed and transported for storage (e.g., geological injection, mineralization) or further utilized or converted.
CO2 conversion technologies seek to transform the captured CO2 into useful chemicals, fuels, and materials. Although conversion processes may not directly lead to a net carbon reduction, it enables a circular utilization of CO2 wastes as feedstock, which may reduce the overall carbon footprint of the corresponding chemical and fuel production, thus leading to more environmentally friendly production processes, as shown in a case study of polyol production for polyurethane [7]. A comprehensive review of sustainable CO2 conversion by incorporating a life cycle assessment (LCA) in various CO2 conversion pathways has been published by Artz et al. [8], who concluded that replacing energy-intensive feedstock with CO2 seems highly promising, particularly when being integrated with renewable energy. In addition, as a C1 building block, the use of CO2 may also create new, greener synthetic pathways [9].
Despite significant progress having been made for CO2 capture and CO2 conversion individually, numerous challenges remain for each technology, most notably the cost effectiveness and the process scalability. Generating high purity of CO2 intermediates for subsequent utilization requires a large amount of energy for CO2 recovery, the regeneration of the capture media, and the subsequent gas compression. The purified CO2 stream then needs to be transported to the location where it will be converted to the desired product, thus further increasing energy demands. To address these issues, combining CO2 capture and utilization in an integrated system, where in situ conversion will also serve to regenerate the capture media in the same reactor, would be a potential solution to eliminate both compression and transportation costs, thus making the whole process more economical and efficient. For example, the total energy required to produce 1 metric ton of methanol from separate direct air capture and hydrogenation reactions is about 49.4 GJ, out of which 27 GJ is consumed in the CO2 capture, recovery, and distribution steps. For comparison, to achieve the same product yield, the integrated CO2 capture and conversion (ICCC) system only requires less than 50% of the total energy requirement, as reported by Freyman et al. [10]. Additionally, ICCC could also reduce capital costs by approximately 38%, as fewer unit operations are required. Freyman et al. coined a term “Reactive Capture” to describe this ICCC system, the concept of which is depicted in Figure 2. To make this system work effectively, the optimization of the reaction conditions, the tuning of the material properties, as well as designing an appropriate reactor system configuration are necessary to ensure a good ICCC performance with good product selectivity and catalyst stability.
In this review paper, we will focus on the development of mixed metal oxides (MMO) as dual function materials (DFMs). To perform both CO2 capture and CO2 conversion, DFMs typically are synthesized as a combination of a CO2 sorbent and a catalytic component. In the presence of CO2 and other secondary reactants (e.g., H2, for the methanation of CO2), the sorbent component first captures CO2, which then reacts with secondary reactants on the active catalytic sites, converting it into final products [10]. A schematic diagram of ICCC with DFMs is illustrated in Figure 3. The efficiency of the DFM would therefore depend on the CO2 capture capacity and selectivity, as well as its catalytic activity. Among various solid sorbents, the choice of metal oxides, particularly alkali and alkaline earth oxides, as the sorbent component of a DFM, is convenient, in that the catalytic component would typically also be a transition or noble metal, which can be facilely incorporated into the DFM during its synthesis, such as via co-precipitation or incipient wetness impregnation.
To give an overview of the recent trends in this field, we have carried out a literature search on the Web of Science database with the following keywords:
- “dual function material” AND “CO2 capture” AND “CO2 conversion”
- “dual function material” AND “CO2 capture” AND “CO2 utilization”
It was found that about 60 relevant papers were published in the span of 10 years, between 2012 and 2022. Farrauto et al. were among the pioneering research groups who first published their work on the development of a DFM in 2015, which comprised ruthenium as the methanation catalyst and nano-dispersed CaO as the CO2 sorbent, which are both supported on a porous γ-Al2O3 carrier [11]. The synthesized material was able to adsorb CO2 and convert it in the same reactor into synthetic natural gas by using H2 gas as the co-reactant at 320 °C. Upon the introduction of steam into the reactor, the material showed a stable methanation performance with more than 99% CH4 purity after 20 cycles. The initial success of this experiment has since gathered increasing interests to explore DFM systems, as indicated by the increasing number of publications in recent years (Figure 4). We have seen efforts being made to improve the CO2 capture capacity, conversion, and selectivity while reducing the precious metal content. The first review paper on DFMs was published in 2019 by Melo Bravo and Debecker [12], which focused on combined CO2 capture and methanation, followed by several others in 2020 [6] and 2021 [13,14,15,16]. The highlights of these review papers are summarized in Table 2.
To continue building upon the previous reviews, herein we aim to cover the latest developments on the application of DFMs that employ solid metal oxides for ICCC, particularly from 2021–2023. We will begin our discussion by first giving an overview of the principles of CO2 capture with metal oxides as solid sorbents, as well as the catalytic conversion of CO2 from thermodynamic and material perspectives, followed by the typical synthetic methods of DFMs. Thereafter, the performance of various DFMs in ICCC systems will be further assessed for key reactions, such as methanation, RWGS, and DRM, by evaluating key metrics such as the CO2 capture capacity, selectivity, reaction conversion, and stability. We will correlate these metrics with material design parameters, such as the types of metal oxides used as adsorbents, metallic active sites as catalysts, and the synthetic methods. Mechanistic studies on the molecular interaction between CO2 and the adsorption and catalytic active sites will also be presented to shed some light on the relationship between the materials’ physicochemical properties and their performance. As a conclusion, we will present our perspectives, as well as an outlook on the future opportunities with the emerging technologies of ICCC with DFMs.
2. Metal Oxides as Sorbents for CO2 Capture
Various CO2 capture methods have been extensively studied and explored, and several excellent reviews have been recently published [17,18,19,20,21,22,23,24,25,26,27,28]. An overview of these technologies is presented in Figure 5, among which, liquid absorption in organic amines, such as methanolamine (MEA) and n-methyldiethanolamine (MDEA), have seen the most implementation in large demonstration plants worldwide [29] due to its high absorption capacity. However, this system requires a high energy consumption for amine regeneration, which is prone to degradation. It is also highly corrosive, thus leading to high operating and capital costs.
As potential alternatives to liquid absorption, solid sorbents, such as zeolites [30], hydrotalcites [17,20,31,32], magnesium oxides [33,34,35,36], metal organic frameworks (MOFs) [23,37], and porous organic polymers (POPs) [38,39,40], have been studied extensively due to their easy regeneration and tunable pore size and surface properties. Given their wide variety, the suitability of solid adsorbents depends on their application temperatures, typically ranging from low temperatures (<200 °C) for MOFs, POPs, and zeolites to medium temperatures (200–400 °C) for hydrotalcites and MgO-based sorbents. An overview of commonly used solid sorbents and their typical application temperatures is presented in Figure 6.
In principle, their CO2 capture capability primarily relies on the physical adsorption of CO2 without forming a chemical bond. Physisorption is typically mildly exothermic (−25 to −40 kJ/mol) and requires less energy for regeneration as compared to chemisorption processes. Owing to its exothermic nature, the adsorption of CO2 decreases at a higher temperature, making it more suitable for low-to-medium operating temperatures (<400 °C). However, physisorption processes are usually less selective, leading to the adsorption of other gases, such as N2 and water vapor, thus lowering the adsorption capacity of CO2, which is the primary challenge that needs to be solved in order to scale up their application. A common strategy to improve the CO2 adsorption capacity is by increasing the basicity of the materials via the incorporation of alkaline metals, such as K and Na, though their amounts should be carefully studied as high metal loading may decrease the surface area of the material. For example, for hydrotalcite-based materials, many attempts have been made to modify Mg/Al-based hydrotalcites with K [41,42,43,44,45], Na [43,46], and Cs [42,43,47], with typical optimum K loading of about 20 wt% to achieve the maximum sorption capacity at temperatures between 300 and 400 °C.
On the other hand, for high-temperature CO2 adsorption (see Figure 6), calcium looping has been found to be a promising technology [25,48]. It utilizes CaO to react with CO2 to form calcium carbonate (carbonation), which can be calcined back to CaO for reuse, while recovering high-purity CO2, as shown in Equation (1).
Owing to its low cost and high theoretical sorption capacity of up to 78.6 wt% (17.8 mmol CO2/g sorbent), CaO has been extensively studied, and a pilot scale of this technology has been implemented in cement production [49,50]. An economic feasibility study by MacKenzie et al. shows that calcium looping is highly competitive with liquid amine-based systems [51]. However, to fully realize the industrial applications of the CaO sorbent, its material stability upon prolonged use at a high temperature needs to be improved as it is susceptible to sintering and surface carbonate poisoning [52], which may significantly decrease its CO2 sorption capacity and limit its recyclability over time.
Extensive amounts of work have been devoted to the study and development of CaO-based sorbents for CO2 capture, the kinetic and thermodynamic perspectives on the mechanisms of CO2 sorption, as well as methods to improve the performance of the sorbents by mitigating their drawbacks [52,53,54,55]. The forward, highly exothermic carbonation of CaO with CO2 and the backward, endothermic calcination of CaCO3 as described in Equation (1) occur at temperature ranges of 600–900 °C [53,55]. The calcination reaction typically takes place at a higher temperature than the carbonation reaction. However, at such temperature ranges, CaO-based CO2 adsorbents may suffer from sintering, a phenomenon of the agglomeration of adsorbent particles when operating temperatures exceed the Tammann temperature (TT), at which the mobility of the grains/crystallites in the solid material becomes appreciable, leading to major changes in the particle morphology. Given that TT of CaCO3 is typically about 533 °C [56], CaO crystallite growth would primarily occur during the calcination step of CaCO3. As a result, particle sintering is inevitable under repeated carbonation–calcination cycles [57].
Lysikov et al. studied the behavior of CO2 uptake by different CaO precursors in a series of carbonation–calcination cycles in the temperature range of 750–850 °C, where they observed that post-calcination CaO does not fully re-carbonate, and that the amount of unreacted CaO increases from cycle to cycle until it forms a rigid interconnected CaO skeleton after multiple cycles, as shown in Figure 7 [58]. They, therefore, proposed a model in which, upon repeated cycles, small unreacted CaO grains agglomerate, grow into larger, denser clusters, and ultimately form a stable, non-porous recalcitrant CaO skeleton that is resistant to further sintering, as illustrated in Figure 8.
The sintering phenomenon shows to be destructive to the internal structure of the CaO-based sorbent, as it significantly reduces the particle surface area and pore volume due to the large, non-porous aggregate formation, which will eventually adversely affect the CO2 sorption capacity with each carbonation–calcination cycle [59].
The common method of investigating the CO2 sorption capacity of an adsorbent is thermogravimetric analysis (TGA), as depicted in Figure 9, which records the adsorbent’s mass increase during carbonation and mass decrease during calcination. It can be seen that CO2 uptake drastically decreases in the first few cycles, as reflected by the rapid decline of the sample weight after repeated carbonation–calcination reactions. CaO conversion to CaCO3 decreased from an initial conversion of about 60% to a residual conversion of about 7–8%. This observation is in agreement with the model proposed by Lysikov et al. where the unreacted fraction of CaO increases with every cycle and only the outer layer of the non-porous recalcitrant CaO skeleton is carbonated, leading to a relatively constant residual conversion after sufficient cycles.
Meanwhile, kinetic analysis reveals that CO2 adsorption characteristically takes place in two stages [52,54,55]—a fast, reaction-controlled stage, reflected by a spike in Figure 10 (first stage), followed by a slow, diffusion-controlled stage, reflected by a plateau in Figure 10 (second stage). As illustrated in Figure 11, in the reaction-controlled stage, carbonation may occur wherever CO2 molecules come into contact with the exposed CaO. The rate of carbonation is then dependent on the rate at which CO2 molecules reach the CaO surface, which in turn might be affected by internal characteristics such as pore structures and porosity. As the carbonation proceeds, a layer of CaCO3 forms on the unreacted CaO particle. Following this, the reaction then transitions into the diffusion-controlled stage where incoming CO2 molecules must diffuse through the CaCO3 layer to be in contact with any unreacted CaO. Consequently, the rate of carbonation decreases drastically as it now becomes limited by mass transfer.
The observed phenomena above allow us to glean important governing principles in the design of novel CO2 sorbents, which can be incorporated into the synthesis of DFMs. Firstly, an ideal sorbent should be resistant to sintering at the required sorption temperatures to ensure its stability and prolonged use. In the case of CaO, metal supports such as the oxides of Al [61,62,63,64], Mg [65,66,67], and Yb [68,69], among many others, are commonly employed. These supports enhance an adsorbent’s performance in numerous ways. For instance, certain metal oxides with high TT can be used to decorate CaO particles [67], as in the case of MgO (TT = 1276 °C), which helps enhance the stability of the material. Others form stable oxides that disperse CaO particles and thus physically retard the sintering process, as in the case of Yb2O3 [68]. Some other metals aid in dispersion by forming composite metal oxides with CaO, as in the case of Ca12Al14O33 and Ca3Al2O6 [64]. These improvements allow supported sorbents to withstand severe reaction conditions while retaining CO2 sorption capacities.
Secondly, an ideal sorbent should have a high internal surface area to allow for large sorption sites. The specific surface area measured via the Brunauer–Emmett–Teller (BET) method and the pore size measured with the Barrett–Joyner–Halenda (BJH) model are two common characteristics used to provide some insight into the internal morphology and microstructure of a sorbent. Primarily, the choice of synthetic method strongly influences the internal structures, and consequently, the specific surface area, of a sorbent [64]. Several studies evaluate the effect of various preparation methods on the sorbent’s performance and show that sol–gel methods and flame spray pyrolysis, for example, produce sorbents with higher surface areas compared to those synthesized via simpler methods, such as the co-precipitation or mixing of metal precursor solutions (e.g., metal nitrate solutions). The choice of precursor chemicals seems also to influence the surface areas of a sorbent. It is expected that different chemical species will have varying interactions with CaO, giving rise to a variety of internal structures by the end of the synthesis depending on the chemical species present [55,70,71,72].
3. Overview of CO2 Catalytic Conversion
Owing to its molecular structure, CO2 is thermodynamically stable, as reflected in Figure 12, which compares the Gibbs free energy of formation ( for different carbon-containing compounds. Having the highest carbon oxidation state and lowest Gibbs free energy state, the chemical transformation of CO2 will require considerable amounts of energy and, therefore, can only be achieved under harsh reaction conditions, such as a high temperature and pressure, leading to large energy penalties.
A strategy to make CO2 transformation more thermodynamically favorable is by reacting it with a co-reactant that has a higher Gibbs free energy. For instance, the direct production of CO from CO2 as a single reactant requires a large positive Gibbs free energy change (Equation (2)), whereas reacting it with H2 significantly reduces the Gibbs free energy change of the reaction (Equation (3)). Consequently, most CO2 transformations have been carried out by using a co-reactant, usually H2, to produce a wide range of products, from small oxygenates to long-chain hydrocarbons.
Another approach for a feasible process is through the addition of a solvent. For example, the gaseous phase conversion of CO2 to formic acid is thermodynamically unfavorable (Equation (3a)), while conducting it in an aqueous phase in the presence of water makes the reaction slightly exergonic (Equation (3b)) [73].
Given its inertness, extensive research efforts have been focused on tackling the high reaction energy barrier by designing more effective catalysts that convert CO2 with good activity, selectivity, and stability. These metrics are important in designing the catalytic component of a DFM, which is usually in the form of supported noble metal or metal oxide nanoparticles that serve as the active sites for CO2 activation. The typical reactions include the methanation of CO2 (also known as the Sabatier reaction; Equation (4)) [74,75,76], a reverse water–gas shift reaction (RWGS, Equation (5)) [77,78,79], and the dry reforming of methane (DRM, Equation (6)) [80,81,82]. The resulting CO produced in the RWGS and DRM reactions can be further hydrogenated to produce methanol (Equation (7)) or further converted into long-chain paraffinic and olefinic hydrocarbons via Fischer–Tropsch synthesis (FTS, Equation (8a–c)) [83,84,85].
Fischer–Tropsch synthesis (FTS):
Different types of transition metals are found to catalyze these reactions. For instance, Ni is commonly used for methanation and DRM reactions, Cu for the RWGS and subsequent hydrogenation to methanol, and Fe or Co for FTS. Owing to their low costs and high abundance, they are usually favored over their noble metal counterparts. However, many of these catalysts are prone to severe deactivation due to carbon deposition and sintering. Additionally, product selectivity would be an issue as the above reactions may all occur in parallel, which brings its complications, especially in industrial settings, where a certain reaction may be preferred to another. For example, the production of methanol via the RWGS pathway might be compromised by methanation occurring as an unwanted side reaction. Therefore, noble metals such as Ru, Rh, Pd, and Pt, though being more expensive, have received considerable attention because of their resistance to deactivation as well as high selectivity toward the desired products, which allow for more efficient downstream processes [86,87,88,89].
Despite major progress having been made in the development of various catalysts, significant challenges still exist in optimizing robust and economical catalysts with a good catalytic performance that can be utilized on an industrial scale. Through the better understanding of the reaction mechanisms, we can identify key descriptors that influence the performance of the catalysts [90], which eventually determine the effectiveness of DFMs in capturing CO2 and catalyzing reactions.
4. Dual Function Materials: Synthetic Methods
A typical investigation into the sorption and catalytic performances of a metal oxide-based DFM starts with the choice of the desired precursors. This depends largely on the independent variables and the goals of the research, where some works aim to compare the efficacies of various adsorbent components [91,92,93], some seek to elucidate the mechanisms underlying CO2 sorption and conversion [94,95], and others seek to evaluate the real-world applicability of DFMs by subjecting them to experiments that mimic realistic conditions.
The choice of adsorbent, catalytic components, and the support are known to influence the quality and performance of the as-synthesized DFM. For example, in an investigation by Bermejo-López et al. [96], supported CaO and Na2CO3 on Al2O3 were used as the adsorbent components of DFMs, with Ru as the catalytic metal. Various such DFMs were synthesized with varying CaO or Na2CO3 loadings. In general, higher adsorbent loadings promote CO2 adsorption and hydrogenation. However, since CaO exhibits stronger basicity than Na2CO3, the former exhibits higher CO2 capture, as shown in Figure 13. Consequently, the former requires higher calcination and hydrogenation temperatures, as compared to the latter.
Upon deciding on the suitable precursors, the next step would be to employ an appropriate method to synthesize the DFM. Several synthetic methods are already well-known, which produce DFMs with certain desired characteristics, such as a high surface area and porosity, homogeneous particle dispersion, and unique morphology, with each of them having their respective benefits and disadvantages. A host of other factors also needs to be considered in selecting a synthetic method, including time, material, and labor costs, as well as ease of implementation, in addition to the typical structural characteristics. In this section, we will explore four commonly used methods, presented in an approximate order of their prevalence in the literature.
4.1. Incipient Wetness Impregnation
Incipient wetness impregnation (IWI) is a well-known and highly popular synthetic method for producing heterogeneous catalysts, and is, unsurprisingly, the method that is employed in most of the works on DFMs [11,91,92,93,94,95,96,97,98,99,100,101]. IWI is favored for its ease of implementation and relatively low costs [102]. This procedure aims to impregnate adsorbent and/or catalyst species onto a support by adding small amounts of adsorbent/catalyst precursor solution to a powdered support. The wet powder is then dried and may further be subjected to thermal activation treatment, e.g., calcination or reduction. The process is then repeated until the total volume of precursor solutions added equals the total pore volume of the support [103]. The final product is ideally a porous material with adsorbent and catalyst particles distributed throughout the surface area of the pores. Sietsma et al. found that the pore size distribution and drying rate affect the redistribution of the particles on the support, which eventually determines the final crystal size [102]. For supports with large pore size distribution, a fast drying rate leads to a larger crystal size due to a higher degree of particle redistribution. On the contrary, for supports with narrow pore size distribution, the effect of the drying rate is minimal.
Since surface area is one of the determinants in designing an effective DFM, the careful selection of the support materials, preferably ones with a larger surface area and well-defined pore structures, is an important consideration in optimizing the properties of DFMs after IWI post-treatment. Additionally, optimum loadings of adsorbent/catalyst components should be considered as well.
4.2. Sol–Gel Method
In order to gain greater control over the particles’ morphology and porosity, sol–gel synthesis is an alternative method that can reliably ensure the formation of microstructures favorable to CO2 capture, as well as uniform particulate dispersion and the minimal granulation of adsorbent/catalyst particles, owing to its ability to produce a solid-state material from a chemically homogeneous liquid precursor [104]. Sol–gel chemistry conventionally involves the hydrolysis and condensation of metal alkoxides, with reaction rates depending on the carbon length of the alkoxides (e.g., methoxide, ethoxide, propoxide, etc.) and the solvent/alkoxide ratio. A major limitation of the alkoxide-based synthetic method is the instability of the metal alkoxides itself, especially in the presence of water and moisture, which makes it difficult to handle. As a result, alternative methods have been developed to employ metal salts instead of alkoxides in aqueous solution, together with small organic molecules as chelating agents, such as hydroxycarboxylic acids, to modify the hydrolysis chemistry of metal ions in aqueous solution.
One of the most commonly used organic acids is citric acid. Given its low price and wide availability, the sol–gel method with citric acid has been widely used to synthesize various metal oxides, including binary and tertiary oxides [105]. The key feature of this method is the formation of a metal-citrate complex, which ensures the homogeneity of the starting liquid solution. The presence of a citrate organic matrix is believed to help maintain the uniform mixing and dispersion of the metal ions on the atomic scale, resulting in small crystallite sizes.
A modification to the citrate sol–gel method was developed by Pechini in 1967 [106], where polyhydroxy alcohol, such as ethylene glycol, was added into the aqueous citrate solution. This results in a polyesterification reaction, which forms a covalent polymeric network that entraps metal ions [104], allowing two or more metals to be dispersed homogeneously throughout the network, as shown in Figure 14. The intermediate product here is a brittle solid, which is subsequently subjected to thermal treatment, e.g., calcination, to drive off organic components and obtain the final metal oxide powder [107].
Further variations and modifications have been made since Pechini’s initial proposition. For example, Pechini suggested metal oxides, hydroxides, and carbonates as possible cation precursor choices, but due to a need to control the amount of organic material in the final product, as well as economic reasons, nitrate salts have been a popular choice. Additionally, a modified pathway that substitutes the polyhydroxy alcohol for water subverts the esterification step and forms a gel-like substance instead.
Representative works on DFMs synthesized via a sol–gel method exhibit an expected degree of variability in terms of the synthetic pathways used. Nitrate precursors were common, though we note a study by Radfarnia et al. that used calcium acetate and aluminum isopropoxide as the metal cation precursors [108]. A study by Naeem et al. used ethylene glycol for polyesterification [69], as in the original Pechini method, while the aqueous citric acid variation is more common [109,110,111].
Notably, a study by Zhang et al. demonstrated that a sol–gel synthetic method produces DFMs with superior CO2 capture capacities in comparison with other methods like co-precipitation and mixing [112]. Their study investigated the usage of CaO-based DFMs in the oxidative dehydrogenation of ethane. The sorbent component was comprised of ceria-doped CaO, which was synthesized via four methods—dry and wet mixing, co-precipitation, as well as the modified Pechini method with nitrate precursors of the calcium and cerium cations, and aqueous citric acid serving as the chelating agent. It was found that the sol–gel method produced a sample with an initial CO2 capture capacity of 0.58 g/g, almost five times higher than that prepared by the dry mixing method, which exhibited an initial CO2 capture capacity of 0.12 g/g. It was concluded that multiple factors influenced the CO2 capturing abilities of a particular sorbent. While the available surface area is important, larger crystallite sizes severely impede the carbonation reaction when a surface layer of CaCO3 forms around the sorbent material, causing the carbonation reaction to transit into the diffusion-controlled phase [52,54,55]. Therefore, a balance between maximizing the surface area and optimizing the crystallite sizes is necessary for ensuring excellent CO2 capturing capacities, which can be obtained by fine-tuning the sol–gel synthesis parameters.
4.3. Co-Precipitation Method
Co-precipitation is a widely employed method in heterogeneous catalysis to synthesize materials such as mixed metal oxides [113,114]. The general method involves dissolving the precursor chemicals for the adsorbent, catalyst, and/or support/promoter (typically nitrate salts) in either deionized water or polar solvent under rigorous stirring to ensure homogeneity. A precipitating agent, e.g., NaOH, NH4OH, etc., is then added dropwise to induce precipitation. Subsequently, aging to enhance crystallinity is practiced, though not necessary. The precipitate is collected via filtration followed by washing cycles in deionized water or another solvent. Lastly, the precipitate is subjected to thermal treatment, e.g., calcination, to obtain the final product.
As a synthetic method, co-precipitation is straightforward and inexpensive. The material’s stoichiometry can be easily controlled by using appropriate amounts of precursors during the precipitation reaction. Depending on the aging conditions, adsorbents produced via this method typically exhibit a homogeneous particulate distribution with good crystallite sizes. However, disadvantages of this method include generally lower surface areas compared to adsorbents synthesized via IWI or sol–gel methods [105]. The process is also fairly laborious, with repeated washing being commonly necessary. Further, Li et al. note that the structural properties of the resulting precipitate are sensitive and heavily dependent on reaction conditions and equipment setup, which makes reproducibility an issue [115].
Like the sol–gel pathways, there are several variations to the co-precipitation procedure. These include the types of precipitating agent, the solvent used to dissolve the precursors and wash the precipitate, any mechanical assistance to the reaction (e.g., stirring, sonication, microwave, etc.), as well as aging temperature and time, to enhance the crystallinity via Ostwald ripening [114,115] at a high temperature, e.g., hydrothermal treatment. With the careful selection and optimization of the reaction conditions, the wide variation of methods under the family of co-precipitation procedures could be used to overcome certain shortcomings or to reinforce certain desirable qualities in a DFM. For example, Molina-Ramírez et al. synthesized a DFM comprising of a Ni-Ba unsupported catalyst via a co-precipitation method assisted by sonication [116], where nitrate precursors were used and ammonia was employed as the precipitant. The as-synthesized DFM showed an exceptionally high BET surface area of 112 m2/g, though we note that colloidal silica was used as a surface area promoter. Meanwhile, Karami and Mahinpey investigated the effect of synthetic methods on several Ca-alumina sorbents [117], such as the precipitation of a Ca salt solution onto colloidal gelled alumina, the co-precipitation of Ca and Al salt solutions with Na2CO3 or sodium aluminate as the precipitants, and the physical mixing of a Ca precipitate with a peptized alumina gel. It was found that co-precipitation produced a sorbent with the poorest CO2 uptake performance, possibly due to the low surface area and porosity.
4.4. Mixing Method
The mixing of adsorbent/catalytic/support precursors under varying conditions to produce a DFM precursor is arguably the simplest and most straightforward synthetic method. However, it clearly lacks precision in terms of producing the most favorable microstructures required for the ICCC process. Slightly varied methods of mixing exist depending on the relative solubilities of the precursors used [53]. For example, the wide usage of nitrate precursors as described in the preceding sections lends itself to facile dissolution in water. The precursor solutions are then simply mixed under stirring for a few hours to ensure homogeneity, the resultant solution is then dried to obtain a powder, followed by calcination to obtain the DFM.
Mixing is more commonly used in conjunction with other synthetic methods to produce a DFM. For example, the adsorbent component might be synthesized by mixing, while the catalytic component is synthesized via another method, e.g., co-precipitation or impregnation. For example, in a work by Huang et al., who investigated the performance of NaNO3-promoted Ni/MgO DFMs for integrated CO2 capture and methanation, the unpromoted Ni/MgO catalysts were synthesized via a facile one-pot wet mixing method [118], whereas the alkali metal salt promoter NaNO3 was subsequently impregnated upon the bare catalyst. When mixing is employed as the sole synthetic method for a DFM, its characteristics are decidedly inferior. Wu et al. demonstrated this in a comparative study investigating the use of Ni-CaO-CeO2 DFMs for ICCC via the RWGS reaction [105]. Different DFMs were prepared using acetate/nitrate precursors of Ni, Ca, and Ce via wet mixing, co-precipitation, and sol–gel methods. It was found that the wet mixing method produced a sample with the lowest CO2 capture capacity and product yield as compared to the other synthetic methods. The poor performance by wet mixing is mainly due to the low BET surface area, inferior pore structure, large Ni crystallite sizes, low surface basicity, and low reducibility.
5. Performance Evaluation of Dual Function Materials
