
Use of the Asymmetrical Chelating N-Donor 2-Imino-Pyridine as a Redox [Fe4S4] Cubane Surrogate at a Di-Iron Site Related to [FeFe]-Hydrogenases
 and
and
Abstract
:
1. Introduction
2. Results and Discussion
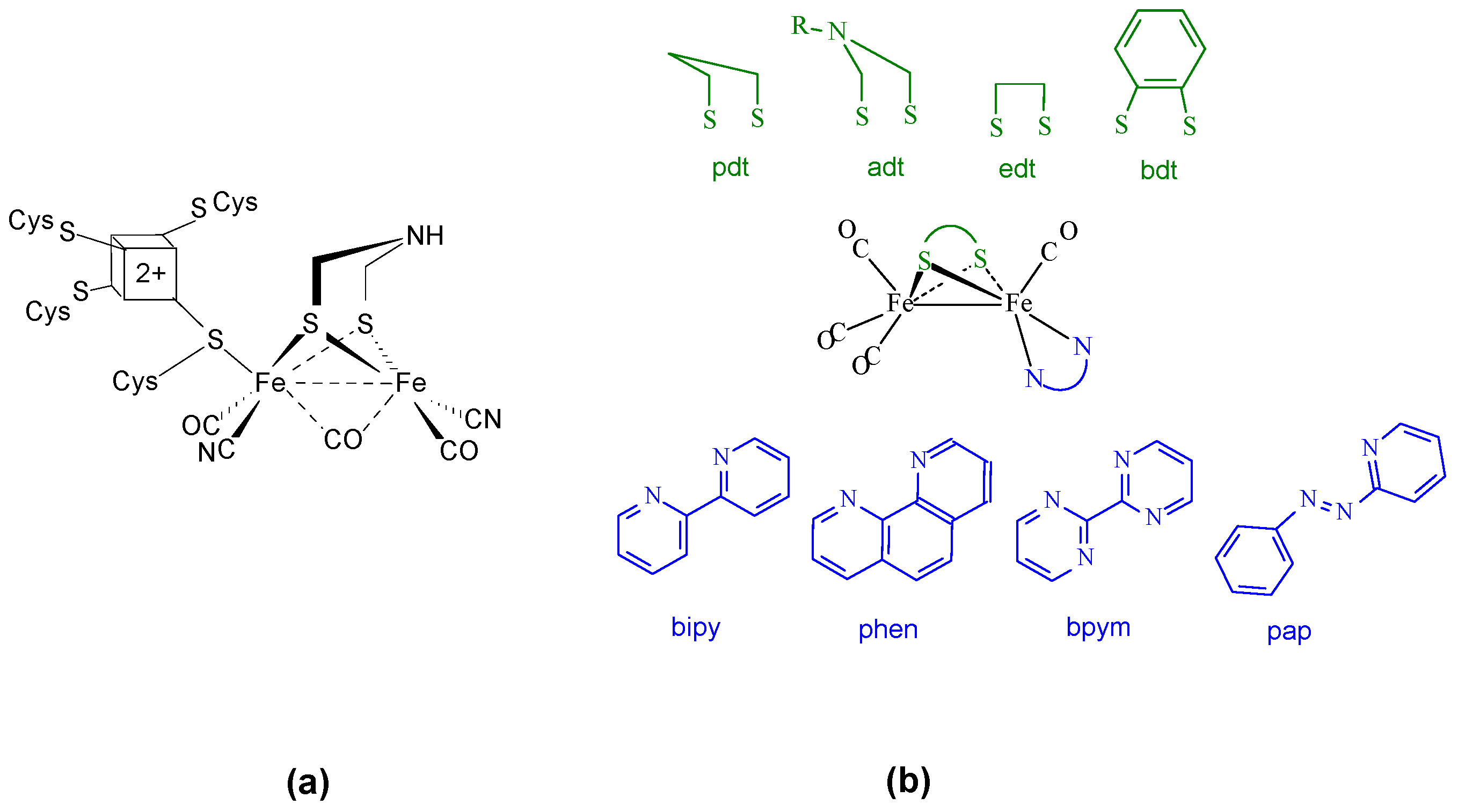