With a general understanding of the kinetics and thermodynamics of CO2 capture and conversion, as well as the typical synthetic methods of DFMs that enable the ICCC process, we proceed to evaluate the performance of DFMs across the literature. This section catalogues recent works according to the key ICCC reactions that the DFM catalyzes, namely ICCC with a methanation reaction, a reverse water–gas shift (RWGS) reaction, and the dry reforming of methane (DRM). With each case study, we present examples of the synthetic, operating, and performance parameters of the DFMs, as well as the characteristics and mechanistic studies of the interaction between CO2 and the adsorption and catalytic sites of the DFMs. Given the dual function characteristic of the materials, the efficacy of DFMs first depends on their ability to efficiently capture CO2 with a high capacity, which becomes the reactant for the subsequent reactions. Therefore, the material selection for a DFM has to satisfy the specific operating conditions required for both CO2 capture and reaction. As DFMs are subjected to repeated cycles of CO2 capture and reaction, the robustness of the materials is also an important factor to be considered in the design and evaluation of DFMs.
5.1. ICCC with Methanation Reaction
The majority of studies on ICCC using DFMs focus on the methanation of CO2, which takes place primarily via the Sabatier reaction (Equation (9)):
The reaction is typically carried out at temperatures around 300–350 °C and pressures of 5–20 MPa [119]. Given its highly exothermic nature, the reaction releases copious amounts of heat, which has to be managed accordingly to prevent the thermal degradation (e.g., sintering) of the DFMs. This can be achieved via the engineering of more thermally resistant DFMs, or by harnessing the heat of the reaction via efficient heat transfer and heat integration, e.g., powering endothermic reactions. In this view, CO2 methanation offers an isothermal solution to the DFMs system because the exothermic reaction can supply the required heat for the decarbonation reaction. Additionally, the usage of real flue gas in industrial settings might lead to the poisoning and deactivation of the Ni-based DFMs typically used for methanation [119,120]. Practical solutions include the pre-scrubbing of the flue gas or considering the usage of alternative catalysts like Ru, which can be activated via pre-reduction at lower temperatures in a H2 environment. The vast body of work studying the ICCC–methanation process is summarized in Table 3. It can be seen that Ru- and Ni-based DFMs supported on alkaline metal oxides, such as Na2O, MgO, and CaO, are the most commonly studied materials. In fact, as shown in Figure 15, Na2O, MgO, and CaO are among the most common sorbents for ICCC with methanation [14], due to their high CO2 sorption capacity and methanation activity at moderate temperatures, given the thermodynamic limitation of the reaction.
With a wide range of variables that are at play in the synthesis of a DFM, an extensive amount of work is naturally devoted to finding the best synthetic routes that produce DFMs with optimized makeups [11,95,101,110,117,121,122,123,125,126,127,128,129,130,131,134,135,136]. In a pioneering work by Duyar et al., CaO-based DFMs with Ru as the methanation catalyst supported on alumina were synthesized via incipient wetness impregnation (IWI) with varying adsorbent/catalyst loadings [11]. It was observed that the order of impregnation (i.e., Ru impregnated on CaO-alumina vs. CaO impregnated on Ru-alumina) affected the CO2 capture and methanation performances at 320 °C. In particular, a DFM comprising 5 wt% Ru impregnated on 10 wt% CaO-γAl2O3 showed a single-pass methanation capacity of 0.5 mmol/g. Upon subjecting to cyclic testing, an average of 0.41 mmol/g of CO2 capture could be obtained, with an average CO2 conversion of 76.17% and a methanation capacity of 0.31 mmol CH4/g over 19 cycles. The authors concluded that the order of impregnation was significant where the impregnation of Ru was found to be better than that of CaO due to the good dispersion of Ru. Should the order be reversed, the active Ru sites could be blocked by CaO particles, thus decreasing the catalytic activity of the DFM. Upon varying the CaO:Ru ratio, a high methane turnover (CH4 yield/Ru) was observed at a high CaO:Ru ratio, indicating CO2 spillover from the CaO to Ru sites where the methanation reaction takes place. It also suggests that the close proximity of CaO and Ru was an important factor in the DFM performance. However, low Ru loadings might not generate enough heat from the exothermic methanation reaction to liberate CO2 chemisorbed to CaO, since methanation begins primarily where CO2 is chemisorbed to Ru, thus compromising the overall CH4 yield. Lastly, testing under simulated flue gas conditions showed that, in the presence of steam, the methanation capacity decreased to 0.27 mmol CH4/g over 20 cycles, although the purity of the effluent gas improved to 99.9% CH4 by volume, as compared to 83.6% CH4 (balance CO2) in the absence of steam, suggesting the competing adsorption of H2O and CO2 on the DFM surface. A follow-up study by Duyar et al. presented two new DFMs with similar adsorbent/catalyst loadings, but with K2CO3 or Na2CO3 substituting CaO as the adsorbent component [93]. These DFMs showed improved methanation capacities of 0.91 and 1.05 mmol CH4/g for K and Na, respectively, owing to the increasing CO2 adsorption capacity in the same order: Na2CO3 > K2CO3 > CaO. The results, therefore, show the importance of optimizing the adsorbent component of DFMs in future attempts to engineer more efficient materials.
In another study, Porta et al. assessed the CO2 capture and methanation performances at 350 °C of Ru-based DFMs with six different adsorbent components (Li/Na/K/Mg/Ca/Ba) supported on alumina [123]. They found that metals capable of forming carbonates with high thermal stability led to the production of the highest amounts of CH4, namely, DFMs promoted with K, Ca, and Ba. On the other hand, Arellano-Treviño et al. evaluated Ru-based DFMs supported on γ-Al2O3 containing different alkali and alkaline earth oxides, such as Na2O, K2O, CaO, and MgO, and found that Na2O-promoted DFM (5%Ru-6.1%Na2O/Al2O3) exhibited a high CO2 capture capacity (0.651 mmol/g) and methanation rate (0.614 mmol CH4/g) when tested in 10%CO2/N2 atmosphere [92]. Although the CH4 yield significantly decreased upon exposure to a simulated flue gas condition (7.5%CO2, 4.5%O2, 15%H2O, balance N2), the DFM remained active, producing 0.291 mmol CH4/g at 320 °C. In another experiment using a Rh-Na2O-based DFM, the CO2 capture capacity and catalytic activity of 0.5%Rh was compared with those of 5%Ru in order to obtain a similar material price. While both DFMs exhibited a similar CO2 capture capacity (0.625 vs. 0.651 mmol/g, respectively), 5%Ru yielded higher methanation (0.614 mmol/g) than the 0.5%Rh counterpart (0.422 mmol/g), although one could argue that the methane yield per unit of metal is higher for Rh than that for Ru. As an alternative to noble metals, Ni-Na2O-based DFM was also synthesized and tested, which produced 0.276 mmol CH4/g under a 10%CO2/N2 atmosphere. However, the material was deactivated when exposed to flue gas containing H2O, with no methane being produced, as shown in Figure 16. Further analysis suggested that Ni atoms could not be completely reduced to the active metallic state in the presence of O2 and H2O, thus losing its catalytic activity.
Further possible variables during the synthesis of a DFM include the choice of adding promoters that can improve the CO2 capture capacity and CH4 selectivity, or the choice of precursor chemicals, which might affect the interactions between different components in the synthesized DFM. One study by Cimino et al. [135] found that promoting Ru/Al DFM with Li resulted in a four-to-five-fold increase in the CO2 capture capacity compared to the unpromoted material. In addition, stable cyclic carbonation/methanation was possible at temperatures around 230 °C, instead of the commonly utilized temperatures of 300 to 320 °C. In another study, the incorporation of Cs into Ni-hydrotalcite-based DFMs was found to increase the CO2 capture capacity up to 0.48 mmol/g and a CH4 yield of 0.33 mmol/g at 350 °C [130]. These examples show the promotional effect of alkali metal as it increases the material’s basicity, in particular, the number of medium and strong basic sites. Besides enhancing the basicity of the material, the addition of a promoter can also improve the stability of the material. For example, Ma et al. investigated the effect of doping Ni-CaO-based DFMs with various metal oxides, such as Mg, Al, Mn, Y, Zr, La, and Ce [110]. Among the synthesized DFMs, Zr-doped Ni/CaO exhibited the best performance by maintaining a stable CO2 capture capacity of 9 mmol/g and 74% CH4 selectivity after 20 cycles. It was attributed to the formation of CaZrO3, which helped improve the thermal resistance to sintering.
To investigate the effect of catalyst loading, Bermejo-López et al. varied the Ni content on CaO- or Na2CO3-based DFMs with alumina support through the impregnation method [101]. Notably, it was found that increasing Ni loading would increase the particulate sizes, catalyst reducibility, and consequently, methanation rates, with 0.142 mmol CH4/g produced by a DFM comprising 15 wt% Ni on 15% CaO/Al2O3 at 520 °C.
Lastly, the choice of the synthetic method has also been known to significantly influence the final microstructures and other physicochemical characteristics of a DFM, and a range of works have been devoted to comparatively evaluating the various possible synthetic routes [105,112,117,125]. For example, Zhang et al. noted that sol–gel synthetic methods tend to produce DFMs with physicochemical characteristics more favorable for ICCC processes, including an optimal balance of the BET surface area and crystallite sizes [112]. Sun et al. noted that the dispersion between catalyst and sorbent particles could even be tuned via different synthetic methods, thus giving rise to varying carbonation and methanation capabilities [125].
In addition to optimizing the materials design and synthesis, careful attention should also be given to the experimental parameters of the carbonation and methanation reaction, as well as the simulated flue gas conditions, as they can help uncover the possibility of translating ideal laboratorial conditions to a more real-life (e.g., industrial) setting, and provide plausible avenues for future work [98,100,122,124,137]. In one such study by Wang et al., a DFM consisting of Ru-Na2O impregnated on an alumina support was subjected to parametric and cyclic tests [98]. Specifically, the effect of three parameters—gas space velocity, reaction temperature, and exposure to oxygen during adsorption—on performance indicators such as CO2 uptake and CH4 generation, were investigated. From the cyclic studies, a CO2 uptake and CH4 generation capacities of 0.40 mmol/g and 0.32 mmol/g, respectively, were reported after 50 cycles at 300 °C. Through parametric studies, it was found that gas space velocities and temperature would affect the rates and the extents of carbonation and methanation in accordance with kinetic and thermodynamic principles. For instance, increasing the gas space velocity would increase the rates of CO2 uptake and CH4 production due to a more efficient mass transfer, whereas increasing the temperature would decrease CO2 adsorption and CH4 generation due to the exothermic nature of the reactions. The oxygen exposure tests also showed that, minimally, 15% H2/N2 for methanation was necessary for the reduction of oxidized Ru due to exposure to oxygen-containing flue gas during adsorption. The presence of precious metals such as Ru in overcoming the limitations of Ni as a catalytic metal is further displayed by studies such as one by Arellano-Treviño et al., which suggests that Ru doping aids in the reduction and re-activation of Ni after oxygen exposure, leading to the possibility of developing DFMs which are more resistant to industrial reaction conditions at cheaper costs [100].
Meanwhile, other authors have studied reaction parameters such as optimal carbonation/methanation durations, feed gas concentrations, and process pressures. Kosaka et al., for example, investigated Ni-based DFMs promoted with alkali metals including Na/K/Ca supported on alumina for their carbonation and methanation at 450 °C [122]. It was found that increasing operational pressures (a range of 0.1 MPa to 0.9 MPa was used) could increase the CO2 capture capacity of a Ni/Na-Al2O3 DFM from 0.209 mmol CO2/g to 0.299 mmol CO2/g, and its methanation capacity from 0.188 mmol CH4/g to 0.266 mmol CH4/g, in agreement with thermodynamics as the methanation reaction involves a decrease in the number of moles. For these tests, a gas feed containing 5% CO2/N2 was used, but it was additionally reported that increasing pressures enhanced carbonation and methanation at lower CO2 concentrations of 100 and 400 ppm CO2/N2 as well. Jeong-Potter and Farrauto also attempted an investigation of the effectiveness of a Ru/Na-Al2O3 DFM at a CO2 concentration of 400 ppm in air to assess the feasibility of utilizing the DFM for direct air capture purposes [124].
Considering that ICCC and methanation have been extensively studied, a good understanding of the kinetics and mechanisms of carbonation and methanation is equally important [94,95,96,138,139,140]. The mechanistic study is typically performed via investigative techniques including Fourier transform–infrared (FT-IR) spectroscopy, which is capable of measuring the gas composition at the reactor outlet and its changes with time, or by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), which detects the presence of chemical bonds that suggest the occurrence of possible reaction intermediates.
For instance, a study by Bermejo-López et al. uses FT-IR to study the temporal evolution of the gas composition within a reactor [96]. Their work involved the usage of Ru-based CaO and Na2CO3 DFMs supported on alumina. They proposed a mechanism where CaCO3 formed through the carbonation of Ca(OH)2 (Equations (10) and (11)) is decomposed upon the addition of H2 during the methanation step (Equation (12)), releasing CO2 which is subsequently converted on Ru sites according to the Sabatier reaction (Equation (13)). Additional observations include the delayed evolution of CO2 itself during the carbonation step, suggesting the saturation of the CO2 uptake sites, and the delayed evolution of H2O during methanation (even though the Sabatier reaction produces H2O), suggesting the hydration of CaO to form Ca(OH)2 (Equation (14)). A similar reaction scheme is suggested for Na2CO3-based DFMs.
Conversely, other studies utilize DRIFTS to detect the presence of reaction intermediates, and suggest possible reaction mechanisms [94,95,140]. One such study by Proaño et al. with Ru-based DFM [94,95] suggests the formation of bidentate carbonate species during carbonation and formate species during methanation (see Figure 17 for their detailed mechanism), but there exists a range of works that find corroborating [139] and disputing [140] results. Proaño’s group has since conducted further studies into DFMs with a similar makeup (10% Ni, 6.1% Na2O/Al2O3 DFMs enhanced with 1% Pt/Ru), utilizing in situ DRIFTS to study carbonation and methanation under oxidizing and non-oxidizing conditions, yielding largely corroborative results.
5.2. ICCC with Reverse Water–Gas Shift (RWGS) Reaction
RWGS is another key reaction in C1 chemistry that takes place according to the following reversible reaction (Equation (15)):
RWGS is an important reaction for producing syngas with desired H2/CO ratios, where CO can be further valorized into various chemicals, such as methanol via the CAMERE (carbon dioxide hydrogenation to form methanol via a reverse-water–gas-shift reaction) process or hydrocarbons via Fischer–Tropsch synthesis. However, although CO is industrially more useful than methane, the development of an effective DFM system to generate CO has been much less explored than that for methane generation. This is partly because the ability of DFMs to achieve a practical H2/CO ratio remains a major challenge. The accurate tuning of syngas composition can be tricky because of other secondary reactions that may occur in parallel with the RWGS reaction, such as the Sabatier reaction (Equation (10)) and the methanation of CO (Equation (16)):
Considering the endothermic nature of RWGS, increasing operating temperatures might reduce the extent of the exothermic side reactions and favor the RWGS reaction. Indeed, in a review by González-Castaño et al. [141], where the equilibrium concentrations of chemical species involved in the RWGS, Sabatier, and CO methanation reactions were investigated, it was found that the equilibrium concentration of CH4 decreases to near zero above 700 °C, as shown in Figure 18. In addition, the CO2/H2 molar ratio also plays a critical role in adjusting the CO composition, with a low CO2/H2 ratio being thermodynamically favorable for the RWGS reaction. These considerations are therefore important in maximizing the selectivity of CO, which turns out to be a key metric in evaluating DFMs used in ICCC processes involving RWGS.
Apart from thermodynamic considerations, ensuring a high selectivity of CO is also dependent on a prudent catalyst design, which is aided by the understanding of the kinetics and mechanisms of RWGS. Conventionally, Cu is favored for its CO selectivity and low methanation rates, while Pt is preferred for higher CO2 conversions [78]. With regards to DFM selection for the RWGS reaction, a variety of materials have been developed, including those containing transition metals, such as Fe, Co, Ni, or combinations thereof [142,143]. Recently, there have also been studies that do not involve any transition metals, such as supported Na on CaO and Al2O3 [144,145]. Table 4 summarizes the DFMs that catalyze the RWGS reaction.
In a recent work by Wu et al., Ni-CaO-CeO2 DFMs were synthesized via their acetate/nitrate precursors, using several methods including wet mixing, co-precipitation, and a citric acid-based sol–gel pathway [105]. Among the synthesized samples, DFMs prepared by sol–gel method exhibited superior physicochemical properties and hence displayed the highest CO2 capture capacities and conversion, as well as CO yields and selectivity. In particular, the sol–gel-derived material with acetate precursors showed a CO2 uptake of 15.34 mmol/g and CO2 conversion of 92.4%, with CO selectivity of 89.1% at 650 °C. Meanwhile, the material prepared with nitrate precursors displayed a lower CO2 uptake of 13.15%, but higher CO2 conversion of 96% and higher CO selectivity (87–96%). It was concluded that, firstly, the sol–gel synthesis resulted in an optimal morphology and internal structure for the CO2 capture and conversion. For instance, large BET surface areas and pore volumes facilitated CO2 diffusion and adsorption, whereas an enhanced surface basicity promoted CO2 affinity. Meanwhile, a smaller NiO grain size, uniform Ni dispersion, and higher reducibility could have led to improved catalytic activity. Secondly, the choice of acetate/nitrate precursors could affect catalyst–support interactions, which in turn result in variations in CO2 uptake and conversion, CO selectivity, as well as the material’s stability upon recycle.
Similar DFMs consisting of Ni over Ce-modified CaO were reported earlier by Sun et al., who employed a sol–gel method with citric acid [111]. The incorporation of CeO2 was found to enhance the material’s stability due to the formation of well-dispersed CeO2 that can effectively prevent the growth and agglomeration of CaO and NiO crystallites, resulting in a stable performance after 20 cycles. This is reflected by the synthesized material with Ca/Ni/Ce molar ratio of 1:0.1:0.033, which exhibited a CO2 uptake of 14.1 mmol/g, 51.8% CO2 conversion, and almost 100% CO selectivity at 650 °C. The authors proposed possible reaction mechanisms which are based on a redox (Scheme 1) or associative formate (Scheme 2) mechanism, as illustrated in Figure 19. According to the redox mechanism, upon switching to the hydrogenation step, the adsorbed CO2 could spill over to the Ni active sites, which may result in the formation of CO and NiO. In the presence of CeO2, the oxygen vacancy on the CeO2 surface may also interact with CO2, leading to the formation of CO and the oxidation of Ce3+ species. The introduction of H2 into the reactor would subsequently reduce NiO back to Ni and release H2O molecules. Likewise, the adsorbed H2 species on the CeO2 surface extracts the lattice oxygen from ceria, releasing a H2O molecule along with the reduction of the catalyst. Alternatively, the associative formate mechanism suggests that the adsorbed CO2 on the CeO2 surface may form bidentate formate as a reaction intermediate, which decomposes to form CO and terminal hydroxyl groups. Subsequently, the adsorbed H2 reacts with the hydroxyl group, releasing a H2O molecule.
In another study by Guo et al., ZrO2 was used as a dopant for Ni/CaO, where it enhanced the surface basicity and reducibility of the DFM, resulting in an increased CO2 uptake and catalytic activity [146]. At the optimum 12 wt% ZrO2 loading, the material exhibited a high CO2 uptake of 16.7 mmol/g with 63.2% CO2 conversion and a 10.5 mmol/g CO yield at 650 °C. The formation of CaZrO3 was also found to reduce the sintering of CaO and NiO crystals. The further incorporation of CeO2 into Ni/CaO-ZrO2 resulted in increased CO2 conversion to 72.1% due to the increase of an oxygen vacancy, although the CO2 uptake capacity decreased to 11.9 mmol/g, which could be attributed to a lower surface area. A similar strategy was also implemented by Sun et al. who incorporated Fe into a Ni/CaO DFM. An optimum CO yield of 11.3 mmol/g was obtained by using Ni1Fe9-CaO at 650 °C. It was observed that the formation of Ca2Fe2O5 acted as an oxygen carrier that promoted CO production and helped improve the stability of the DFM by acting as a physical barrier that slowed down CaO sintering [143].
As alternatives to transition-metal-based materials, Sasayama et al. prepared DFMs that only contain alkali/alkaline earth metals (e.g., Na, K, and Ca) supported on γ-Al2O3 via the impregnation method [144]. They were then tested with 5 vol% and 400 ppm CO2 in N2 to investigate the production of syngas under model flue gas or direct air capture modes, respectively. Upon testing with 5 vol% CO2 at 500 °C, Na/Al2O3 exhibited a 0.135 mmol/g CO2 uptake with 77% CO2 conversion and 99.8% CO selectivity. On the other hand, with 400 ppm CO2, its CO2 uptake was about 0.09 mmol/g with 77.7% CO2 conversion and 94.3% CO selectivity. The results therefore indicate the potential use of transition-metal-free DFMs for ICCC process, although further works are needed to improve the materials’ performance and to study the reaction mechanism as well as to understand the catalytic active sites.
In another study, the further incorporation of Pt on Na/Al2O3 was found to be selective for CO formation at lower temperatures of 350 °C [145], which is advantageous in reducing the typically high temperature requirement for the RWGS reaction. The DFM was prepared via the sequential impregnation method of Al2O3, first with Na, followed by Pt. The CO2 capture was evaluated with 1% CO2/10% O2 in N2, and its uptake capacity was found to be 0.19 mmol/g, comparable to that of Na/Al2O3 (0.21 mmol/g), and much higher than that of Pt/Al2O3 (0.08 mmol/g), indicating that Na serves as the CO2 capture site. Upon hydrogenation, CO2 was converted to CO with 89% conversion and 93% CO selectivity. In a control experiment, a physical mixture of Na/Al2O3 and Pt/Al2O3 was tested, and it was found that CO2 conversion and CO selectivity decreased significantly to only 57% and 41%, respectively. The EDX spectroscopy (Figure 20) suggests that a core–shell structure of Pt-Na nanoparticles was formed on Pt-Na/Al2O3, with Pt as the core. As a result of the close interaction between the two metals, the Na species not only serves as the CO2 capture site, but also as a promoter to enhance CO formation. Through in situ FTIR measurements, it was found that the adsorbed CO species was hardly observed over the material’s surface, indicating that the presence of the Na species inhibited the adsorption of the generated CO, leading to high selectivity for CO.
5.3. ICCC with Dry Reforming of Methane (DRM)
Besides RWGS, syngas can also be produced via the dry reforming of methane (DRM), where two potent greenhouse gases, CH4 and CO2, react with each other according to the following equation (Equation (17)):
Despite being favored for its “greening” capability, ICCC with DRM faces several disadvantages at industrial scales. First, the highly endothermic reaction requires a large energy input (Equation (17)). Second, the DFMs that catalyze the DRM reaction typically contain Ni in the catalytic component, which is susceptible to poisoning and deactivation via coking in the absence of steam. It has, therefore, been proposed that continued research in this field would focus on developing Ni-based DFMs that exhibit stronger coking resistance, e.g., by including alkaline promoters like Mg, or considering the use of Rh- or Ru-based DFMs to catalyze the reaction, in addition to uncovering further insights into the mechanisms of DRM so as to strengthen and optimize its industrial applicability [134,148]. The summary of DFMs that catalyze the DRM reaction is presented in Table 5 and Table 6. The two tables categorize works according to their presentation of the ICCC performances of DFMs. In the former, the conversions of CO2 and CH4 are presented, whereas the yields of H2 and CO are presented in the latter.
In an earlier work, Kim et al. first demonstrated the calcium looping process combined with a catalytic system by using a physical mixture of CaO and Ni/MgO-Al2O3 as the CO2 sorbent and DRM catalyst, respectively, in a single fluidized bed reactor [150]. The two-step process was operated in a cyclic manner where CO2 was first captured on CaO during the carbonation step. Upon gas switching to CH4, the CaO sorbent was regenerated while releasing CO2, which instantaneously reacted with CH4 on the Ni/MgO-Al2O3 catalyst, as depicted in Figure 21. CaO was derived from limestone via calcination at 800 °C whereas the Ni catalyst was derived from a hydrotalcite precursor that was synthesized via the co-precipitation method. At 720 °C, the CO2 capture capacity was 14.1 mmol/g in the first cycle with an almost 100% conversion of CO2 and CH4. The resulting syngas thus had a H2:CO ratio of 1.06:1. Due to the sintering of CaO, there was a significant loss of CO2 uptake, which decreased to 9 mmol/g after 10 cycles of the reaction. Although, initially, Ni catalyst deactivation was observed due to coke formation, the carbon deposition amount was decreasing with increasing cycle numbers. It was found that the deposited carbon was removed during the carbonation step while the Ni metallic state was preserved, which could be due to the reverse Boudouard reaction that is favorable at high temperatures [152]. Therefore, CO2 and CH4 conversion can be maintained above 95% throughout the 10 cycles.
In subsequent works by Tian et al., a Ni/CaO-based DFM was prepared via citrate-based sol–gel synthesis which produced an average syngas yield of around 9 mmol/g throughout 10 cycles [152] of CO2 capture (at 600 °C) and conversion with CH4 (at 800 °C). The experimental H2:CO ratio was about 1.1, higher than the theoretical ratio of 1. As the average CH4 conversion (86%) was higher than that of CO2 (65%), it could be suggested that the Ni-CaO interface in the DFM was more active in dissociating CH4 to yield H2 than reducing CO2 to yield CO. On the other hand, despite using a Ni/CaO DFM prepared with a similar method, Jo et al. obtained syngas with a high H2:CO ratio of 6.52 at 700 °C, indicating that the CO2 reduction reaction was suppressed [153]. It was observed that a large amount of carbon deposit was formed on the DFM, as well as the hydration of CaO to form Ca(OH)2.
A further improvement of the Ni/CaO DFM was reported by Hu et al. where porous CeO2-modified CaO microparticles were used as the support for Ni impregnation [154]. The introduction of CeO2 into the material enhanced the DFM performance in two aspects, in that it acted as (i) a promoter that enhanced the CO2 affinity towards CaO through the increase in lattice oxygen, thus enabling a high CO2 uptake, and (ii) a physical stabilizer that enhanced the sintering resistance of CaO, improved Ni particle dispersion through the formation of small Ni crystallites, as well as increased Ni reducibility, thus enabling high and stable catalytic activity. Among the investigated samples, the material with 85%CaO:15%CeO2 (Ni/Ca85Ce15) showed a constant CO2 uptake and retained about 80–90% of its initial syngas time-averaged space time yield (STY) over nine cycles of CO2 sorption and conversion at 650 °C, as shown in Figure 22. In another experiment, a layer of ZrO2 was coated on CaCO3 nanoparticles, followed by the co-impregnation of Ni and Ce [149]. During the CO2 capture–DRM cycle tests at 720 °C, 5% CO2 was used as the gas feed to mimic flue gas composition where over 40% conversion of CO2 and CH4 can be achieved. The presence of ZrO2 was also found to enhance material stability by preventing sintering. Through the in situ DRIFTS analysis (Figure 23), monodentate carbonate was observed on the CaO surface during the carbonation step, whereas polydentate carbonate was observed on ZrO2 and bidentate carbonate on the CeO2 surface. CO2 dissociation to CO was also detected on NiCe/Ca-Zr oxide due to the presence of abundant oxygen vacancies on the reduced CeO2. Upon gas switching to CH4, the characteristic IR peaks of formate species and gaseous CO were observed, while carbonate peak intensities were decreased. These suggested that the occurrence of CaCO3 decarbonation was coupled with the DRM reaction, during which the adsorbed species produced from CH4 decomposition reacted with the species to form formate species, which further decomposed to CO [149].
The combination of Ni with Ru supported on CeO2/Al2O3 containing Na2O, K2O, or CaO was reported by Merkouri et al. [155]. The DFMs contained 15%Ni and 1%Ru, and were mostly active for ICCC with DRM at 650 °C, where the CaO-containing material produced 0.338 mmol/g CO and 32.6 mmol/g H2, yielding H2-rich syngas. The large formation of H2 during DRM was mainly due to methane cracking and its decomposition into carbon, evidenced by CH4-TPSR experiments which showed that methane cracking peaked at around 635 °C. By using an operando DRIFTS-MS method, the authors elucidated the CO2 capture and reaction mechanism over the Ni-Ru-Na2O/CeO2-Al2O3 DFM through alternating successive CO2 and CH4 cycles at 550 °C. In agreement with Tian et al.’s previous work [152], CH4 reduction on the metallic sites produced a H2 and graphitic carbon layer, which further gasified with CO2 during the capture step to yield CO through a reverse Bourdouard reaction. However, it was noted that the coke gasification kinetics were slower than the coke formation, resulting in the rapid deactivation of the catalyst. This mechanistic study thus highlights the importance of controlling CH4 cracking during the DRM reaction via optimizing the material’s design and reaction engineering in order to slow down the catalyst deactivation, as well as to obtain syngas with a favorable H2:CO ratio.
Further alternatives that include the dry reforming of other hydrocarbons, such as ethane (DRE), to produce syngas (Equation (18)) was reported by Al-Mamoori et al. [91], who investigated the use of CaO- and MgO-based double salts promoted by K/Ca, supported on alumina, and impregnated with Ni as the catalyst at 650 °C.
The material’s synthetic methods consisted of a sol–gel method for the alumina support, while the adsorbent and catalyst components were incorporated via wet impregnation. The CaO-based DFMs comprising 20 wt% Ni and a 1:1 ratio of adsorbent to support showed the best ICCC performance, with CO2 adsorption capacities of 0.99 mmol/g and 0.63 mmol/g for the K-Ca-based and Na-Ca-based DFMs, respectively, a 65% and 75% conversion of CO2 for the K-Ca-based and Na-Ca-based DFMs, respectively, and a 100% C2H6 conversion for both DFMs. Additionally, yields of approximately 45% and 37.5% for CO and H2, respectively, were reported for the K-Ca-based DFM when subjected to a sustained DRE process (up to 10 h) at 650 °C. Despite a relatively stable performance, coke formation (9 wt%) was observed due to a high Ni content. From their investigations, it was concluded that the adsorbent/catalyst contents directly influenced CO2 capture and conversion performances, and that reaction conditions strongly influenced the selectivity toward DRE or the occurrence of side reactions, such as oxidative dehydrogenation, ethane cracking, RWGS, and coking. Therefore, it is imperative to optimize their chemical and physical properties.
6. Perspectives and Future Outlook
It is evident that DFMs have increasingly played an important role in decarbonization efforts through CO2 recycling and reutilization. Through numerous studies in the past decade, DFMs have shown to offer feasibility and versatility in capturing CO2 from both model flue gas and air (direct air capture), and subsequently converting it to other C1 species, such as methane and syngas. The recent literature has reported extensive DFM formulations and configurations for ICCC that primarily include Ni, Ru, and Rh as the catalytic metals, basic oxides such as Na2O, K2O, CaO, and MgO as the CO2 sorbents, and Al2O3 as a common support. From the various given examples, the combinations of these materials are shown to form flexible DFMs that catalyze CO2 methanation, RWGS, and DRM, each of which has its own optimum operating conditions. For example, CO2 methanation is favored at an intermediate temperature range of 300–350 °C, whereas RWGS and DRM are favored at high temperatures (600–750 °C).
The effectiveness of DFMs is determined by both their CO2 capture capacity and catalytic activity. The CO2 uptake principally depends on (i) the basicity of the materials, with medium to strong basic sites favorable for CO2 adsorption, and (ii) the surface area and porosity. On the other hand, the catalytic activity depends on several factors, such as (i) the crystallite size and dispersion of the active metals, with a small size and high metal dispersion attributed to high catalytic activity, (ii) the reducibility of the active metals, which is determined by the interaction between the active metal particles and the oxide support, (iii) the basicity of the material, where weak basic sites are beneficial for CO2 methanation and medium basic sites for the RWGS and DRM [155], and (iv) the availability of oxygen vacancies, which can often be enhanced by incorporating a dopant, such as CeO2. In fact, there is a recent review by Hussain et al. that discusses the importance of oxygen vacancies for CO2 methanation and its tuning strategies [157].
Given the complexity in developing and tuning DFMs with the desired properties, major progress has been achieved at the laboratory scale, leading to several promising candidates with good CO2 conversion and product selectivity. However, there are still many challenges to implement ICCC with DFMs on a larger scale. For instance, the stability and recyclability of the materials need to be improved as DFMs are susceptible to sintering, coking, and poisoning. It, therefore, requires careful material engineering to synthesize robust DFMs that can withstand harsh conditions upon prolonged and repeated cycles. In view of this, lifetime studies of DFMs need to be carried out under real flue gas conditions that typically contain moisture, O2, and other impurities, such as NOx, to guarantee their industrial viability. To accelerate the screening process, the use of computational DFT modeling would be highly encouraged to aid in the fine-tuning of the material’s properties while studying the reaction mechanisms to control the desired product selectivity as well as to minimize catalyst deactivation.
Considering that ICCC with DFMs is an emerging field, there are plenty of research opportunities in the future to develop versatile and robust DFMs that enable the production of other chemicals, such as alcohols and longer-chain hydrocarbons, which could offer the greater flexibility of producing different products within the same production facility. We believe that the advancement of this technology will continue promoting sustainable chemical manufacturing and a low carbon economy.
Author Contributions
Conceptualization, W.J.T. and P.G.; Methodology, W.J.T. and P.G.; Writing—original draft preparation, W.J.T. and P.G.; Writing—review and editing, W.J.T. and P.G.; Project administration, P.G.; Funding acquisition, P.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the C. N. Yang Scholars Program at Nanyang Technological University AY2022-2023.
Acknowledgments
This research was made possible by the C. N. Yang Scholars Program at Nanyang Technological University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- An IPCC Special Report on the Impacts of Global Warming of 1.5 °C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2018.
- Koytsoumpa, E.I.; Bergins, C.; Kakaras, E. The CO2 economy: Review of CO2 capture and reuse technologies. J. Supercrit. Fluids 2018, 132, 3–16. [Google Scholar] [CrossRef]
- Leeson, D.; Mac Dowell, N.; Shah, N.; Petit, C.; Fennell, P.S. A Techno-economic analysis and systematic review of carbon capture and storage (CCS) applied to the iron and steel, cement, oil refining and pulp and paper industries, as well as other high purity sources. Int. J. Greenh. Gas Control 2017, 61, 71–84. [Google Scholar] [CrossRef]
- Naims, H. Economics of carbon dioxide capture and utilization—A supply and demand perspective. Environ. Sci. Pollut. Res. 2016, 23, 22226–22241. [Google Scholar] [CrossRef] [PubMed]
- Wilberforce, T.; Baroutaji, A.; Soudan, B.; Al-Alami, A.H.; Olabi, A.G. Outlook of carbon capture technology and challenges. Sci. Total Environ. 2019, 657, 56–72. [Google Scholar] [CrossRef]
- Omodolor, I.S.; Otor, H.O.; Andonegui, J.A.; Allen, B.J.; Alba-Rubio, A.C. Dual-Function Materials for CO2 Capture and Conversion: A Review. Ind. Eng. Chem. Res. 2020, 59, 17612–17631. [Google Scholar] [CrossRef]
- von der Assen, N.; Bardow, A. Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: Insights from an industrial case study. Green Chem. 2014, 16, 3272–3280. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Huang, C.-H.; Tan, C.-S. A Review: CO2 Utilization. Aerosol Air Qual. Res. 2014, 14, 480–499. [Google Scholar] [CrossRef]
- Freyman, M.C.; Huang, Z.; Ravikumar, D.; Duoss, E.B.; Li, Y.; Baker, S.E.; Pang, S.H.; Schaidle, J.A. Reactive CO2 capture: A path forward for process integration in carbon management. Joule 2023, 7, 631–651. [Google Scholar] [CrossRef]
- Duyar, M.S.; Treviño, M.A.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168–169, 370–376. [Google Scholar] [CrossRef]
- Melo Bravo, P.; Debecker, D.P. Combining CO2 capture and catalytic conversion to methane. Waste Dispos. Sustain. Energy 2019, 1, 53–65. [Google Scholar] [CrossRef]
- Merkouri, L.-P.; Reina, T.R.; Duyar, M.S. Closing the Carbon Cycle with Dual Function Materials. Energy Fuels 2021, 35, 19859–19880. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Williams, P.T.; Wu, C. Recent advances in integrated CO2 capture and utilization: A review. Sustain. Energy Fuels 2021, 5, 4546–4559. [Google Scholar] [CrossRef]
- Sabri, M.A.; Al Jitan, S.; Bahamon, D.; Vega, L.F.; Palmisano, G. Current and future perspectives on catalytic-based integrated carbon capture and utilization. Sci. Total Environ. 2021, 790, 148081. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dang, C.; Yang, G.; Cao, Y.; Wang, H.; Peng, F.; Yu, H. Bi-functional particles for integrated thermo-chemical processes: Catalysis and beyond. Particuology 2021, 56, 10–32. [Google Scholar] [CrossRef]
- Bhatta, L.K.G.; Subramanyam, S.; Chengala, M.D.; Olivera, S.; Venkatesh, K. Progress in hydrotalcite like compounds and metal-based oxides for CO2 capture: A review. J. Clean. Prod. 2015, 103, 171–196. [Google Scholar] [CrossRef]
- Khalilpour, R.; Mumford, K.; Zhai, H.; Abbas, A.; Stevens, G.; Rubin, E.S. Membrane-based carbon capture from flue gas: A review. J. Clean. Prod. 2015, 103, 286–300. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Krishnamurthy, A.; Rownaghi, A.A.; Rezaei, F. Carbon Capture and Utilization Update. Energy Technol. 2017, 5, 834–849. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Wei, J.-J.; Zeng, G.-M.; Zhang, H.-Q.; Tan, X.-F.; Ma, C.; Li, X.-C.; Li, Z.-H.; Zhang, C. A review on strategies to LDH-based materials to improve adsorption capacity and photoreduction efficiency for CO2. Coord. Chem. Rev. 2019, 386, 154–182. [Google Scholar] [CrossRef]
- Xie, K.; Fu, Q.; Qiao, G.G.; Webley, P.A. Recent progress on fabrication methods of polymeric thin film gas separation membranes for CO2 capture. J. Membr. Sci. 2019, 572, 38–60. [Google Scholar] [CrossRef]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial carbon dioxide capture and utilization: State of the art and future challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- Younas, M.; Rezakazemi, M.; Daud, M.; Wazir, M.B.; Ahmad, S.; Ullah, N.; Inamuddin; Ramakrishna, S. Recent progress and remaining challenges in post-combustion CO2 capture using metal-organic frameworks (MOFs). Prog. Energy Combust. Sci. 2020, 80, 100849. [Google Scholar] [CrossRef]
- Zheng, J.; Chong, Z.R.; Qureshi, M.F.; Linga, P. Carbon Dioxide Sequestration via Gas Hydrates: A Potential Pathway toward Decarbonization. Energy Fuels 2020, 34, 10529–10546. [Google Scholar] [CrossRef]
- Dunstan, M.T.; Donat, F.; Bork, A.H.; Grey, C.P.; Müller, C.R. CO2 Capture at Medium to High Temperature Using Solid Oxide-Based Sorbents: Fundamental Aspects, Mechanistic Insights, and Recent Advances. Chem. Rev. 2021, 121, 12681–12745. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ren, Z.; Si, W.; Ma, Q.; Huang, W.; Liao, K.; Huang, Z.; Wang, Y.; Li, J.; Xu, P. Research progress on CO2 capture and utilization technology. J. CO2 Util. 2022, 66, 102260. [Google Scholar] [CrossRef]
- Dubey, A.; Arora, A. Advancements in carbon capture technologies: A review. J. Clean. Prod. 2022, 373, 133932. [Google Scholar] [CrossRef]
- Peres, C.B.; Resende, P.M.R.; Nunes, L.J.R.; Morais, L.C.d. Advances in Carbon Capture and Use (CCU) Technologies: A Comprehensive Review and CO2 Mitigation Potential Analysis. Clean Technol. 2022, 4, 1193–1207. [Google Scholar] [CrossRef]
- Idem, R.; Supap, T.; Shi, H.; Gelowitz, D.; Ball, M.; Campbell, C.; Tontiwachwuthikul, P. Practical experience in post-combustion CO2 capture using reactive solvents in large pilot and demonstration plants. Int. J. Greenh. Gas Control 2015, 40, 6–25. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Yu, J. Applications of Zeolites in Sustainable Chemistry. Chem 2017, 3, 928–949. [Google Scholar] [CrossRef]
- Suescum-Morales, D.; Jiménez, J.R.; Fernández-Rodríguez, J.M. Review of the Application of Hydrotalcite as CO2 Sinks for Climate Change Mitigation. ChemEngineering 2022, 6, 50. [Google Scholar] [CrossRef]
- Veerabhadrappa, M.G.; Maroto-Valer, M.M.; Chen, Y.; Garcia, S. Layered Double Hydroxides-Based Mixed Metal Oxides: Development of Novel Structured Sorbents for CO2 Capture Applications. ACS Appl. Mater. Interfaces 2021, 13, 11805–11813. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhou, T.; Gao, Y.; Louis, B.; O’Hare, D.; Wang, Q. Molten salts-modified MgO-based adsorbents for intermediate-temperature CO2 capture: A review. J. Energy Chem. 2017, 26, 830–838. [Google Scholar] [CrossRef]
- Kwak, J.-S.; Kim, K.-Y.; Oh, K.-R.; Kwon, Y.-U. Performance enhancement of all-solid CO2 absorbent based on Na2CO3-promoted MgO by using ZrO2 dispersant. Int. J. Greenh. Gas Control 2019, 81, 38–43. [Google Scholar] [CrossRef]
- Joo, H.; Cho, S.J.; Na, K. Control of CO2 absorption capacity and kinetics by MgO-based dry sorbents promoted with carbonate and nitrate salts. J. CO2 Util. 2017, 19, 194–201. [Google Scholar] [CrossRef]
- Jeon, H.; Triviño, M.L.T.; Hwang, S.; Moon, J.H.; Yoo, J.; Seo, J.G. Unveiling the carbonation mechanism in molten salt-promoted MgO-Al2O3 sorbents. J. CO2 Util. 2020, 39, 101153. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Y.; Shah, B.B.; Zhao, D. CO2 Capture in Metal–Organic Framework Adsorbents: An Engineering Perspective. Adv. Sustain. Syst. 2019, 3, 1800080. [Google Scholar] [CrossRef]
- Song, K.S.; Fritz, P.W.; Coskun, A. Porous organic polymers for CO2 capture, separation and conversion. Chem. Soc. Rev. 2022, 51, 9831–9852. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Wang, Y.; Zhang, D.; Xiao, Q.; Huang, J.; Liu, Y.-N. Recent progress in porous organic polymers and their application for CO2 capture. Chin. J. Chem. Eng. 2022, 42, 91–103. [Google Scholar] [CrossRef]
- Luo, R.; Chen, M.; Liu, X.; Xu, W.; Li, J.; Liu, B.; Fang, Y. Recent advances in CO2 capture and simultaneous conversion into cyclic carbonates over porous organic polymers having accessible metal sites. J. Mater. Chem. A 2020, 8, 18408–18424. [Google Scholar] [CrossRef]
- Silva, J.M.; Trujillano, R.; Rives, V.; Soria, M.A.; Madeira, L.M. High temperature CO2 sorption over modified hydrotalcites. Chem. Eng. J. 2017, 325, 25–34. [Google Scholar] [CrossRef]
- Miguel, C.V.; Trujillano, R.; Rives, V.; Vicente, M.A.; Ferreira, A.F.P.; Rodrigues, A.E.; Mendes, A.; Madeira, L.M. High temperature CO2 sorption with gallium-substituted and promoted hydrotalcites. Sep. Purif. Technol. 2014, 127, 202–211. [Google Scholar] [CrossRef]
- Faria, A.C.; Trujillano, R.; Rives, V.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. Alkali metal (Na, Cs and K) promoted hydrotalcites for high temperature CO2 capture from flue gas in cyclic adsorption processes. Chem. Eng. J. 2022, 427, 131502. [Google Scholar] [CrossRef]
- Lee, J.M.; Min, Y.J.; Lee, K.B.; Jeon, S.G.; Na, J.G.; Ryu, H.J. Enhancement of CO2 Sorption Uptake on Hydrotalcite by Impregnation with K2CO3. Langmuir 2010, 26, 18788–18797. [Google Scholar] [CrossRef]
- Halabi, M.H.; de Croon, M.H.J.M.; van der Schaaf, J.; Cobden, P.D.; Schouten, J.C. High capacity potassium-promoted hydrotalcite for CO2 capture in H2 production. Int. J. Hydrogen Energy 2012, 37, 4516–4525. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.B. Impregnation of hydrotalcite with NaNO3 for enhanced high-temperature CO2 sorption uptake. Chem. Eng. J. 2019, 356, 964–972. [Google Scholar] [CrossRef]
- Oliveira, E.L.G.; Grande, C.A.; Rodrigues, A.E. CO2 sorption on hydrotalcite and alkali-modified (K and Cs) hydrotalcites at high temperatures. Sep. Purif. Technol. 2008, 62, 137–147. [Google Scholar] [CrossRef]
- Perejón, A.; Romeo, L.M.; Lara, Y.; Lisbona, P.; Martínez, A.; Valverde, J.M. The Calcium-Looping technology for CO2 capture: On the important roles of energy integration and sorbent behavior. Appl. Energy 2016, 162, 787–807. [Google Scholar] [CrossRef]
- Benhelal, E.; Shamsaei, E.; Rashid, M.I. Challenges against CO2 abatement strategies in cement industry: A review. J. Environ. Sci. 2021, 104, 84–101. [Google Scholar] [CrossRef]
- Plaza, M.G.; Martínez, S.; Rubiera, F. CO2 Capture, Use, and Storage in the Cement Industry: State of the Art and Expectations. Energies 2020, 13, 5692. [Google Scholar] [CrossRef]
- MacKenzie, A.; Granatstein, D.L.; Anthony, E.J.; Abanades, J.C. Economics of CO2 Capture Using the Calcium Cycle with a Pressurized Fluidized Bed Combustor. Energy Fuels 2007, 21, 920–926. [Google Scholar] [CrossRef]
- Geng, Y.-q.; Guo, Y.-x.; Fan, B.; Cheng, F.-q.; Cheng, H.-g. Research progress of calcium-based adsorbents for CO2 capture and anti-sintering modification. J. Fuel Chem. Technol. 2021, 49, 998–1013. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, H.; Liu, W.; Yang, Y.; Li, H. Incorporation of CaO into inert supports for enhanced CO2 capture: A review. Chem. Eng. J. 2020, 396, 125253. [Google Scholar] [CrossRef]
- Krödel, M.; Landuyt, A.; Abdala, P.M.; Müller, C.R. Mechanistic Understanding of CaO-Based Sorbents for High-Temperature CO2 Capture: Advanced Characterization and Prospects. ChemSusChem 2020, 13, 6259–6272. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, C.; Shen, B.; Zhang, X.; Zhang, Y.; Huang, J. Progress in the development and application of CaO-based adsorbents for CO2 capture—A review. Mater. Today Sustain. 2018; 1–2, 1–27. [Google Scholar] [CrossRef]
- Lu, H.; Khan, A.; Pratsinis, S.E.; Smirniotis, P.G. Flame-Made Durable Doped-CaO Nanosorbents for CO2 Capture. Energy Fuels 2009, 23, 1093–1100. [Google Scholar] [CrossRef]
- Li, L.; King, D.L.; Nie, Z.; Howard, C. Magnesia-Stabilized Calcium Oxide Absorbents with Improved Durability for High Temperature CO2 Capture. Ind. Eng. Chem. Res. 2009, 48, 10604–10613. [Google Scholar] [CrossRef]
- Lysikov, A.I.; Salanov, A.N.; Okunev, A.G. Change of CO2 Carrying Capacity of CaO in Isothermal Recarbonation—Decomposition Cycles. Ind. Eng. Chem. Res. 2007, 46, 4633–4638. [Google Scholar] [CrossRef]
- Grasa, G.S.; Abanades, J.C. CO2 Capture Capacity of CaO in Long Series of Carbonation/Calcination Cycles. Ind. Eng. Chem. Res. 2006, 45, 8846–8851. [Google Scholar] [CrossRef]
- Kierzkowska, A.M.; Pacciani, R.; Müller, C.R. CaO-Based CO2 Sorbents: From Fundamentals to the Development of New, Highly Effective Materials. ChemSusChem 2013, 6, 1130–1148. [Google Scholar] [CrossRef]
- Jing, J.-Y.; Li, T.-Y.; Zhang, X.-W.; Wang, S.-D.; Feng, J.; Turmel, W.A.; Li, W.-Y. Enhanced CO2 sorption performance of CaO/Ca3Al2O6 sorbents and its sintering-resistance mechanism. Appl. Energy 2017, 199, 225–233. [Google Scholar] [CrossRef]
- Zhou, Z.; Qi, Y.; Xie, M.; Cheng, Z.; Yuan, W. Synthesis of CaO-based sorbents through incorporation of alumina/aluminate and their CO2 capture performance. Chem. Eng. Sci. 2012, 74, 172–180. [Google Scholar] [CrossRef]
- Han, R.; Gao, J.; Wei, S.; Su, Y.; Qin, Y. Development of highly effective CaO@Al2O3 with hierarchical architecture CO2 sorbents via a scalable limited-space chemical vapor deposition technique. J. Mater. Chem. A 2018, 6, 3462–3470. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Liu, L.; Guo, X. Effects of preparation methods on the structure and property of Al-stabilized CaO-based sorbents for CO2 capture. Fuel Process. Technol. 2018, 173, 276–284. [Google Scholar] [CrossRef]
- López, J.M.; Grasa, G.; Murillo, R. Evaluation of the effect of inert support on the carbonation reaction of synthetic CaO-based CO2 sorbents. Chem. Eng. J. 2018, 350, 559–572. [Google Scholar] [CrossRef]
- Yu, Y.S.; Liu, W.Q.; An, H.; Yang, F.S.; Wang, G.X.; Feng, B.; Zhang, Z.X.; Rudolph, V. Modeling of the carbonation behavior of a calcium based sorbent for CO2 capture. Int. J. Greenh. Gas Control 2012, 10, 510–519. [Google Scholar] [CrossRef]
- Kurlov, A.; Broda, M.; Hosseini, D.; Mitchell, S.J.; Pérez-Ramírez, J.; Müller, C.R. Mechanochemically Activated, Calcium Oxide-Based, Magnesium Oxide-Stabilized Carbon Dioxide Sorbents. ChemSusChem 2016, 9, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Z.; Peng, Y.; Su, W.; Sun, X.; Li, J. Investigation on a novel CaO–Y2O3 sorbent for efficient CO2 mitigation. Chem. Eng. J. 2014, 243, 297–304. [Google Scholar] [CrossRef]
- Naeem, M.A.; Armutlulu, A.; Imtiaz, Q.; Müller, C.R. CaO-Based CO2 Sorbents Effectively Stabilized by Metal Oxides. ChemPhysChem 2017, 18, 3280–3285. [Google Scholar] [CrossRef]
- Kim, S.M.; Kierzkowska, A.M.; Broda, M.; Müller, C.R. Sol-gel synthesis of MgAl2O4-stabilized CaO for CO2 capture. Energy Procedia 2017, 114, 220–229. [Google Scholar] [CrossRef]
- Koirala, R.; Reddy, G.K.; Smirniotis, P.G. Single nozzle flame-made highly durable metal doped Ca-based sorbents for CO2 capture at high temperature. Energy Fuels 2012, 26, 3103–3109. [Google Scholar] [CrossRef]
- Lu, H.; Smirniotis, P.G. Calcium oxide doped sorbents for CO2 uptake in the presence of SO2 at high temperatures. Ind. Eng. Chem. Res. 2009, 48, 5454–5459. [Google Scholar] [CrossRef]
- De, S.; Dokania, A.; Ramirez, A.; Gascon, J. Advances in the Design of Heterogeneous Catalysts and Thermocatalytic Processes for CO2 Utilization. ACS Catal. 2020, 10, 14147–14185. [Google Scholar] [CrossRef]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Song, C.; Ji, P.; Wang, N.; Wang, W.; Cui, L. Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water-Gas Shift Reaction. Front. Chem. 2020, 8, 709. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Nielsen, D.U.; Hu, X.-M.; Daasbjerg, K.; Skrydstrup, T. Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicals. Nat. Catal. 2018, 1, 244–254. [Google Scholar] [CrossRef]
- le Saché, E.; Reina, T.R. Analysis of Dry Reforming as direct route for gas phase CO2 conversion. The past, the present and future of catalytic DRM technologies. Prog. Energy Combust. Sci. 2022, 89, 100970. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Kim, H.-M.; Yoo, S.-Y.; Roh, H.-S. A review on dry reforming of methane in aspect of catalytic properties. Catal. Today 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Singh, R.; Dhir, A.; Mohapatra, S.K.; Mahla, S.K. Dry reforming of methane using various catalysts in the process: Review. Biomass Convers. Biorefinery 2020, 10, 567–587. [Google Scholar] [CrossRef]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: A review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Tang, R.; Zhu, Z.; Li, C.; Xiao, M.; Wu, Z.; Zhang, D.; Zhang, C.; Xiao, Y.; Chu, M.; Genest, A.; et al. Ru-Catalyzed Reverse Water Gas Shift Reaction with Near-Unity Selectivity and Superior Stability. ACS Mater. Lett. 2021, 3, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Tawalbeh, M.; Javed, R.M.N.; Al-Othman, A.; Almomani, F.; Ajith, S. Unlocking the potential of CO2 hydrogenation into valuable products using noble metal catalysts: A comprehensive review. Environ. Technol. Innov. 2023, 31, 103217. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Ojelade, O.A.; Zaman, S.F. A Review on Pd Based Catalysts for CO2 Hydrogenation to Methanol: In-Depth Activity and DRIFTS Mechanistic Study. Catal. Surv. Asia 2020, 24, 11–37. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Rownaghi, A.A.; Rezaei, F. Combined capture and utilization of CO2 for syngas production over dual-function materials. ACS Sustain. Chem. Eng. 2018, 6, 13551–13561. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Proaño, L.; Arellano-Treviño, M.A.; Farrauto, R.J.; Figueredo, M.; Jeong-Potter, C.; Cobo, M. Mechanistic assessment of dual function materials, composed of Ru-Ni, Na2O/Al2O3 and Pt-Ni, Na2O/Al2O3, for CO2 capture and methanation by in-situ DRIFTS. Appl. Surf. Sci. 2020, 533, 147469. [Google Scholar] [CrossRef]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Mechanism of the CO2 storage and in situ hydrogenation to CH4. Temperature and adsorbent loading effects over Ru-CaO/Al2O3 and Ru-Na2CO3/Al2O3 catalysts. Appl. Catal. B Environ. 2019, 256, 117845. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Riesco-García, J.M.; Penelás-Pérez, G.; Urakawa, A. Enabling continuous capture and catalytic conversion of flue gas CO2 to syngas in one process. J. CO2 Util. 2016, 14, 106–111. [Google Scholar] [CrossRef]
- Wang, S.; Farrauto, R.J.; Karp, S.; Jeon, J.H.; Schrunk, E.T. Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM). J. CO2 Util. 2018, 27, 390–397. [Google Scholar] [CrossRef]
- Hu, L.; Urakawa, A. Continuous CO2 capture and reduction in one process: CO2 methanation over unpromoted and promoted Ni/ZrO2. J. CO2 Util. 2018, 25, 323–329. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; Kanani, N.; Jeong-Potter, C.W.; Farrauto, R.J. Bimetallic catalysts for CO2 capture and hydrogenation at simulated flue gas conditions. Chem. Eng. J. 2019, 375, 121953. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni loading effects on dual function materials for capture and in-situ conversion of CO2 to CH4 using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Sietsma, J.R.A.; Jos van Dillen, A.; de Jongh, P.E.; de Jong, K.P. Application of ordered mesoporous materials as model supports to study catalyst preparation by impregnation and drying. In Studies in Surface Science and Catalysis; Gaigneaux, E.M., Devillers, M., De Vos, D.E., Hermans, S., Jacobs, P.A., Martens, J.A., Ruiz, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 162, pp. 95–102. [Google Scholar]
- Lu, Z.; Kunisch, J.; Gan, Z.; Bunian, M.; Wu, T.; Lei, Y. Gold catalysts synthesized using a modified incipient wetness impregnation method for propylene epoxidation. ChemCatChem 2020, 12, 5993–5999. [Google Scholar] [CrossRef]
- Danks, A.E.; Hall, S.R.; Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef]
- Wu, J.; Zheng, Y.; Fu, J.; Guo, Y.; Yu, J.; Chu, J.; Huang, P.; Zhao, C. Synthetic Ni–CaO–CeO2 dual function materials for integrated CO2 capture and conversion via reverse water–gas shift reaction. Sep. Purif. Technol. 2023, 317, 123916. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to Form a Capacitor. U.S. Patent US304434A, 11 July 1967. [Google Scholar]
- Sunde, T.O.L.; Grande, T.; Einarsrud, M.-A. Modified pechini synthesis of oxide powders and thin films. In Handbook of Sol-Gel Science and Technology; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Radfarnia, H.R.; Sayari, A. A highly efficient CaO-based CO2 sorbent prepared by a citrate-assisted sol–gel technique. Chem. Eng. J. 2015, 262, 913–920. [Google Scholar] [CrossRef]
- Jin, S.; Bang, G.; Liu, L.; Lee, C.-H. Synthesis of mesoporous MgO–CeO2 composites with enhanced CO2 capture rate via controlled combustion. Microporous Mesoporous Mater. 2019, 288, 109587. [Google Scholar] [CrossRef]
- Ma, X.; Li, X.; Cui, H.; Zhang, W.; Cheng, Z.; Zhou, Z. Metal oxide-doped Ni/CaO dual-function materials for integrated CO2 capture and conversion: Performance and mechanism. AIChE J. 2023, 69, e17520. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Zhao, J.; Shen, B.; Shi, J.; Huang, J.; Wu, C. Dual functional catalytic materials of Ni over Ce-modified CaO sorbents for integrated CO2 capture and conversion. Appl. Catal. B Environ. 2019, 244, 63–75. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, W.; Peng, P.; Zhang, Z.; Du, Q.; Shi, J.; Deng, L. A dual functional sorbent/catalyst material for in-situ CO2 capture and conversion to ethylene production. Fuel 2023, 351, 128701. [Google Scholar] [CrossRef]
- Machida, M.; Uto, M.; Kijima, T. Preparation of large surface area MnOx-ZrO2 for sorptive NOx removal. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2000; Volume 143, pp. 855–862. [Google Scholar]
- Virji, M.; Stefaniak, A. A Review of Engineered Nanomaterial Manufacturing Processes and Associated Exposures; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Li, C.; Li, M.; van Veen, A.C. Synthesis of Nano-Catalysts in Flow Conditions Using Millimixers. In Advanced Nanomaterials for Catalysis and Energy; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–28. [Google Scholar]
- Molina-Ramírez, S.; Cortés-Reyes, M.; Herrera, C.; Larrubia, M.; Alemany, L. CO2-SR Cyclic Technology: CO2 Storage and in situ Regeneration with CH4 over a new dual function NiBa unsupported catalyst. J. CO2 Util. 2020, 40, 101201. [Google Scholar] [CrossRef]
- Karami, D.; Mahinpey, N. Study of Al2O3 addition to synthetic Ca-based sorbents for CO2 sorption capacity and stability in cyclic operations. Can. J. Chem. Eng. 2015, 93, 102–110. [Google Scholar] [CrossRef]
- Huang, P.; Chu, J.; Fu, J.; Yu, J.; Li, S.; Guo, Y.; Zhao, C.; Liu, J. Influence of reduction conditions on the structure-activity relationships of NaNO3-promoted Ni/MgO dual function materials for integrated CO2 capture and methanation. Chem. Eng. J. 2023, 467, 143431. [Google Scholar] [CrossRef]
- Wegener Kofoed, M.V.; Jensen, M.B.; Mørck Ottosen, L.D. Chapter 12—Biological upgrading of biogas through CO2 conversion to CH4. In Emerging Technologies and Biological Systems for Biogas Upgrading; Aryal, N., Mørck Ottosen, L.D., Wegener Kofoed, M.V., Pant, D., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 321–362. [Google Scholar] [CrossRef]
- Seemann, M.; Thunman, H. 9—Methane synthesis. In Substitute Natural Gas from Waste; Materazzi, M., Foscolo, P.U., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 221–243. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Guan, S.; Huang, J.; Wu, C. Direct and highly selective conversion of captured CO2 into methane through integrated carbon capture and utilization over dual functional materials. J. CO2 Util. 2020, 38, 262–272. [Google Scholar] [CrossRef]
- Kosaka, F.; Liu, Y.; Chen, S.-Y.; Mochizuki, T.; Takagi, H.; Urakawa, A.; Kuramoto, K. Enhanced Activity of Integrated CO2 Capture and Reduction to CH4 under Pressurized Conditions toward Atmospheric CO2 Utilization. ACS Sustain. Chem. Eng. 2021, 9, 3452–3463. [Google Scholar] [CrossRef]
- Porta, A.; Matarrese, R.; Visconti, C.G.; Castoldi, L.; Lietti, L. Storage Material Effects on the Performance of Ru-Based CO2 Capture and Methanation Dual Functioning Materials. Ind. Eng. Chem. Res. 2021, 60, 6706–6718. [Google Scholar] [CrossRef]
- Jeong-Potter, C.; Farrauto, R. Feasibility Study of Combining Direct Air Capture of CO2 and Methanation at Isothermal Conditions with Dual Function Materials. Appl. Catal. B Environ. 2021, 282, 119416. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y.; Xu, S.; Osman, A.I.; Stenning, G.; Han, J.; Sun, S.; Rooney, D.; Williams, P.T.; Wang, F.; et al. Understanding the interaction between active sites and sorbents during the integrated carbon capture and utilization process. Fuel 2021, 286, 119308. [Google Scholar] [CrossRef]
- Porta, A.; Visconti, C.G.; Castoldi, L.; Matarrese, R.; Jeong-Potter, C.; Farrauto, R.; Lietti, L. Ru-Ba synergistic effect in dual functioning materials for cyclic CO2 capture and methanation. Appl. Catal. B Environ. 2021, 283, 119654. [Google Scholar] [CrossRef]
- Cimino, S.; Russo, R.; Lisi, L. Insights into the cyclic CO2 capture and catalytic methanation over highly performing Li-Ru/Al2O3 dual function materials. Chem. Eng. J. 2022, 428, 131275. [Google Scholar] [CrossRef]
- Onrubia-Calvo, J.A.; Bermejo-López, A.; Pérez-Vázquez, S.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Applicability of LaNiO3-derived catalysts as dual function materials for CO2 capture and in-situ conversion to methane. Fuel 2022, 320, 123842. [Google Scholar] [CrossRef]
- Sun, Z.; Shao, B.; Zhang, Y.; Gao, Z.; Wang, M.; Liu, H.; Hu, J. Integrated CO2 capture and methanation from the intermediate-temperature flue gas on dual functional hybrids of AMS/CaMgO||NixCoy. Sep. Purif. Technol. 2023, 307, 122680. [Google Scholar] [CrossRef]
- Faria, A.C.; Trujillano, R.; Rives, V.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. Cyclic operation of CO2 capture and conversion into methane on Ni-hydrotalcite based dual function materials (DFMs). J. CO2 Util. 2023, 72, 102476. [Google Scholar] [CrossRef]
- Onrubia-Calvo, J.A.; Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ca doping effect on the performance of La1−xCaxNiO3/CeO2-derived dual function materials for CO2 capture and hydrogenation to methane. Appl. Catal. B Environ. 2023, 321, 122045. [Google Scholar] [CrossRef]
- Sakai, M.; Imagawa, H.; Baba, N. Layered-double-hydroxide-based Ni catalyst for CO2 capture and methanation. Appl. Catal. A Gen. 2022, 647, 118904. [Google Scholar] [CrossRef]
- Catarina Faria, A.; Miguel, C.V.; Ferreira, A.F.P.; Rodrigues, A.E.; Madeira, L.M. CO2 capture and conversion to methane with Ni-substituted hydrotalcite dual function extrudates. Chem. Eng. J. 2023, 476, 146539. [Google Scholar] [CrossRef]
- Wang, X.; Economides, M. CHAPTER 7—Gas-To-Liquids (GTL). In Advanced Natural Gas Engineering; Wang, X., Economides, M., Eds.; Gulf Publishing Company: Houston, TX, USA, 2009; pp. 243–287. [Google Scholar] [CrossRef]
- Cimino, S.; Boccia, F.; Lisi, L. Effect of alkali promoters (Li, Na, K) on the performance of Ru/Al2O3 catalysts for CO2 capture and hydrogenation to methane. J. CO2 Util. 2020, 37, 195–203. [Google Scholar] [CrossRef]
- Jeong-Potter, C.; Zangiabadi, A.; Farrauto, R. Extended aging of Ru-Ni, Na2O/Al2O3 dual function materials (DFM) for combined capture and subsequent catalytic methanation of CO2 from power plant flue gas. Fuel 2022, 328, 125283. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Simulation-based optimization of cycle timing for CO2 capture and hydrogenation with dual function catalyst. Catal. Today 2022, 394–396, 314–324. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Modeling the CO2 capture and in situ conversion to CH4 on dual function Ru-Na2CO3/Al2O3 catalyst. J. CO2 Util. 2020, 42, 101351. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Finocchio, E.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3: Catalytic activity and infrared study. Catal. Today 2016, 277, 21–28. [Google Scholar] [CrossRef]
- Dreyer, J.A.H.; Li, P.; Zhang, L.; Beh, G.K.; Zhang, R.; Sit, P.H.L.; Teoh, W.Y. Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts. Appl. Catal. B Environ. 2017, 219, 715–726. [Google Scholar] [CrossRef]
- González-Castaño, M.; Dorneanu, B.; Arellano-García, H. The reverse water gas shift reaction: A process systems engineering perspective. React. Chem. Eng. 2021, 6, 954–976. [Google Scholar] [CrossRef]
- Shao, B.; Hu, G.; Alkebsi, K.A.M.; Ye, G.; Lin, X.; Du, W.; Hu, J.; Wang, M.; Liu, H.; Qian, F. Heterojunction-redox catalysts of FexCoyMg10CaO for high-temperature CO2 capture and in situ conversion in the context of green manufacturing. Energy Environ. Sci. 2021, 14, 2291–2301. [Google Scholar] [CrossRef]
- Sun, S.; He, S.; Wu, C. Ni promoted Fe-CaO dual functional materials for calcium chemical dual looping. Chem. Eng. J. 2022, 441, 135752. [Google Scholar] [CrossRef]
- Sasayama, T.; Kosaka, F.; Liu, Y.Y.; Yamaguchi, T.; Chen, S.Y.; Mochizuki, T.; Urakawa, A.; Kuramoto, K. Integrated CO2 capture and selective conversion to syngas using transition-metal-free Na/Al2O3 dual-function material. J. CO2 Util. 2022, 60, 102049. [Google Scholar] [CrossRef]
- Li, L.; Miyazaki, S.; Yasumura, S.; Ting, K.W.; Toyao, T.; Maeno, Z.; Shimizu, K.-I. Continuous CO2 Capture and Selective Hydrogenation to CO over Na-Promoted Pt Nanoparticles on Al2O3. ACS Catal. 2022, 12, 2639–2650. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, G.; Yu, J.; Huang, P.; Sun, J.; Wang, R.; Wang, T.; Zhao, C. Tailoring the performance of Ni-CaO dual function materials for integrated CO2 capture and conversion by doping transition metal oxides. Sep. Purif. Technol. 2023, 305, 122455. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, C.; Guan, S.; Xu, S.; Williams, P.T.; Wu, C. Ni/support-CaO bifunctional combined materials for integrated CO2 capture and reverse water-gas shift reaction: Influence of different supports. Sep. Purif. Technol. 2022, 298, 121604. [Google Scholar] [CrossRef]
- Bao, Z.; Yu, F. Chapter Two—Catalytic Conversion of Biogas to Syngas via Dry Reforming Process. In Advances in Bioenergy; Li, Y., Ge, X., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 3, pp. 43–76. [Google Scholar]
- Hu, J.; Hongmanorom, P.; Galvita, V.V.; Li, Z.; Kawi, S. Bifunctional Ni-Ca based material for integrated CO2 capture and conversion via calcium-looping dry reforming. Appl. Catal. B Environ. 2021, 284, 119734. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Broda, M.; Hosseini, D.; Copéret, C.; Müller, C. Integrated CO2 Capture and Conversion as an Efficient Process for Fuels from Greenhouse Gases. ACS Catal. 2018, 8, 2815–2823. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Jin, B.; Liang, Z. Layered double hydroxide derived bifunctional Ca-Fe-Mg material for integrated CO2 capture and utilization via chemical looping strategy. Chem. Eng. J. 2022, 431, 133826. [Google Scholar] [CrossRef]
- Tian, S.; Yan, F.; Zhang, Z.; Jiang, J. Calcium-looping reforming of methane realizes in situ CO2 utilization with improved energy efficiency. Sci. Adv. 2019, 5, eaav5077. [Google Scholar] [CrossRef]
- Jo, S.B.; Woo, J.H.; Lee, J.H.; Kim, T.Y.; Kang, H.I.; Lee, S.C.; Kim, J.C. CO2 green technologies in CO2 capture and direct utilization processes: Methanation, reverse water-gas shift, and dry reforming of methane. Sustain. Energy Fuels 2020, 4, 5543–5549. [Google Scholar] [CrossRef]
- Hu, J.; Hongmanorom, P.; Chirawatkul, P.; Kawi, S. Efficient integration of CO2 capture and conversion over a Ni supported CeO2-modified CaO microsphere at moderate temperature. Chem. Eng. J. 2021, 426, 130864. [Google Scholar] [CrossRef]
- Merkouri, L.-P.; Ramirez Reina, T.; Duyar, M.S. Feasibility of switchable dual function materials as a flexible technology for CO2 capture and utilisation and evidence of passive direct air capture. Nanoscale 2022, 14, 12620–12637. [Google Scholar] [CrossRef] [PubMed]
- Law, Z.X.; Pan, Y.-T.; Tsai, D.-H. Calcium looping of CO2 capture coupled to syngas production using Ni-CaO-based dual functional material. Fuel 2022, 328, 125202. [Google Scholar] [CrossRef]
- Hussain, I.; Tanimu, G.; Ahmed, S.; Aniz, C.U.; Alasiri, H.; Alhooshani, K. A review of the indispensable role of oxygen vacancies for enhanced CO2 methanation activity over CeO2-based catalysts: Uncovering, influencing, and tuning strategies. Int. J. Hydrogen Energy 2023, 48, 24663–24696. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of CO2 capture processes from different sources of combustion.
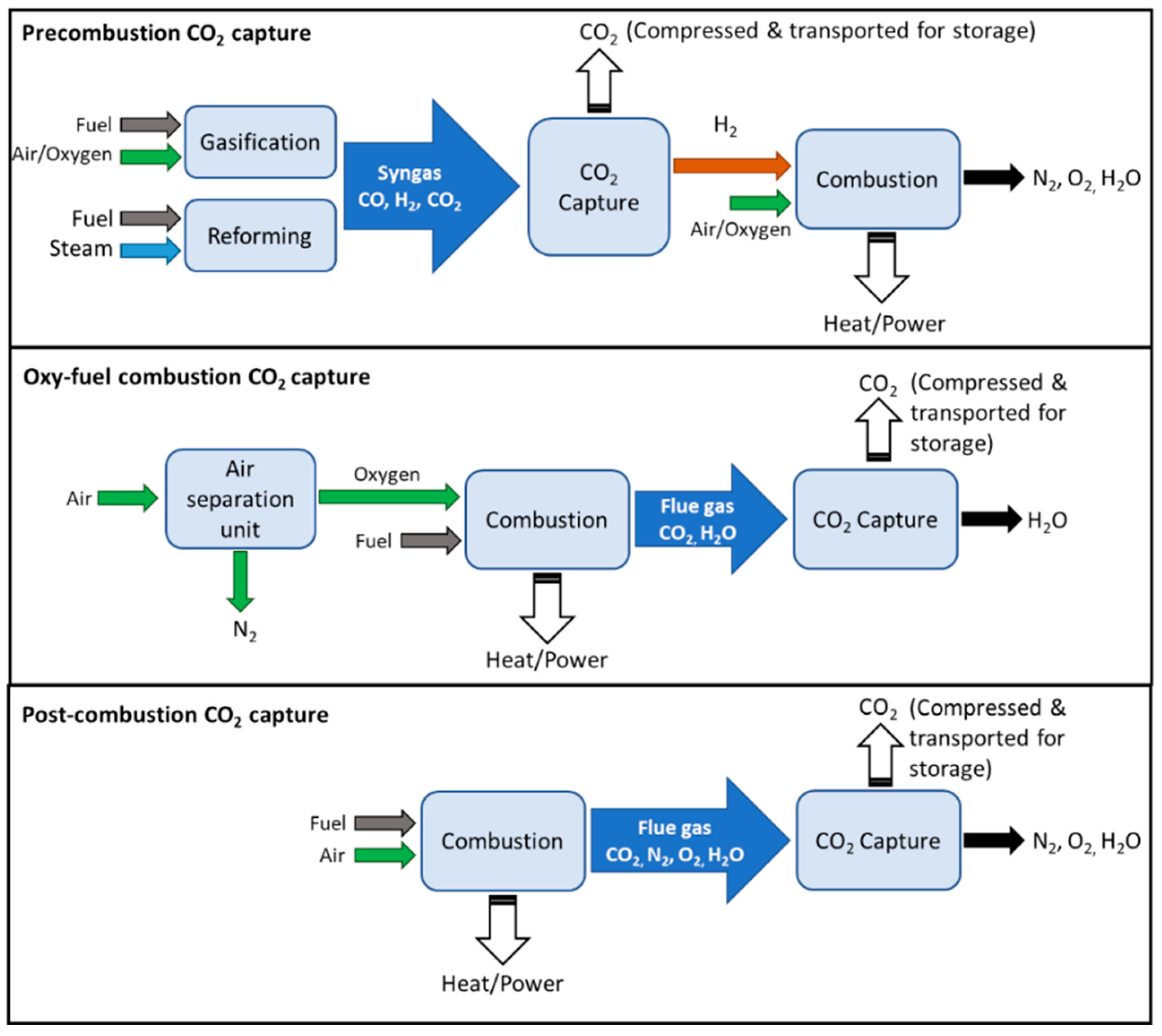
Figure 2.
Integrated CO2 capture and conversion system (Reactive Capture, bottom) in comparison with separate processes. Reprinted from ref. [10] with permission from Elsevier.
Figure 2.
Integrated CO2 capture and conversion system (Reactive Capture, bottom) in comparison with separate processes. Reprinted from ref. [10] with permission from Elsevier.

Figure 3.
Schematic diagram of integrated CO2 capture and conversion using DFMs.
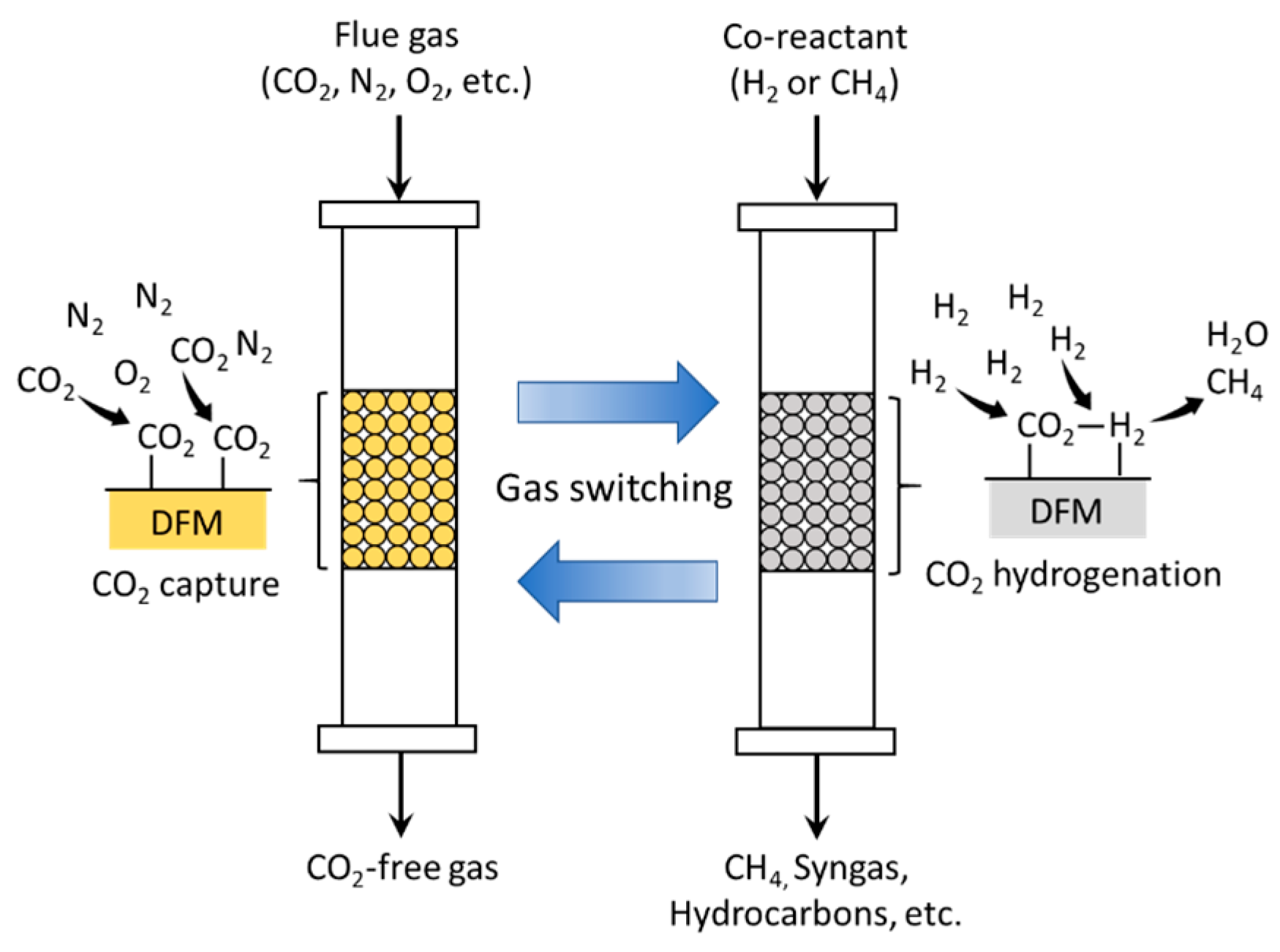
Figure 4.
Annual number of publications in the field of dual function materials for integrated CO2 capture and conversion (2015–2022).
Figure 4.
Annual number of publications in the field of dual function materials for integrated CO2 capture and conversion (2015–2022).

Figure 5.
Overview of CO2 capture technologies.

Figure 6.
Overview of commonly used solid sorbents and their typical application temperatures. Liquid amine system is shown as the basis for comparison. Reprinted from ref. [25] with permission from the American Chemical Society.
Figure 6.
Overview of commonly used solid sorbents and their typical application temperatures. Liquid amine system is shown as the basis for comparison. Reprinted from ref. [25] with permission from the American Chemical Society.

Figure 7.
SEM images of the fresh CaO precursors: (a) MC, (c) MD, and (e) CP; and of the sorbents after carbonation–calcination cycles: (b) MC (211 cycles), (d) MD (86 cycles), and (f) CP (136 cycles). MC: monocrystal-derived sample, MD: monodispersed particles, CP: commercial powder. Reprinted from ref. [58] with permission from the American Chemical Society.
Figure 7.
SEM images of the fresh CaO precursors: (a) MC, (c) MD, and (e) CP; and of the sorbents after carbonation–calcination cycles: (b) MC (211 cycles), (d) MD (86 cycles), and (f) CP (136 cycles). MC: monocrystal-derived sample, MD: monodispersed particles, CP: commercial powder. Reprinted from ref. [58] with permission from the American Chemical Society.

Figure 8.
An illustration of the sintering process—blue spheres represent CaO-based adsorbent particles, and the green layer represents the formation of CaCO3. Adapted from Lysikov et al. [58].
Figure 8.
An illustration of the sintering process—blue spheres represent CaO-based adsorbent particles, and the green layer represents the formation of CaCO3. Adapted from Lysikov et al. [58].

Figure 9.
The profile of weight change vs. time for repeated calcination/carbonation cycles of limestone (CaCO3). Calcination temperature is 850 °C; carbonation temperature is 650 °C. Reprinted from ref. [59] with permission from the American Chemical Society.
Figure 9.
The profile of weight change vs. time for repeated calcination/carbonation cycles of limestone (CaCO3). Calcination temperature is 850 °C; carbonation temperature is 650 °C. Reprinted from ref. [59] with permission from the American Chemical Society.

Figure 10.
Typical carbonation–calcination cycle of CaO: (1) Fast carbonation stage, (2) Slow carbonation stage, and (3) Calcination step, as observed through thermogravimetric analysis (TGA). Reprinted from ref. [60] with permission from Wiley-VCH.
Figure 10.
Typical carbonation–calcination cycle of CaO: (1) Fast carbonation stage, (2) Slow carbonation stage, and (3) Calcination step, as observed through thermogravimetric analysis (TGA). Reprinted from ref. [60] with permission from Wiley-VCH.

Figure 11.
Illustration of the carbonation reaction mechanism of CaO. Adapted from Sun et al. [55] with permission from Elsevier.
Figure 11.
Illustration of the carbonation reaction mechanism of CaO. Adapted from Sun et al. [55] with permission from Elsevier.

Figure 12.
Gibbs free energy of formation ( for different carbon-containing compounds, including that of H2 and H2O as common co-reactants. Reprinted from Ref. [73] with permission from the American Chemical Society.
Figure 12.
Gibbs free energy of formation ( for different carbon-containing compounds, including that of H2 and H2O as common co-reactants. Reprinted from Ref. [73] with permission from the American Chemical Society.

Figure 13.
Concentration distribution profiles during a CO2 adsorption and hydrogenation cycle at 370 °C with Ru10CaO and Ru10Na2CO3. Reprinted from ref. [96] with permission from Elsevier.
Figure 13.
Concentration distribution profiles during a CO2 adsorption and hydrogenation cycle at 370 °C with Ru10CaO and Ru10Na2CO3. Reprinted from ref. [96] with permission from Elsevier.

Figure 14.
Formation of metal–organic gel via Pechini sol–gel synthetic method. Reprinted from ref. [104] with permission from the Royal Society of Chemistry.
Figure 14.
Formation of metal–organic gel via Pechini sol–gel synthetic method. Reprinted from ref. [104] with permission from the Royal Society of Chemistry.

Figure 15.
Typical CO2 adsorption capacity of DFMs for ICCC–methanation. Orange zone: CaO; Red zone: MgO; Green zone: K2O and Na2O. Reprinted from ref. [14] with permission from the Royal Society of Chemistry.
Figure 15.
Typical CO2 adsorption capacity of DFMs for ICCC–methanation. Orange zone: CaO; Red zone: MgO; Green zone: K2O and Na2O. Reprinted from ref. [14] with permission from the Royal Society of Chemistry.

Figure 16.
CO2 adsorption, desorption, and CH4 produced on 10%Ni-6.1%“Na2O”/Al2O3 with and without O2 and steam present in the CO2 feed. CO2 capture and methanation were performed at 320 °C, 1 atm. Reprinted from ref. [92] with permission from Elsevier.
Figure 16.
CO2 adsorption, desorption, and CH4 produced on 10%Ni-6.1%“Na2O”/Al2O3 with and without O2 and steam present in the CO2 feed. CO2 capture and methanation were performed at 320 °C, 1 atm. Reprinted from ref. [92] with permission from Elsevier.

Figure 17.
Proposed carbonation and methanation mechanisms for 5%Ru/Al2O3 (right) and 5%Ru-6.1%“Na2O”/Al2O3 DFMs (left to right). Reprinted from ref. [95] with permission from Elsevier.
Figure 17.
Proposed carbonation and methanation mechanisms for 5%Ru/Al2O3 (right) and 5%Ru-6.1%“Na2O”/Al2O3 DFMs (left to right). Reprinted from ref. [95] with permission from Elsevier.

Figure 18.
Equilibrium mole fractions of chemical species involved in the RWGS and its side reactions. Reprinted from ref. [141] with permission from the Royal Society of Chemistry.
Figure 18.
Equilibrium mole fractions of chemical species involved in the RWGS and its side reactions. Reprinted from ref. [141] with permission from the Royal Society of Chemistry.

Figure 19.
Reaction mechanism of RWGS over CeO2-incorporated Ni/CaO DFM. Reprinted from ref. [111] with permission from the Elsevier.
Figure 19.
Reaction mechanism of RWGS over CeO2-incorporated Ni/CaO DFM. Reprinted from ref. [111] with permission from the Elsevier.

Figure 20.
STEM image and EDX elemental mapping of Pt–Na/Al2O3. Reprinted from ref. [145] with permission from the American Chemical Society.
Figure 20.
STEM image and EDX elemental mapping of Pt–Na/Al2O3. Reprinted from ref. [145] with permission from the American Chemical Society.

Figure 21.
Cyclic process of ICCC with DRM over physical mixture of CaO and Ni/MgO-Al2O3 as CO2 sorbent and catalyst, respectively. Reprinted from ref. [150] with permission from the American Chemical Society.
Figure 21.
Cyclic process of ICCC with DRM over physical mixture of CaO and Ni/MgO-Al2O3 as CO2 sorbent and catalyst, respectively. Reprinted from ref. [150] with permission from the American Chemical Society.
Figure 22.
Time-averaged space time yield (STY) of H2 (top) and CO (bottom) of the prepared Ni/CeO2-CaO DFMs with various CaO:CeO2 molar ratio throughout 9 cycles of CO2 capture and conversion at 650 °C. Ni/CaO was used as reference material. Reprinted from ref. [154] with permission from Elsevier.
Figure 22.
Time-averaged space time yield (STY) of H2 (top) and CO (bottom) of the prepared Ni/CeO2-CaO DFMs with various CaO:CeO2 molar ratio throughout 9 cycles of CO2 capture and conversion at 650 °C. Ni/CaO was used as reference material. Reprinted from ref. [154] with permission from Elsevier.

Figure 23.
In situ DRIFTS spectra of the reduced NiCe/Ca@Zr in CO2 and CH4 environment at 550 °C and 1 bar, with different color to represent spectrum obtained at different time during exposure in different gas environment. Abbreviation—(g): gas phase; (ad): adsorbed; MCa: Monodentate carbonate on CaO surface; PZr: Polydentate carbonate on ZrO2 surface; BCe: Bidentate carbonate on CeO2 surface. Reprinted from ref. [149] with permission from Elsevier.
Figure 23.
In situ DRIFTS spectra of the reduced NiCe/Ca@Zr in CO2 and CH4 environment at 550 °C and 1 bar, with different color to represent spectrum obtained at different time during exposure in different gas environment. Abbreviation—(g): gas phase; (ad): adsorbed; MCa: Monodentate carbonate on CaO surface; PZr: Polydentate carbonate on ZrO2 surface; BCe: Bidentate carbonate on CeO2 surface. Reprinted from ref. [149] with permission from Elsevier.


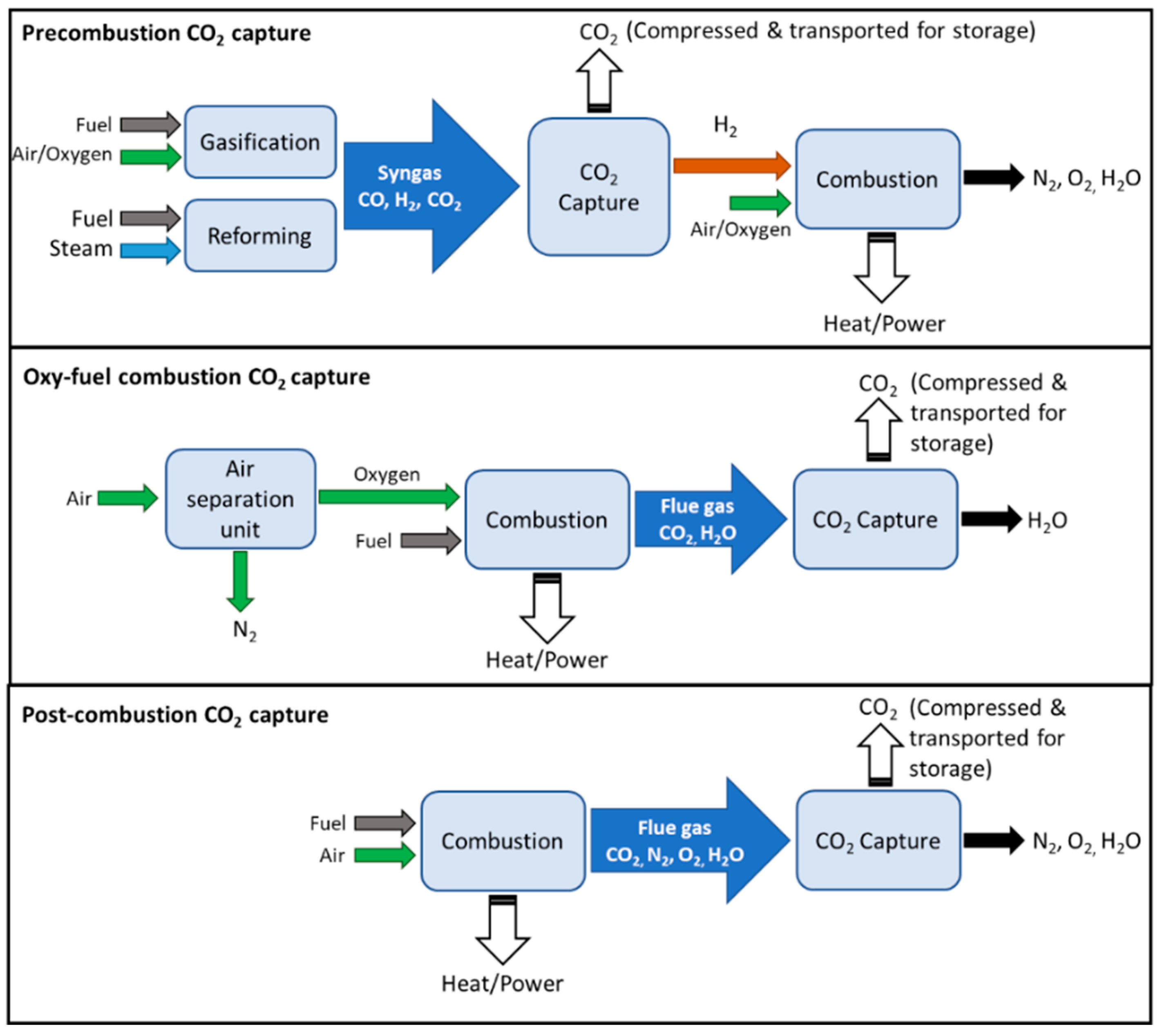{kind=link}
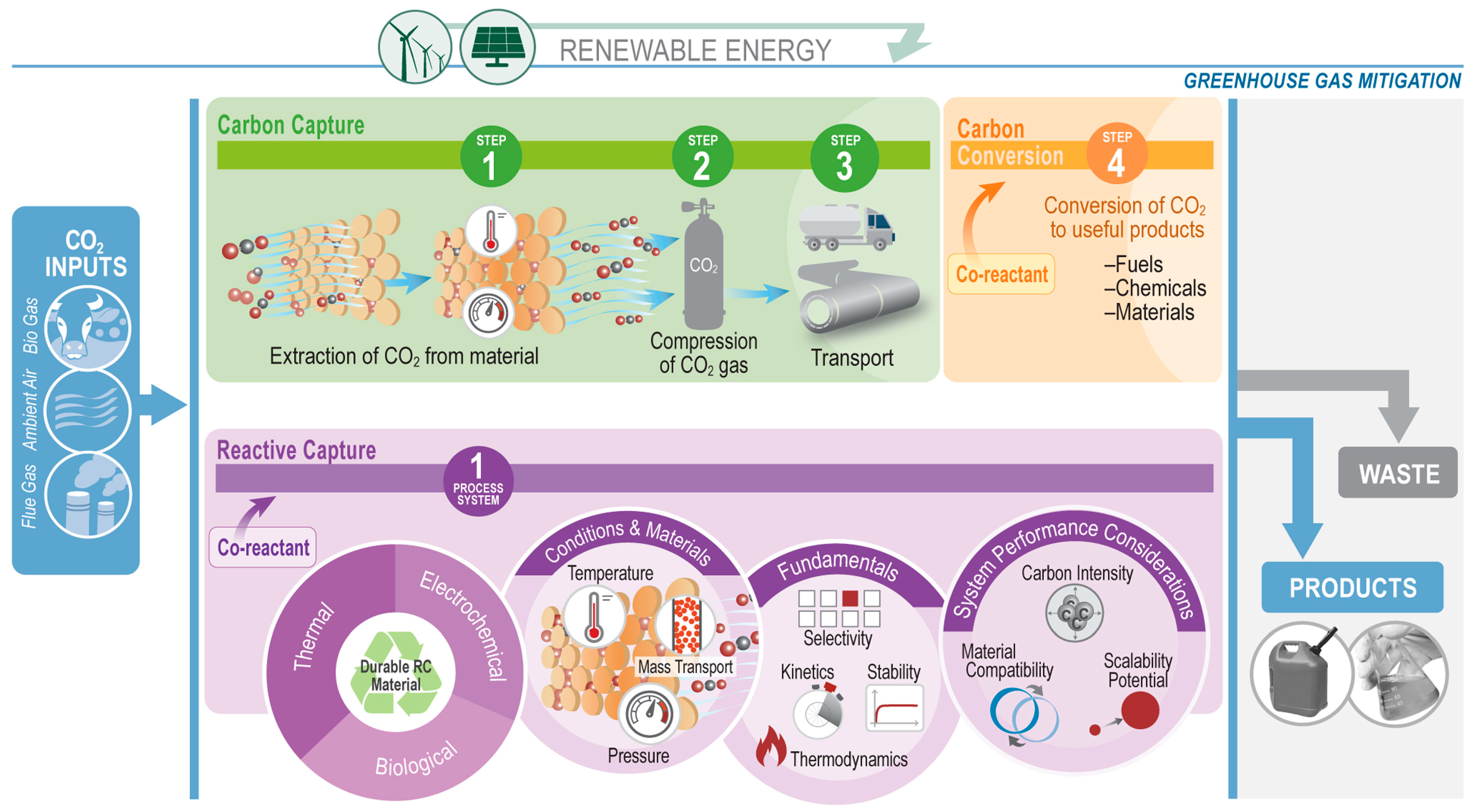{kind=link}
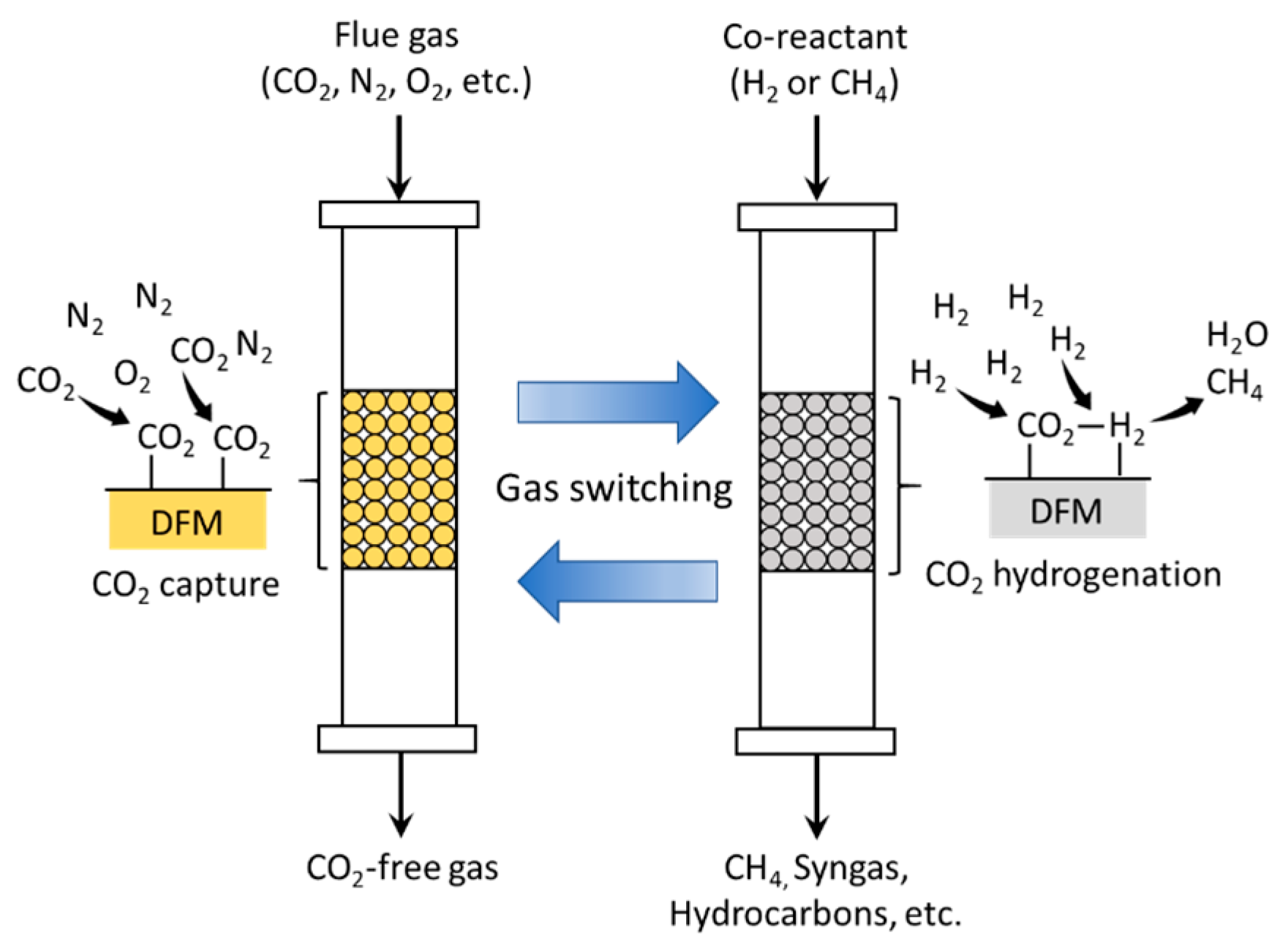{kind=link}
{kind=link}
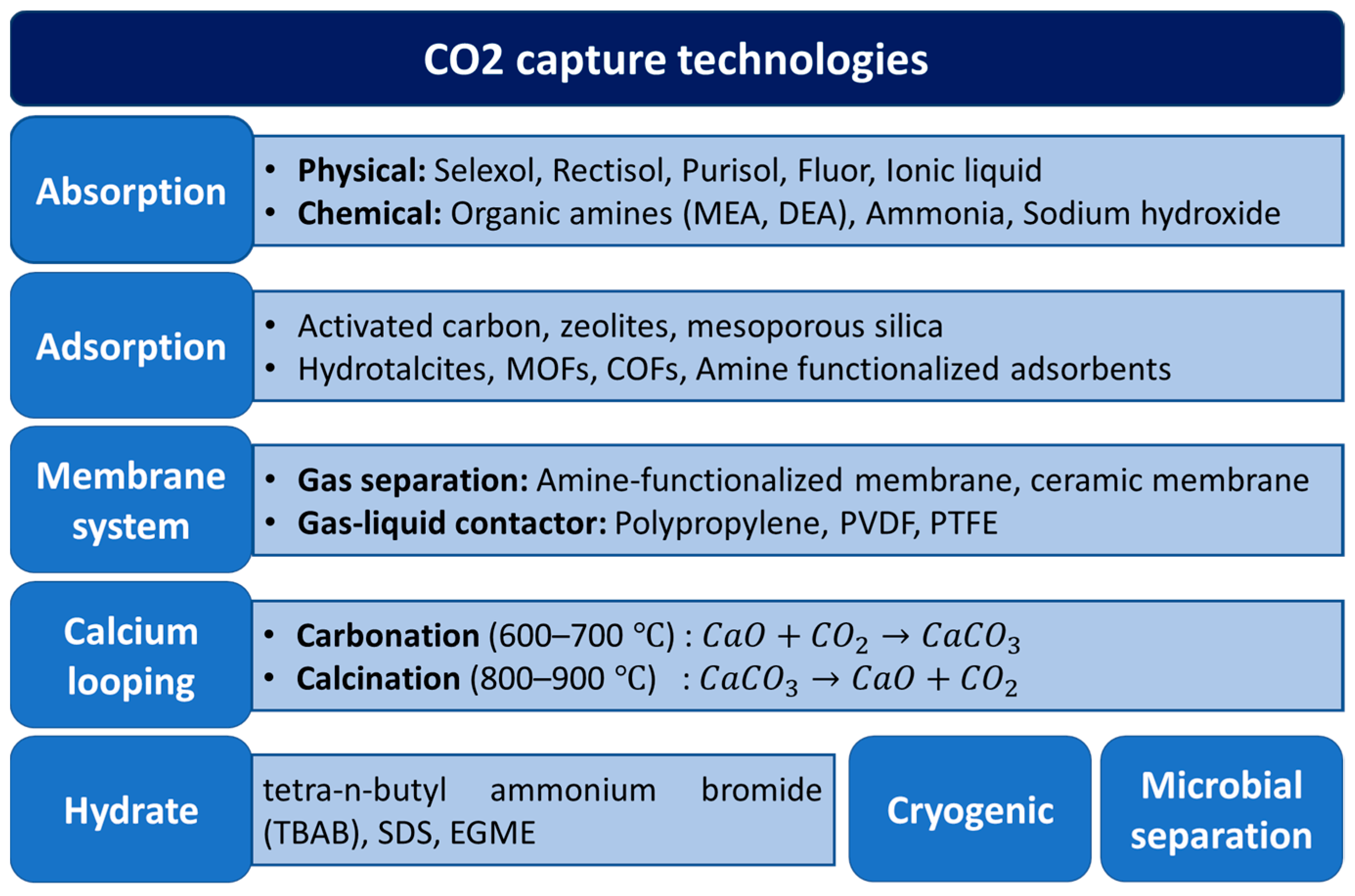{kind=link}
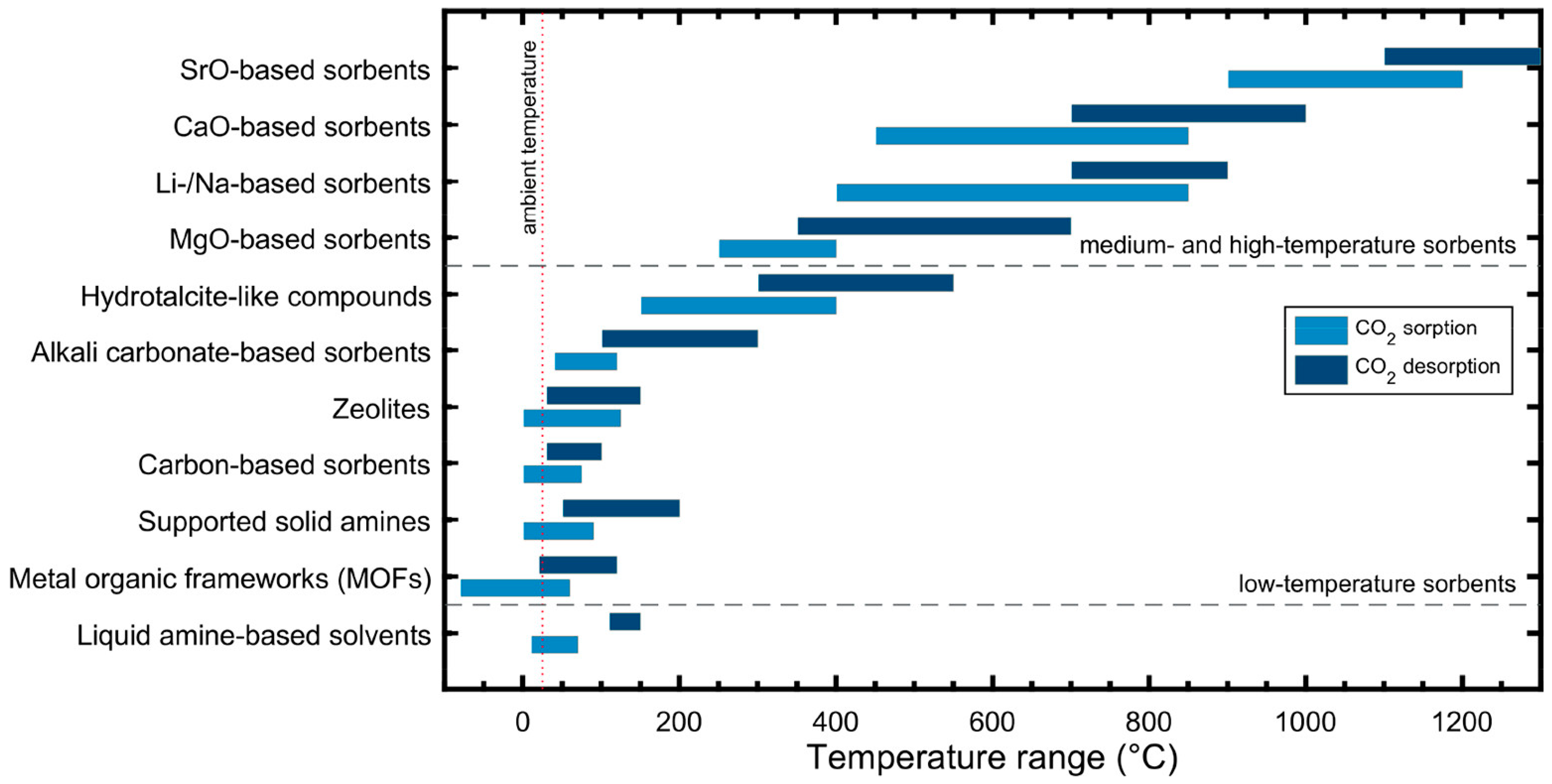{kind=link}
{kind=link}
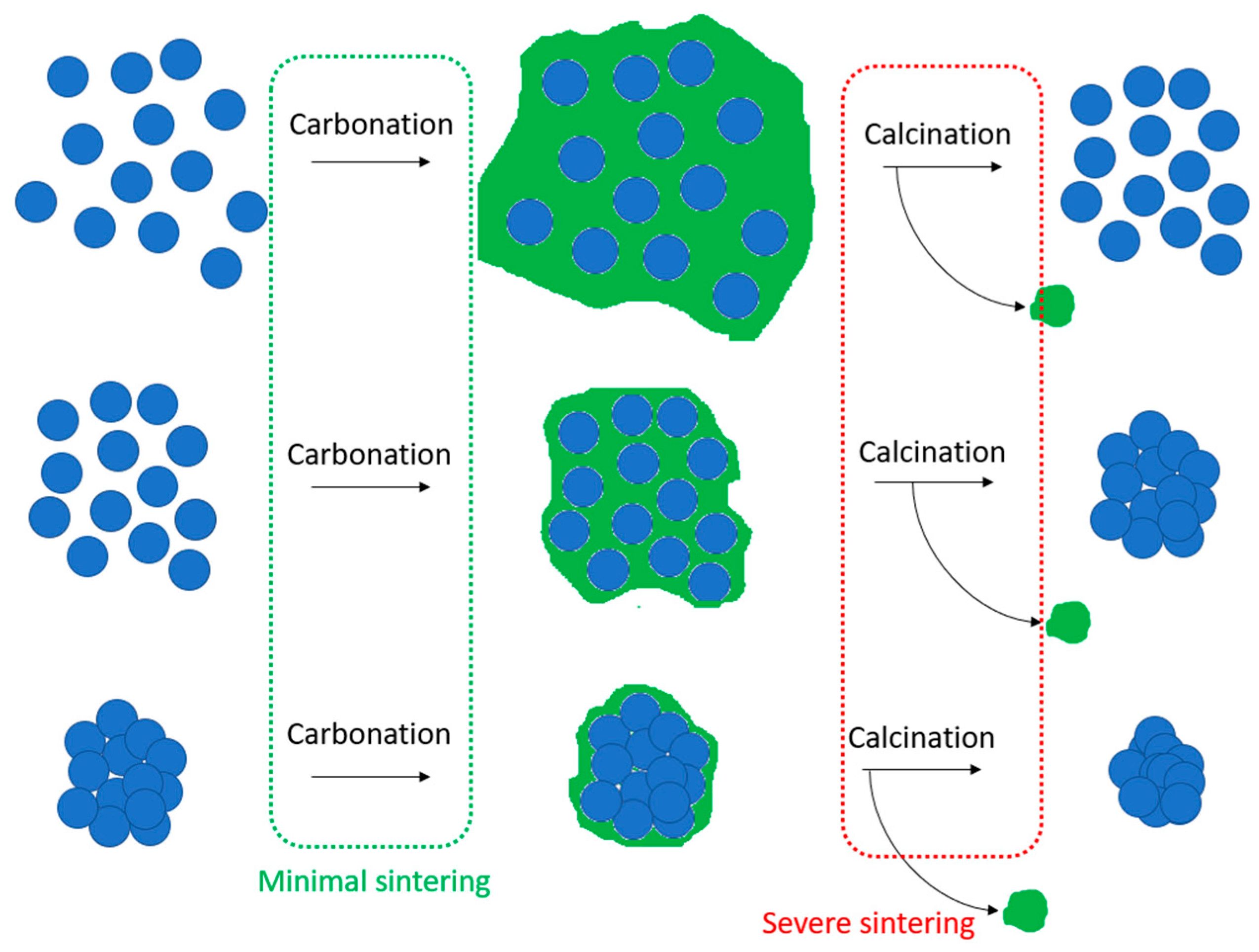{kind=link}
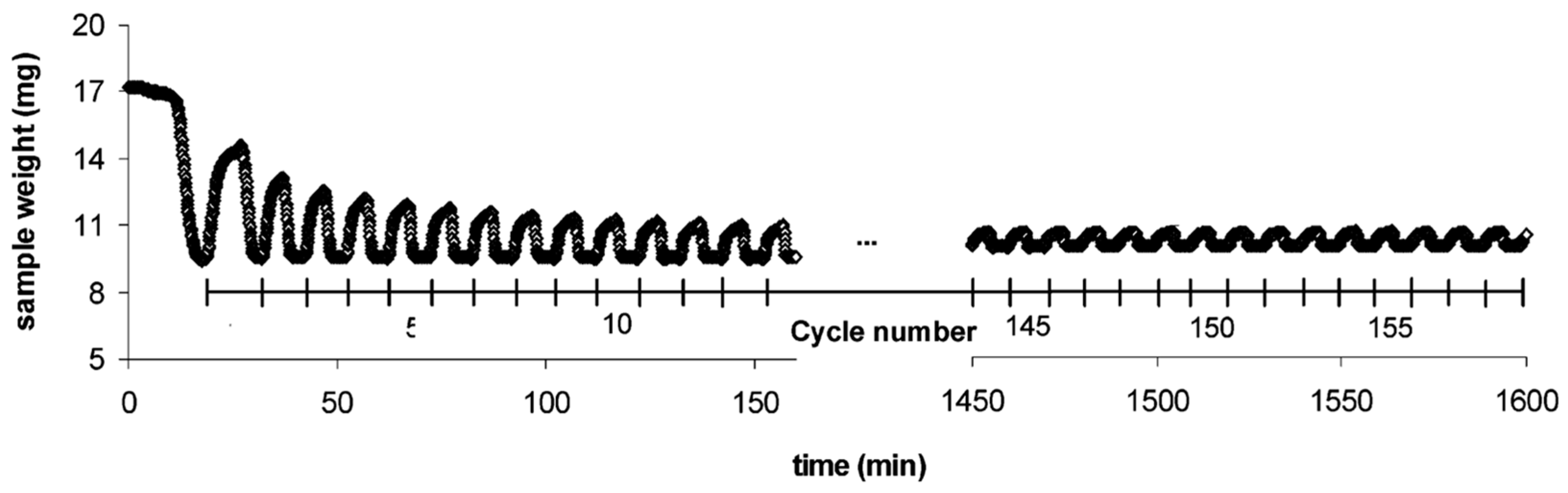{kind=link}
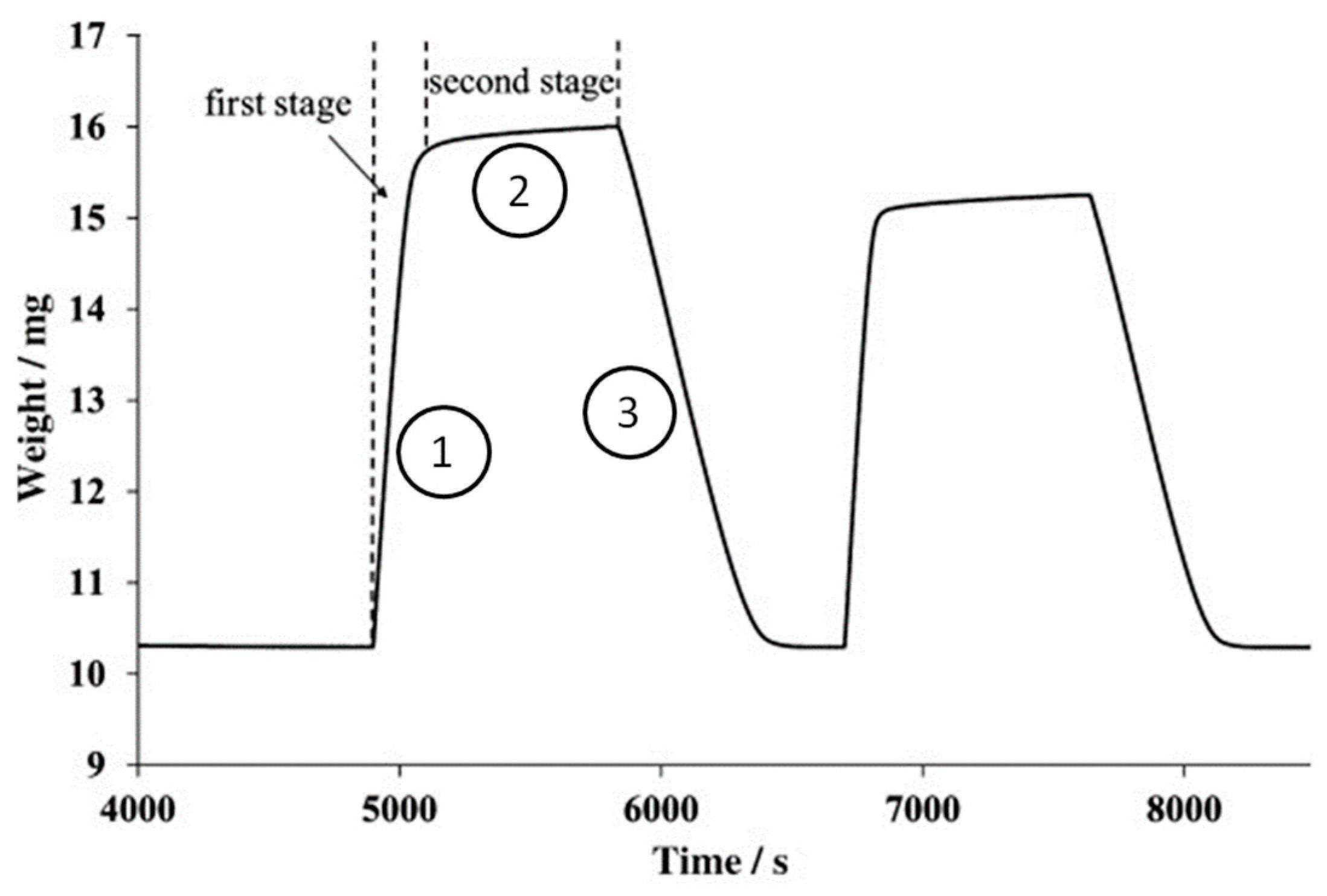{kind=link}
{kind=link}
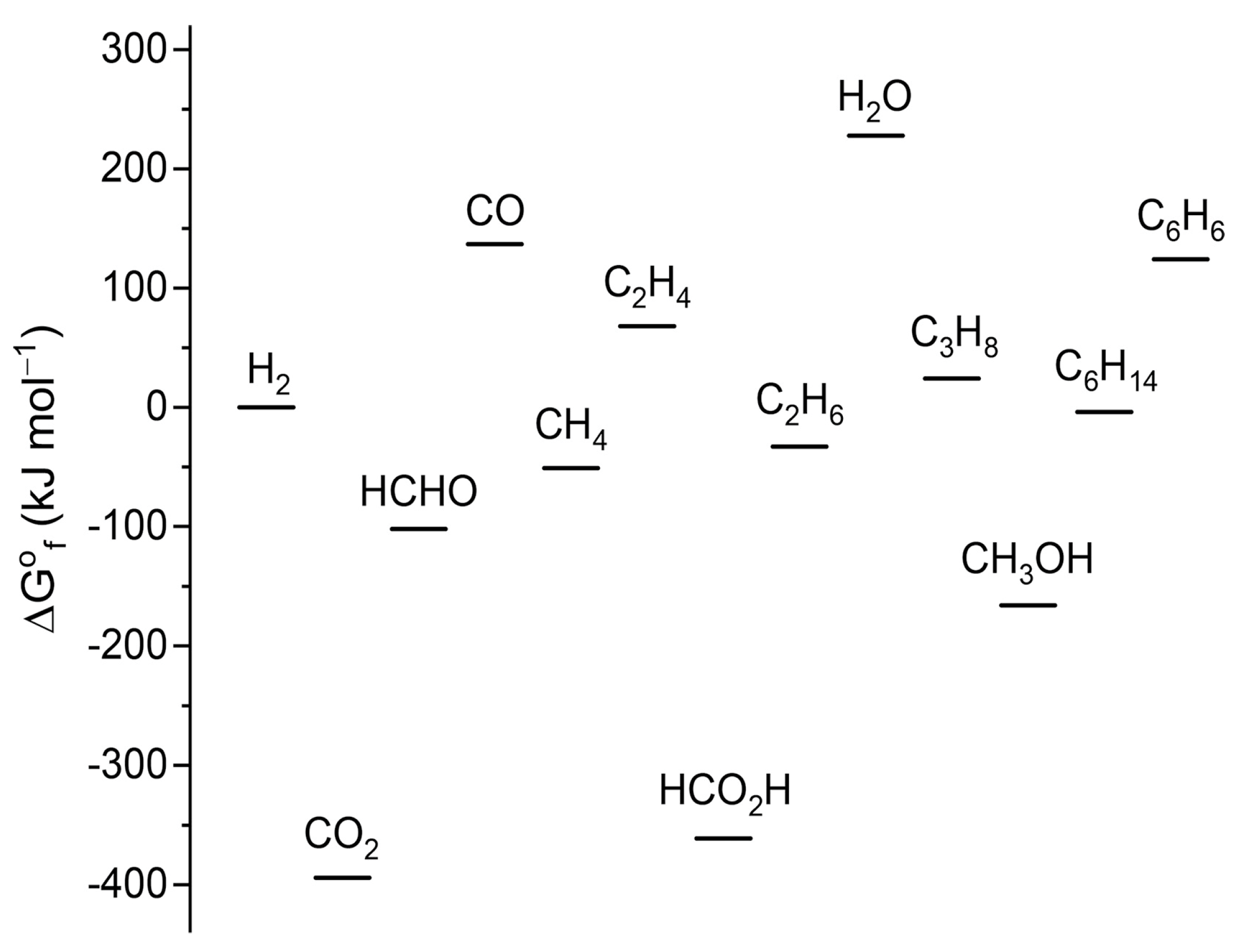{kind=link}
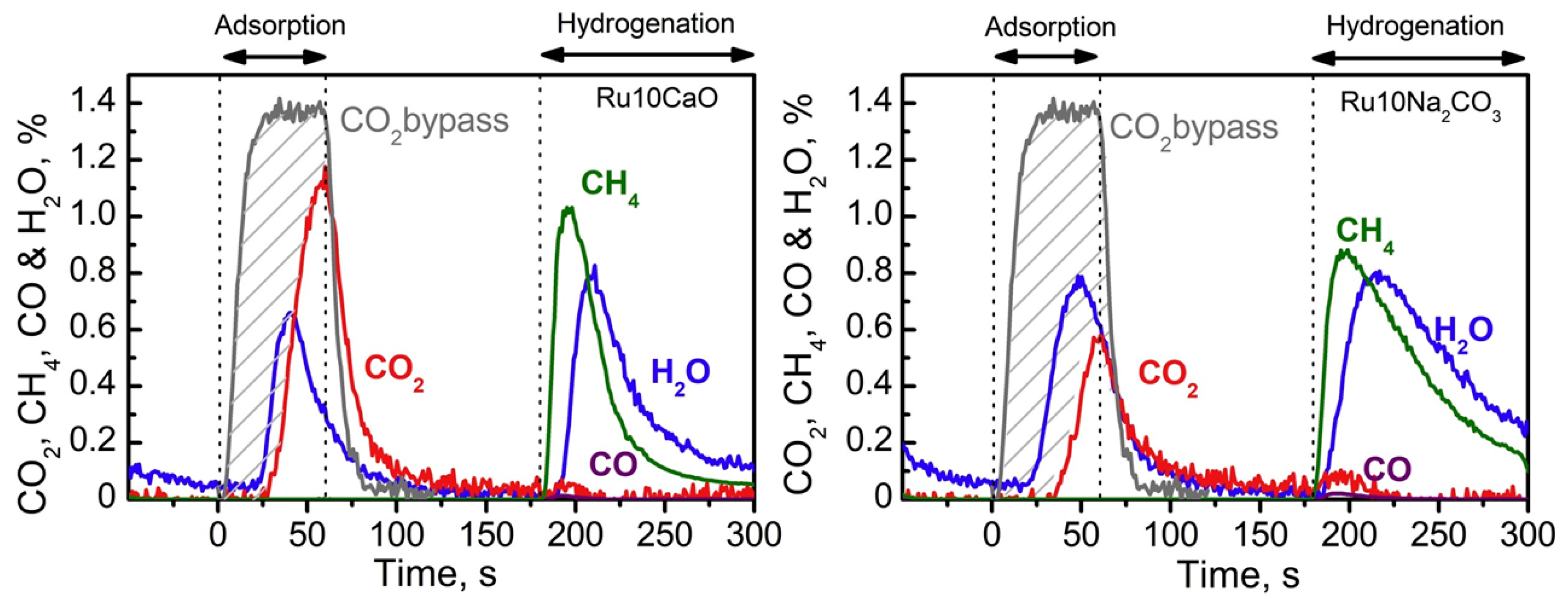{kind=link}
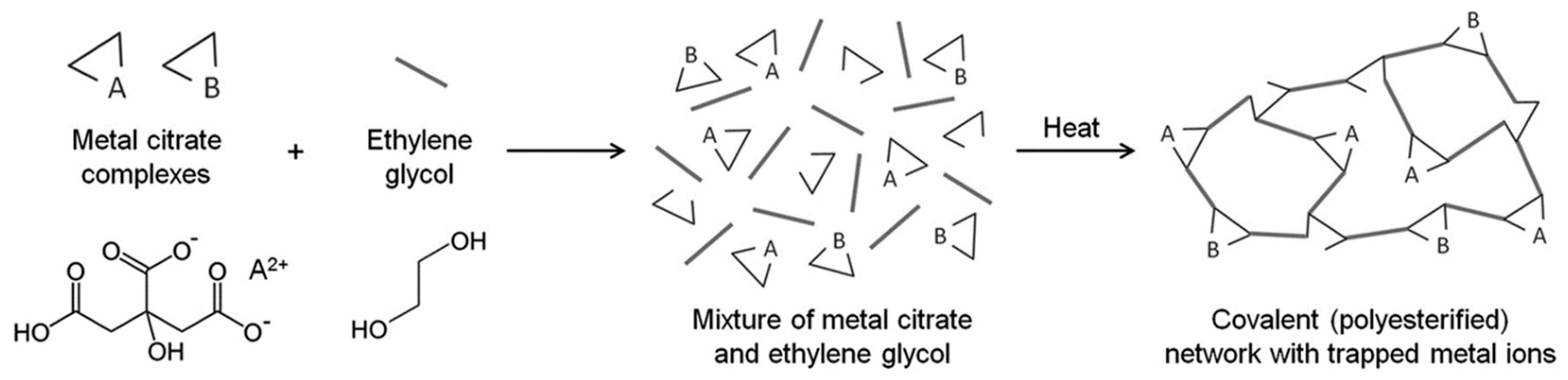{kind=link}
{kind=link}
{kind=link}
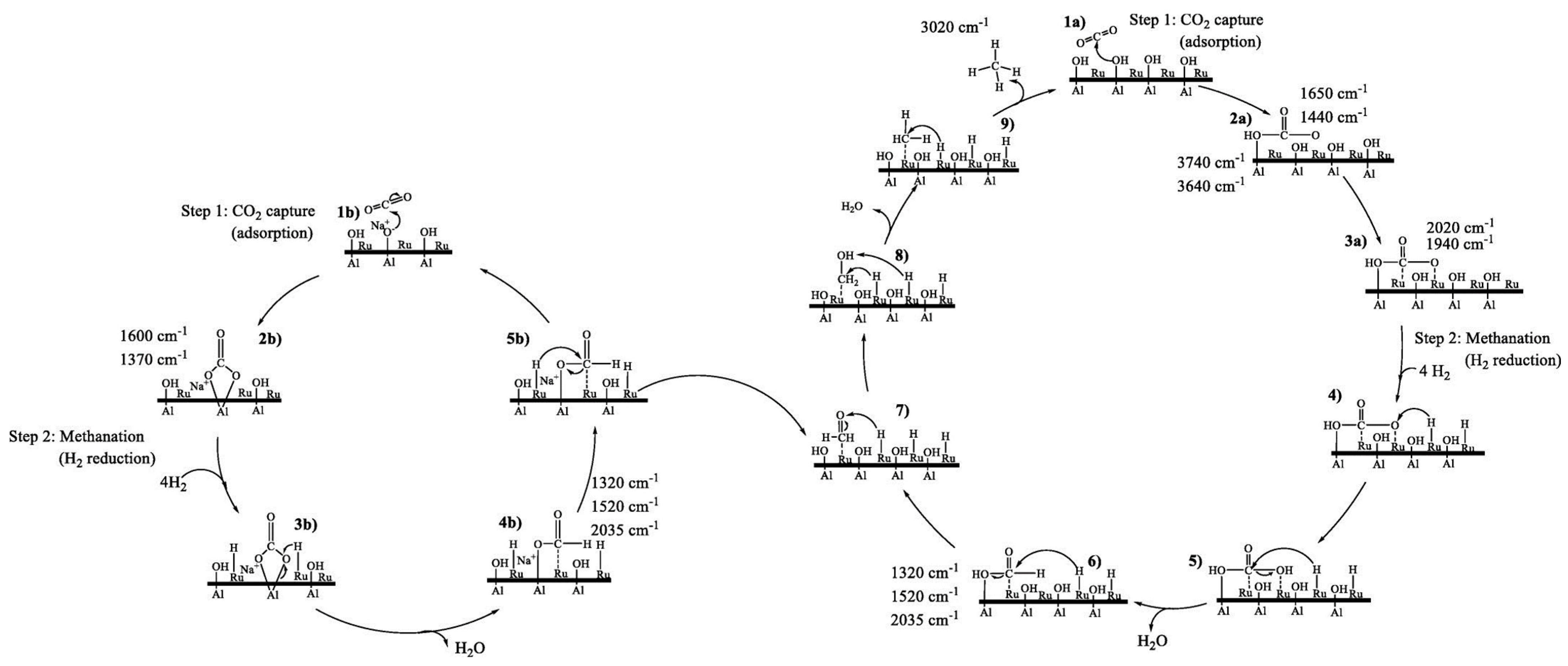{kind=link}
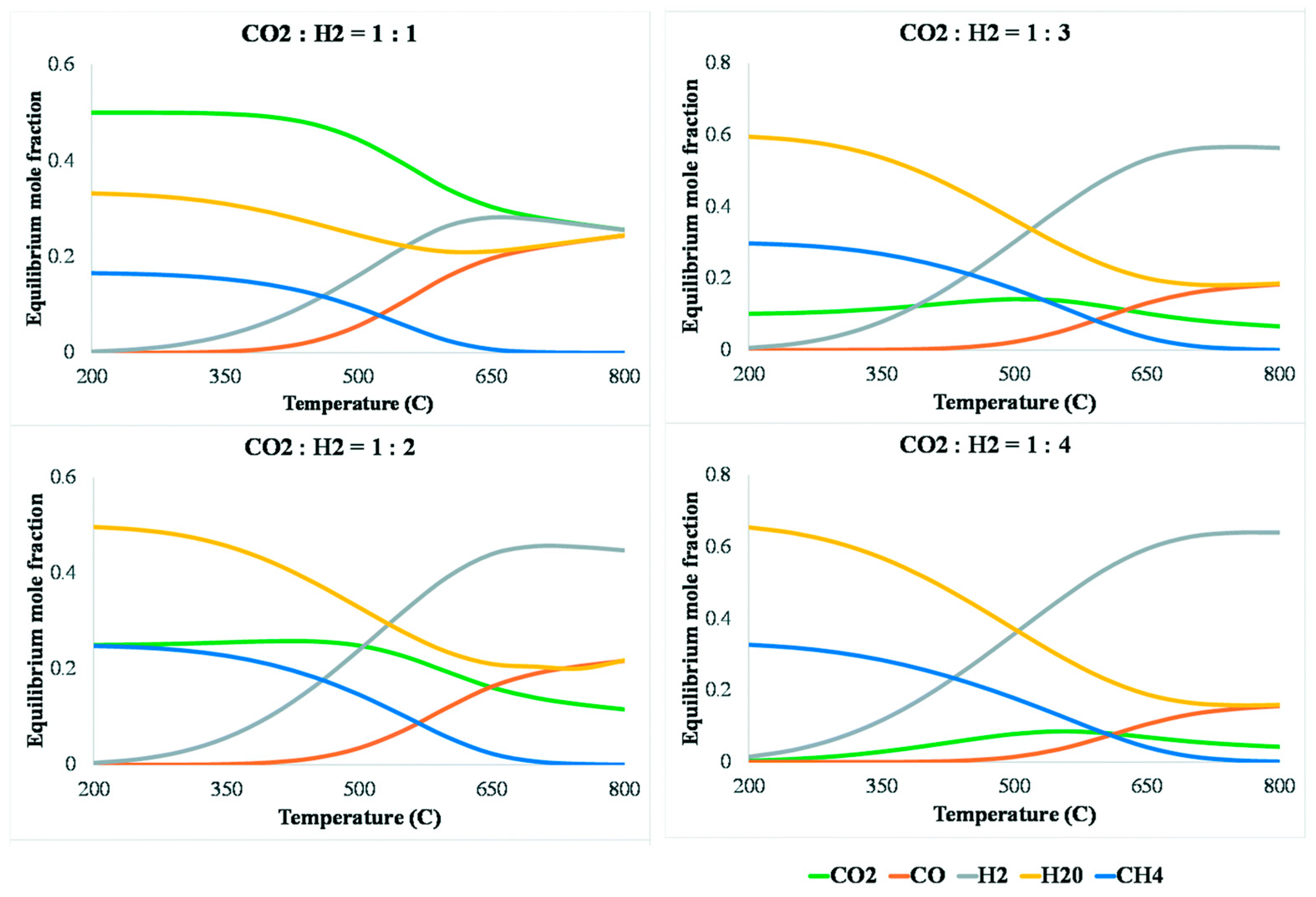{kind=link}
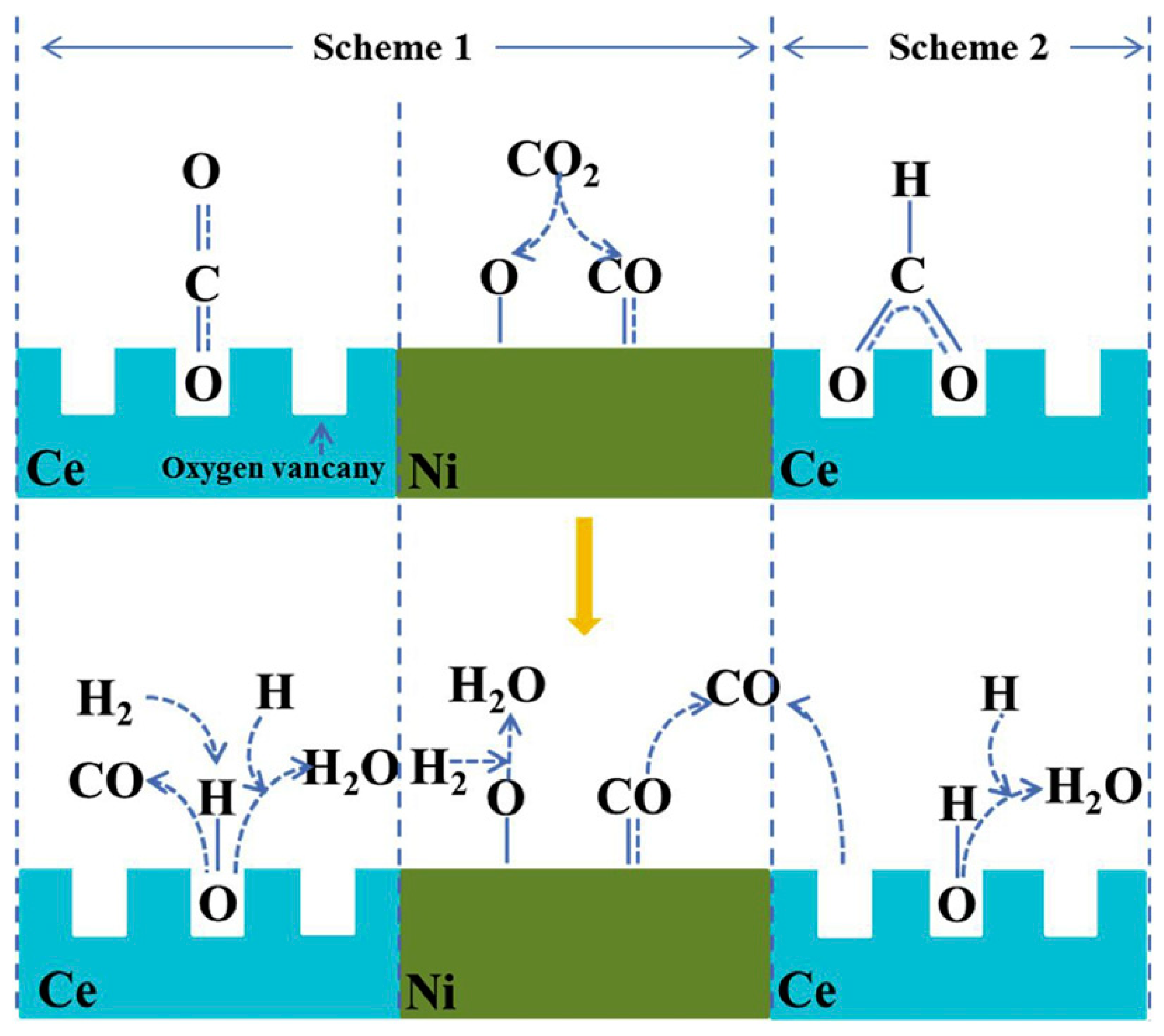{kind=link}
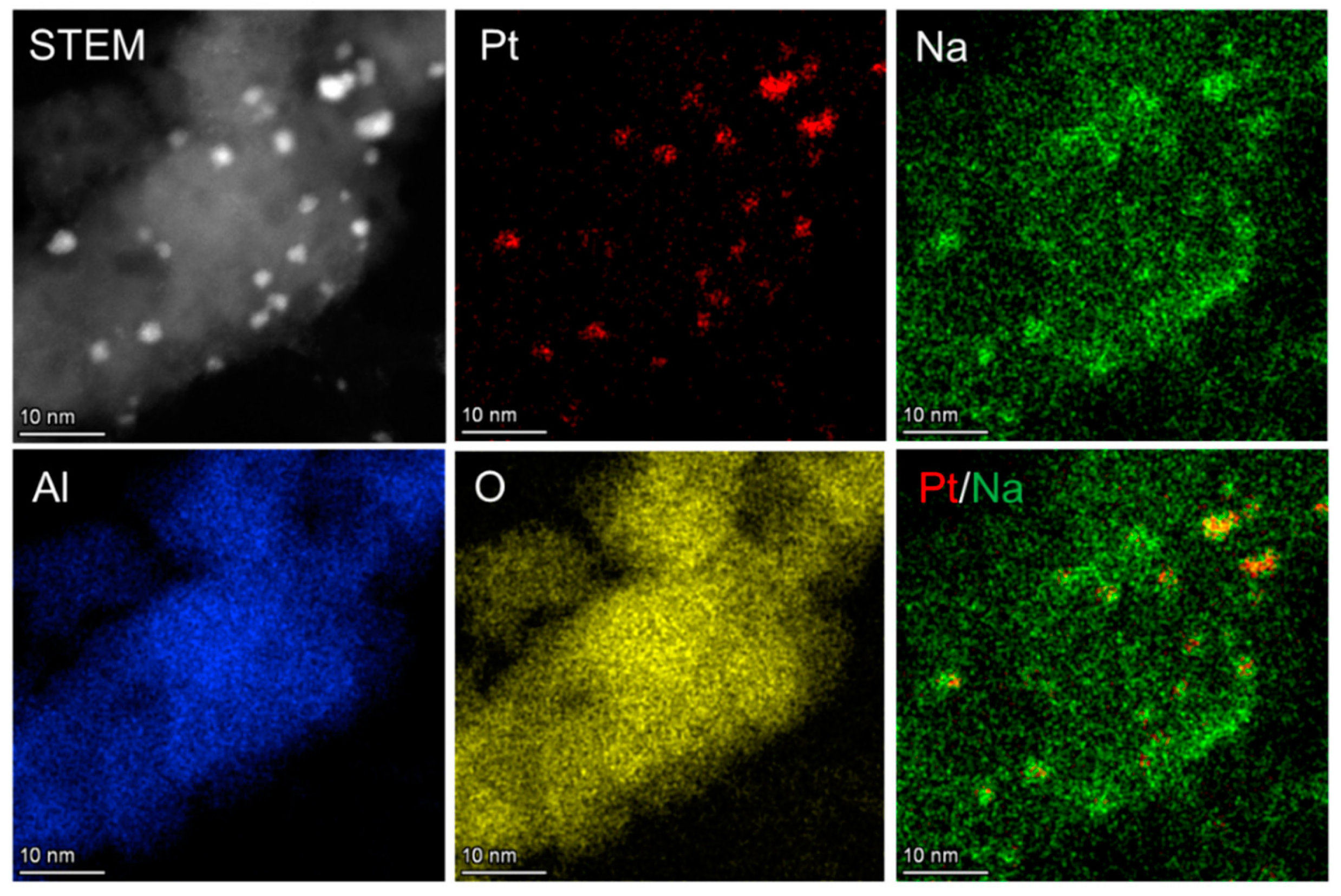{kind=link}
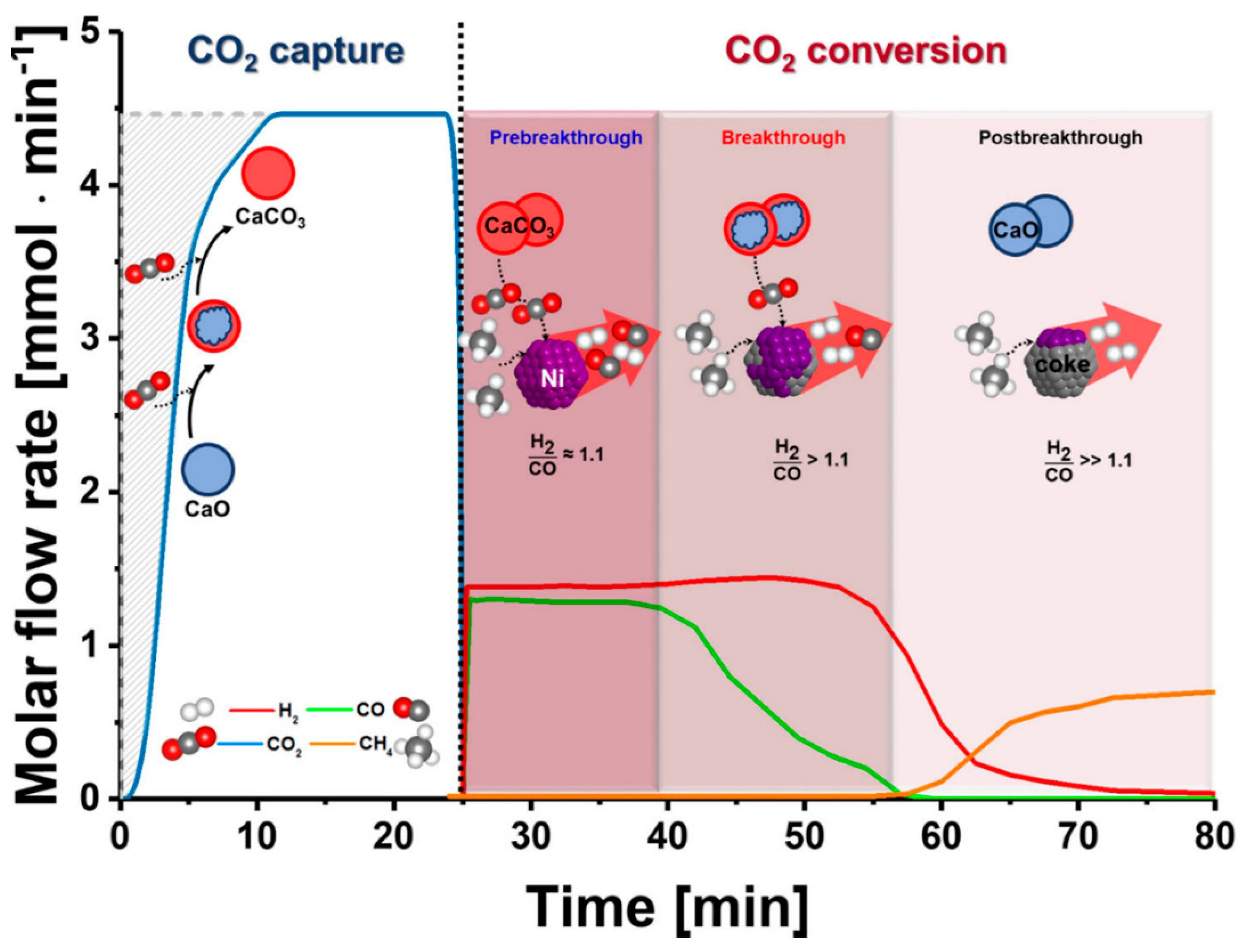{kind=link}
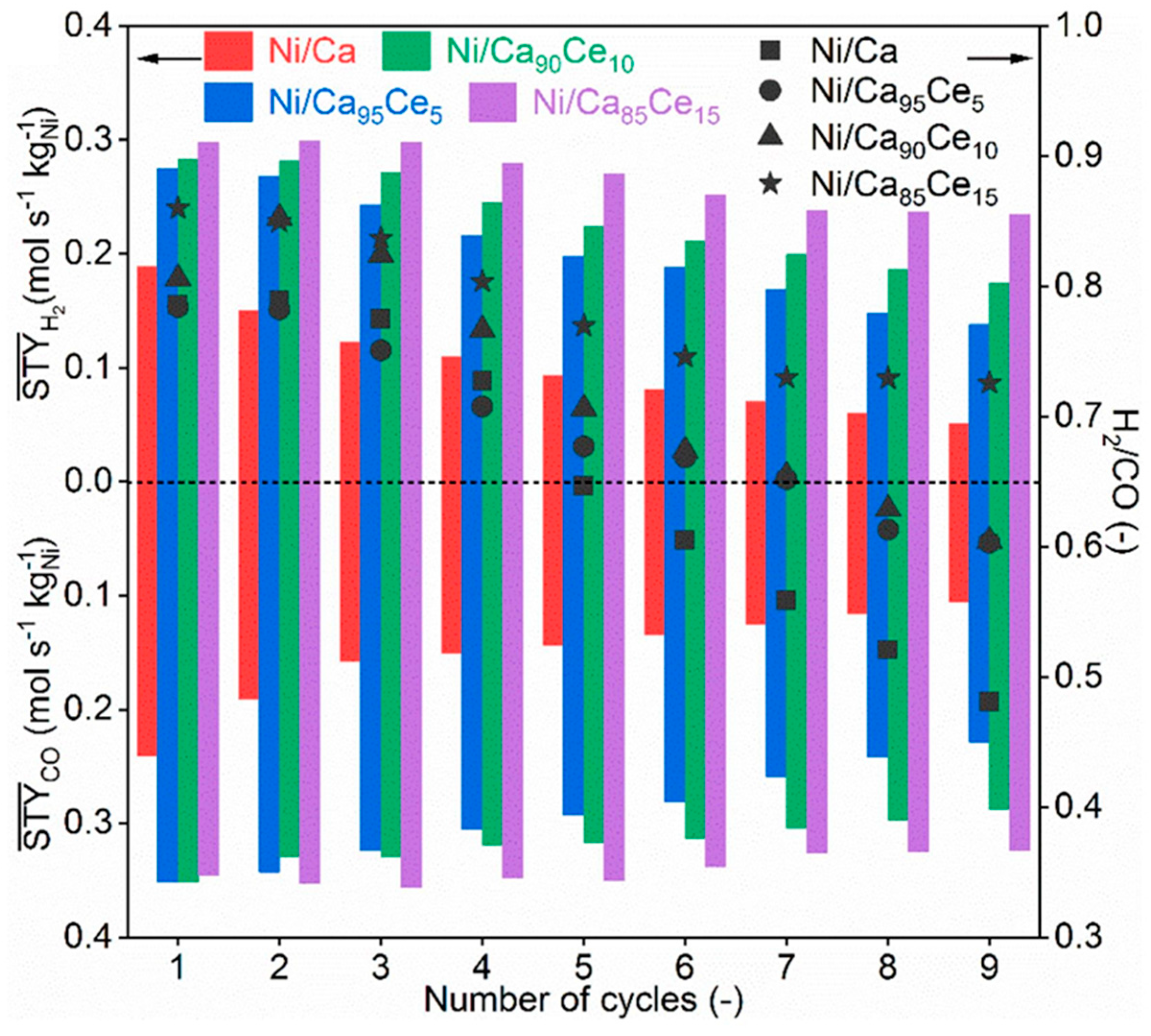{kind=link}
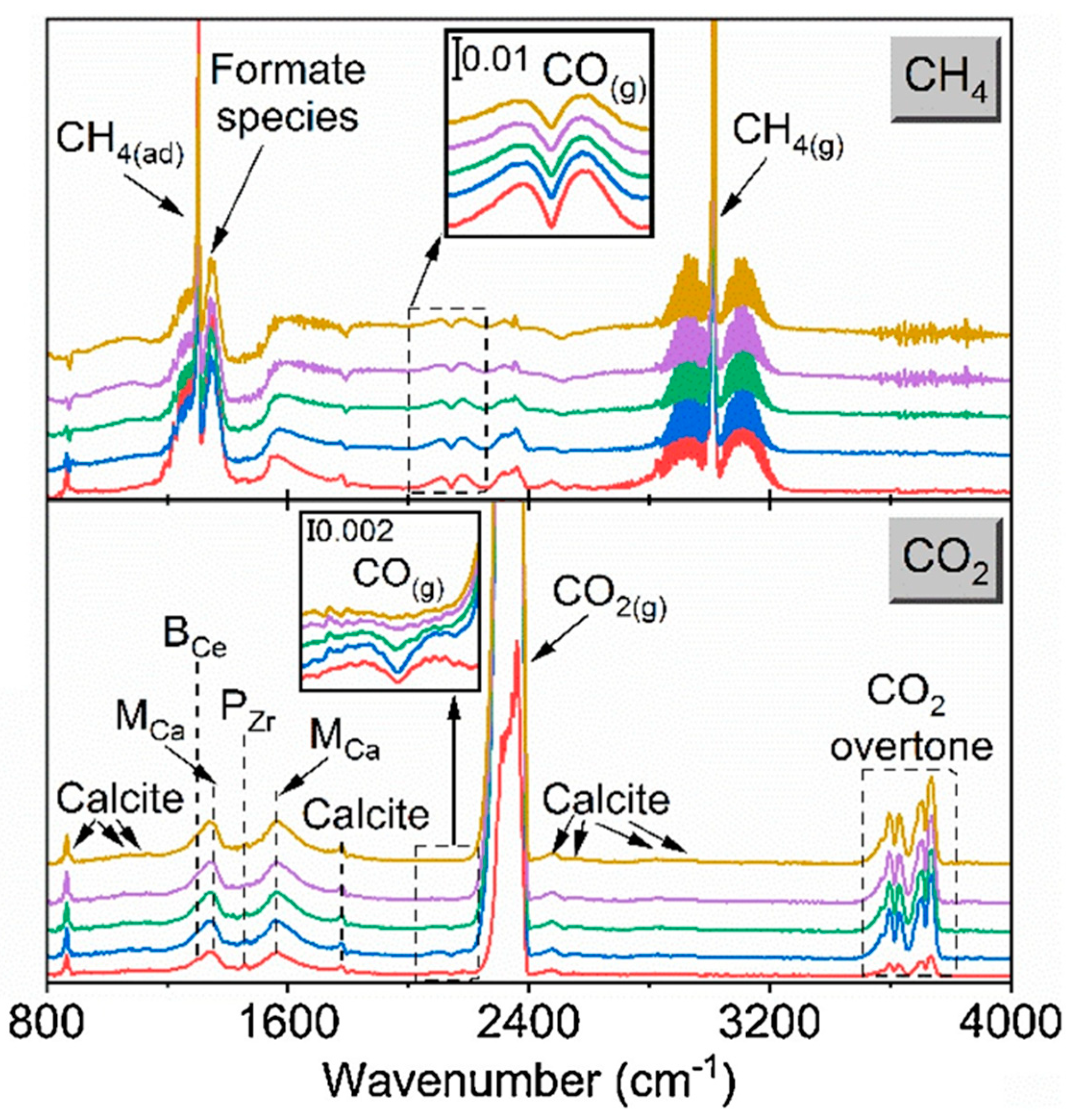{kind=link}
Table 1.
Comparison of different CO2 capture categories. Reproduced from ref. [6] with permission from the American Chemical Society.
Table 1.
Comparison of different CO2 capture categories. Reproduced from ref. [6] with permission from the American Chemical Society.
| Category | Pre-Combustion | Oxyfuel Combustion | Post-Combustion | |
|---|---|---|---|---|
| Amine Absorption | Solid Adsorption | |||
| CO2 recovery | 92–93% | 90–94% | 90–98% | 80–95% |
| Energy requirement | low energy | low energy | high regeneration energy | high regeneration energy |
| Costs | less expensive | moderately expensive | expensive | expensive |
| Challenges | increased process complexity | requires air separation unit | corrosion | solid attrition |
| high CO2 concentration | high CO2 concentration | solvent degradation | high pressure drop | |
| high pressure process | high temperature process | low CO2 concentration | easily poisoned by impurities (NOx, SOx) | |
| low CO2 concentration | ||||
Table 2.
Summary of the published reviews on dual function materials (DFMs) for integrated CO2 capture and conversion.
Table 2.
Summary of the published reviews on dual function materials (DFMs) for integrated CO2 capture and conversion.
| Authors | Year | Brief Description | Reference |
|---|---|---|---|
| Melo Bravo and Debecker | 2019 | Early review of DFMs with focus on CO2 methanation reaction and different types of reactor configurations | [12] |
| Omodolor et al. | 2020 | Overview of oxide- and carbonate-based CO2 adsorbents | [6] |
| Investigation of the active metals, material characteristics, and reaction conditions for Ni-, Ru-, and Rh-based DFMs in CO2 methanation, DRM, and RWGS reactions | |||
| Brief discussion on hydrotalcite-supported Fe-Cr-Cu catalyst as well as promoted Cu supported on alumina for syngas production | |||
| Merkouri et al. | 2021 | Chronological review of advances in DRM, RWGS, and CO2 methanation, as well as evaluation of the reaction mechanism of DFMs, by relating their performances with their physicochemical properties | [13] |
| Sun et al. | 2021 | Investigation of the process parameters and adsorbent–catalyst interaction on high-temperature CO2 capture combined with in situ DRM, RWGS, and methanation reactions | [14] |
| Sabri et al. | 2021 | Environmental and economic evaluation of CO2 capture and utilization process integration, as well as catalyst development for CO2 conversion in various systems, such as photocatalysis, electrocatalysis, and thermocatalysis | [15] |
| Li et al. | 2021 | Discussion on the performances of dual-function oxide particles for CO2 capture integrated with DRM, CO2 hydrogenation, and chemical looping | [16] |
DRM: dry reforming of methane; RWGS: reverse water–gas shift.
Table 3.
Literature summary of DFMs that catalyze the methanation reaction.
| DFM | Synthesis Method | Carbonation Conditions | Methanation Conditions | CO2 Sorption Capacity (mmol/g) | CO2 Conversion (%) | Methanation Capacity (mmol/g) | Ref. |
|---|---|---|---|---|---|---|---|
| 5% Ru 10% CaO/Al2O3 | IWI | 320 °C, 10%CO2/air | 320 °C, 5%H2/N2 | 0.41 | 76.17% | 0.31 | [11] |
| 5% Ru 10% CaO/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 4%H2/N2 | - | - | 0.5 | [93] |
| 5% Ru 10% K2CO3/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 4%H2/N2 | - | - | 0.91 | [93] |
| 5% Ru 10% Na2CO3/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 4%H2/N2 | - | - | 1.05 | [93] |
| 5% Ru 6.1% Na2O/γ-Al2O3 | IWI | 300 °C, simulated flue gas | 300 °C, 15%H2/N2 | 0.4 (50-cycle average) | 77% | 0.32 | [98] |
| 5% Ru-6.1% “Na2O”/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 10%H2/N2 | 0.651 | 96% | 0.614 | [92] |
| 0.5% Rh-6.1% “Na2O”/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 10%H2/N2 | 0.626 | 69% | 0.422 | [92] |
| 15% Ni 15% CaO/Al2O3 | IWI | 280–520 °C, 10%CO2/Ar | 280–520 °C, 10%H2/Ar | - | - | 0.142 | [101] |
| 15% Ni 10% Na2CO3/Al2O3 | IWI | 280–520 °C, 10%CO2/Ar | 280–520 °C, 10%H2/Ar | - | - | 0.186 | [101] |
| 4% Ru 10% CaO/Al2O3 | IWI | 370 °C, 1.4%CO2/Ar | 370 °C, 10%H2/Ar | 0.253 | - | 0.272 | [96] |
| 4% Ru 10% Na2CO3/Al2O3 | IWI | 370 °C, 1.4%CO2/Ar | 370 °C, 10%H2/Ar | 0.391 | - | 0.398 | [96] |
| 1% Ru, 10% Ni, 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 7.5%CO2-4.5%O2-15%H2O-balance N2 | 320 °C, 15%H2/N2 | 0.52 (3-cycle average) | 81% (3-cycle average) | 0.38 (3-cycle average) | [100] |
| 1% Ru, 10% Ni, 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 7.5%CO2-4.5%O2-15%H2O-balance N2 | 320 °C, 15%H2/N2 | 0.52 (20-cycle average) | 100% (20-cycle average) | 0.38 (20-cycle average) | [100] |
| 1% Pt 10% Ni 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 7.5%CO2-4.5%O2-15%H2O-balance N2 | 320 °C, 15%H2/N2 | 0.35 (3-cycle average) | 87% (3-cycle average) | 0.25 (3-cycle average) | [100] |
| 2.5Ru/CeO2-MgO | Impregnation/physical mixing | 300 °C, 65%CO2/N2 | 300 °C, 5%H2/N2 | - | 60% (1st cycle); 39% (10th cycle) | 5.73 (1st cycle); 1.13 (10th cycle) | [121] |
| 5Ru/CeO2-MgO | Impregnation/physical mixing | 300 °C, 65%CO2/N2 | 300 °C, 5%H2/N2 | - | 74% (1st cycle); 79% (10th cycle) | 6.6 (1st cycle); 3.36 (10th cycle) | [121] |
| 10Ru/CeO2-MgO | Impregnation/physical mixing | 300 °C, 65%CO2/N2 | 300 °C, 5%H2/N2 | - | 89% (1st cycle); 69% (10th cycle) | 7.07 (1st cycle); 2.31 (10th cycle) | [121] |
| Ni/Na-γ-Al2O3 | IWI | 450 °C, 5%CO2/N2 | 450 °C, H2 | 0.209 | 96% | 0.188 | [122] |
| Ni/Na-γ-Al2O3 | IWI | 450 °C, 5%CO2/N2 | 450 °C, H2 | 0.299 | 92% | 0.266 | [122] |
| 0.95% Ru-5% K/Al2O3 | IWI | 350 °C, 2.5%H2O/3%O2/1%CO2/He | 350 °C, 4%H2/He | 0.028 (3rd cycle) | - | 0.028 (3rd cycle) | [123] |
| 0.95% Ru-5.1% Ca/Al2O3 | IWI | 350 °C, 2.5%H2O/3%O2/1%CO2/He | 350 °C, 4%H2/He | 0.116 (3rd cycle) | - | 0.036 (3rd cycle) | [123] |
| 0.84% Ru-16% Ba/Al2O3 | IWI | 350 °C, 2.5%H2O/3%O2/1%CO2/He | 350 °C, 4%H2/He | 0.165 (3rd cycle) | - | 0.080 (3rd cycle) | [123] |
| 0.5% Ru, 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 400 ppm CO2/air | 320 °C, 15% H2/N2 | 0.2 | - | 0.15 | [124] |
| 1%Ni/CeCaO-imp | One-pot citric acid chelation/wet impregnation | 550 °C, 15%CO2/N2 | 550 °C, H2 | 10.6 | 42% | 3.3 | [125] |
| 1%Ni/CeCaCO3-imp | One-pot citric acid chelation/carbonation/wet impregnation | 550 °C, 15%CO2/N2 | 550 °C, H2 | 14.1 | 52% | 6 | [125] |
| 1%Ni/CeO2-CaO-phy | Hydrothermal/one-pot citric acid chelation/physical mixing | 550 °C, 15%CO2/N2 | 550 °C, H2 | 15.3 | 62% | 8 | [125] |
| Ru-BaO/Al2O3 (intimate mixture) | IWI | 350 °C, 1%CO2/He | 350 °C, 4%H2/He | 0.025 | 20% at 291 °C | 0.151 | [126] |
| BaO/Al2O3 + Ru/Al2O3 (mechanical mixture) | IWI | 350 °C, 1%CO2/He | 350 °C, 4%H2/He | 0.055 | 20% at 362 °C | 0.097 | [126] |
| 5Li-Ru/A | IWI | 263 °C, 10%CO2/N2 | 263 °C, 10%H2/N2 | - | 98% | 0.32 | [127] |
| 5Li-Ru/A | IWI | 293 °C, 10%CO2/N2 | 293 °C, 10%H2/N2 | - | 97.4% | 0.34 | [127] |
| 5Li-Ru/A | IWI | 318 °C, 10%CO2/N2 | 318 °C, 10%H2/N2 | - | 95.4% | 0.29 | [127] |
| 30% LaNiO3/CeO2 | Citric acid/impregnation | 400 °C, 1.4%CO2/Ar, | 400 °C, 10%H2/Ar | 0.089 (3-cycle average) | - | 0.08 (3-cycle average) | [128] |
| AMS/CaMgO||Ni1-Co3 | Sol–gel/solvent evaporation | 350 °C, 95%CO2/N2 | 350 °C, 10%H2/N2 | 14.7 (sorbent mass basis) | 76.4% | 5.46 (catalyst mass basis) | [129] |
| AMS/CaMgO||Ni1-Co1 | Sol–gel/solvent evaporation | 350 °C, 95%CO2/N2 | 350 °C, 10%H2/N2 | 14.4 (sorbent mass basis) | 90% | 9.97 (catalyst mass basis) | [129] |
| AMS/CaMgO||Ni3-Co1 | Sol–gel/solvent evaporation | 350 °C, 95%CO2/N2 | 350 °C, 10%H2/N2 | 14.1 (sorbent mass basis) | 75% | 6.66 (catalyst mass basis) | [129] |
| cDFM-2-0.4Ni-10Cs | Co-precipitation/IWI | 350 °C, 15%CO2/N2 | 350 °C, H2 | 0.48 | - | 0.33 | [130] |
| 20% LaNiO3/CeO2 | Citric acid/impregnation | 480 °C, 10%CO2/Ar | 480 °C, 10% H2/Ar | 0.113 | - | 0.075 | [131] |
| 20% La0.5Ca0.5NiO3/CeO2 | Citric acid/impregnation | 480 °C, 10%CO2/Ar | 480 °C, 10% H2/Ar | 0.175 | - | 0.140 | [131] |
| Ni/CaZrO | Sol–gel | 600 °C, 15%CO2/N2 | 600 °C, 66.7%H2/N2 | 9.9–9 (20 cycles) | 86.3–78% (20 cycles) | 6.7 (6th to 20th cycle) | [110] |
| NiO-MgO | Co-precipitation | 320 °C 10%CO2/5%O2/He | 320 °C, 20%H2/He | ~0.3 (10 cycles) | ~96% (10 cycles) | ~0.26 (10 cycles) | [132] |
| Cs-impregnated Ni-MgO-Al2O3 extrudate (EI) | Co-precipitation/impregnation | 350 °C, 15%CO2/N2 | 350 °C, H2 | 0.24 | 75% (250h on-stream) | 0.18 (250h on-stream) | [133] |
IWI: incipient wetness impregnation.
Table 4.
Literature summary of DFMs that catalyze the RWGS reaction.
| DFM | Synthesis Method | Carbonation Conditions | RWGS Conditions | CO2 Sorption Capacity (mmol/g) | CO2 Conversion (%) | Recycle Study and CO Yield Loss | Ref. |
|---|---|---|---|---|---|---|---|
| Ni-CaO-CeO2 (CP) | Co-precipitation with acetate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 9.81 | 75 | 10 cycles, ~10% loss | [105] |
| Ni-CaO-CeO2 (WM) | Wet mixing with acetate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 8.54 | 84 | 10 cycles, stable | [105] |
| Ni-CaO-CeO2 (SG-A) | Citric acid-based sol–gel with acetate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 15.34 | 92 | 10 cycles, 11.6% loss | [105] |
| Ni-CaO-CeO2 (SG-N) | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 13.15 | 96 | 10 cycles, 3.5% loss | [105] |
| Ce-doped Ni/CaO (Ca1Ni0.1Ce0.033) | Citric acid-based sol–gel with nitrate precursors | 650 °C, 15% CO2/N2 | 650 °C, 5% H2/N2 | 14.1 | 51.8 | 20 cycles, stable | [111] |
| Ni-CaO-ZrO2-12 | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, 45% H2/N2 | 16.72 | 63.2 | 12 cycles, 18.6% loss | [146] |
| Ni-CaO-6ZrO2-6CeO2 | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, 45% H2/N2 | 11.88 | 72.1 | 12 cycles, 12% loss | [146] |
| Ni/CeO2-CaO | Physical mixing | 650 °C, 20% CO2/N2 | 650 °C, 5% H2/N2 | 9.58 | 56.1 | 20 cycles, <5% loss | [147] |
| Na/Al2O3 | Wetness impregnation | 500 °C, 5% CO2/N2 | 500 °C, H2 | 0.135 | 76.9 | 50 cycles at 450 °C, stable | [144] |
| Na/Al2O3 | Wetness impregnation | 500 °C, 400 ppm CO2/N2 | 500 °C, H2 | 0.0907 | 77.7 | - | [144] |
| Na-Pt/Al2O3 | Wetness impregnation | 350 °C, 1% CO2/10% O2/N2 | 350 °C, 5% H2/N2 | 0.19 | 89 | 6000 cycles, stable | [145] |
| Ni1Fe9-CaO | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, H2 | 14.78 | 82.5 | 10 cycles, 20.9% loss | [143] |
| Fe5Co5Mg10CaO | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, H2 | 9.2 | 90 | 10 cycles, stable | [142] |
Table 5.
Literature summary of DFMs that catalyze the dry reforming reaction (with conversions of CO2 and CH4).
Table 5.
Literature summary of DFMs that catalyze the dry reforming reaction (with conversions of CO2 and CH4).
| DFM | Synthesis Method | Carbonation Conditions | Dry Reforming Conditions | CO2 Sorption Capacity | CO2 Conversion | CH4 Conversion | Ref. |
|---|---|---|---|---|---|---|---|
| Ni10-(K-Mg)25/(γ-Al2O3)75 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.22 mmol/g DFM | 14% | 16% (ethane) | [91] |
| Ni10-(Na-Mg)50/(γ-Al2O3)50 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.16 mmol/g DFM | 60% | 47% (ethane) | [91] |
| Ni10-(K-Ca)50/(γ-Al2O3)50 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.99 mmol/g DFM | 65% | 100% (ethane) | [91] |
| Ni10-(Na-Ca)50/(γ-Al2O3)50 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.63 mmol/g DFM | 75% | 100% (ethane) | [91] |
| Ni/Ca-Zr | Precipitation/ammoniacal sol/impregnation | 720 °C, 5%CO2/Ar | 720 °C, 8%CH4/Ar | ~3.64 mmol/g CaO (25 cycles) | - | ~32% | [149] |
| NiCe/Ca-Zr | Precipitation/ammoniacal sol/impregnation | 720 °C, 5%CO2/Ar | 720 °C, 8%CH4/Ar | ~5 mmol/g CaO (25 cycles) | - | ~45% | [149] |
| Ni/MgO-Al2O3 (mixed with CaO) | Co-precipitation | 720 °C, 20%CO2/N2 | 720 °C, 2.4%CH4/N2 | 14.1 mmol/g DFM (initial) | 99.92% | 99.94% | [150] |
| Ca-Fe-Mg oxide | Co-precipitation | 600 °C, 100%CO2 | 900 °C, 4.5%CH4/N2 | 0.64 mmol/g DFM | - | ~50% | [151] |
Table 6.
Literature summary of DFMs that catalyze the dry reforming reaction (with yields of H2 and CO).
Table 6.
Literature summary of DFMs that catalyze the dry reforming reaction (with yields of H2 and CO).
| DFM | Synthesis Method | Carbonation Conditions | Dry Reforming Conditions | CO2 Sorption Capacity | H2 Yield | CO Yield | Ref. |
|---|---|---|---|---|---|---|---|
| CaO/Ni4 | Sol–gel | 600 °C, 20%CO2/N2 | 800 °C, 20%CH4/N2 | ~5.7 mmol/g CaO (initial) | 5 mmol/g DFM | 4.3 mmol/g DFM | [152] |
| CaO/Ni9 | Sol–gel | 600 °C, 20%CO2/N2 | 800 °C, 20%CH4/N2 | ~5.1 mmol/g CaO (initial) | 5 mmol/g DFM | 4.5 mmol/g DFM | [152] |
| CaO/Ni19 | Sol–gel | 600 °C, 20%CO2/N2 | 800 °C, 20%CH4/N2 | ~4.5 mmol/g CaO (initial) | 3.5 mmol/g DFM | 3.5 mmol/g DFM | [152] |
| Ni-CaO catal-sorbents | Sol–gel | 700 °C, 10%CO2/10%H2O/N2 | 700 °C, 10%CH4/N2 | 14.8 mmol/g DFM | 131.7 mmol/g DFM | 20.2 mmol/g DFM | [153] |
| Ni/Ca85Ce15 | Hydrothermal (sorbent); impregnation (DFM) | 650 °C, 10%CO2/Ar | 650 °C, 6%CH4/Ar | ~13 mmol/g CaO (initial) | - | - | [154] |
| 15% Ni-1% Ru, 10% Na2O/CeO2–Al2O3 | Sequential impregnation | 650 °C, 10%CO2/N2 | 650 °C, 10%CH4/N2 | 0.16 mmol/g DFM | 25.774 mmol/g DFM | 0.153 mmol/g DFM | [155] |
| 15% Ni-1% Ru, 10% K2O/CeO2–Al2O3 | Sequential impregnation | 650 °C, 10%CO2/N2 | 650 °C, 10%CH4/N2 | 0.18 mmol/g DFM | 22.512 mmol/g DFM | 0.239 mmol/g DFM | [155] |
| 15% Ni-1% Ru, 10% CaO/CeO2–Al2O3 | Sequential impregnation | 650 °C, 10%CO2/N2 | 650 °C, 10%CH4/N2 | 0.26 mmol/g DFM | 32.639 mmol/g DFM | 0.338 mmol/g DFM | [155] |
| CaO-0.05Ni-0.05CeO2 | Co-precipitation | 600 °C, 2%CO2/N2 | 800 °C, 2%CH4/N2 | 10.3 mmol/g CaO (initial); 7.9 mmol/g CaO (10th cycle) | 754.4 mmol/g Ni (max, 2nd cycle) | 454.6 mmol/g Ni (max, 2nd cycle) | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tan, W.J.; Gunawan, P. Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook. Inorganics 2023, 11, 464. https://doi.org/10.3390/inorganics11120464
AMA Style
Tan WJ, Gunawan P. Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook. Inorganics. 2023; 11(12):464. https://doi.org/10.3390/inorganics11120464
Chicago/Turabian StyleTan, Wei Jie, and Poernomo Gunawan. 2023. "Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook" Inorganics 11, no. 12: 464. https://doi.org/10.3390/inorganics11120464
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.