2.1. Synthesis and Characterization
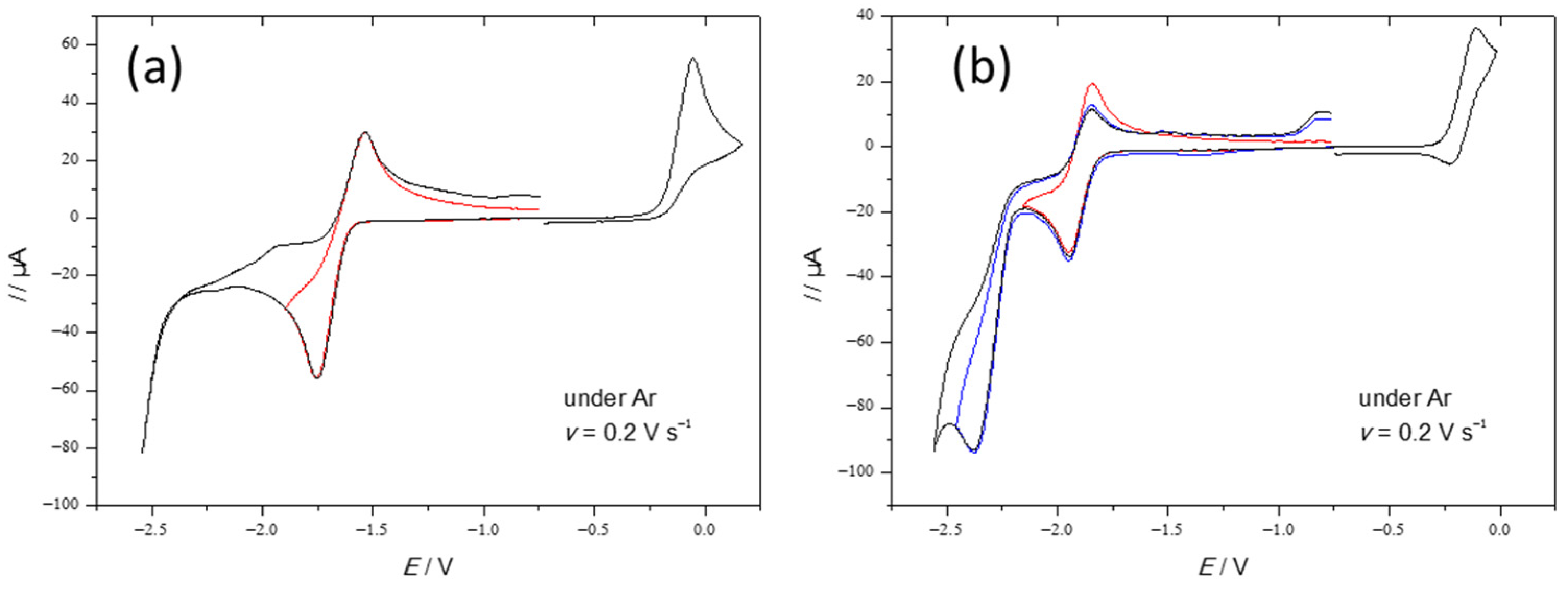
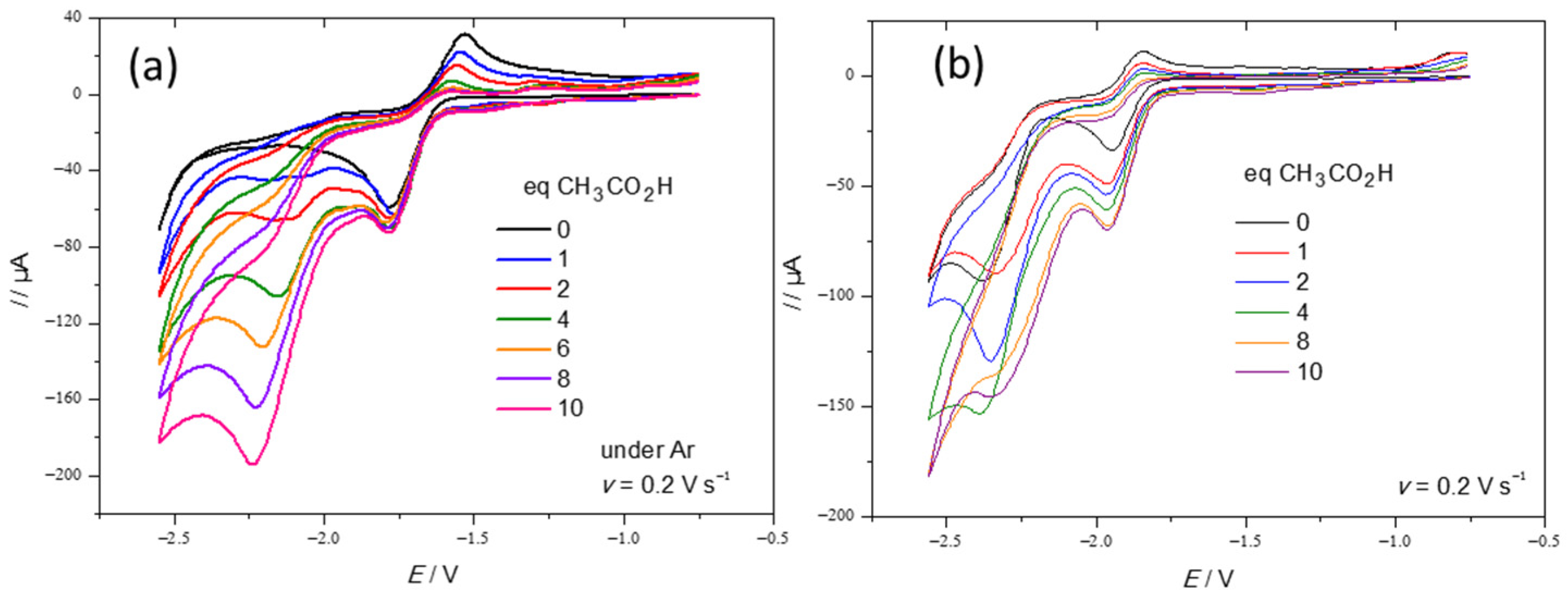
2.2. Electrochemical Behaviour of Complexes 1 and 2 in the Absence and in the Presence of Acid
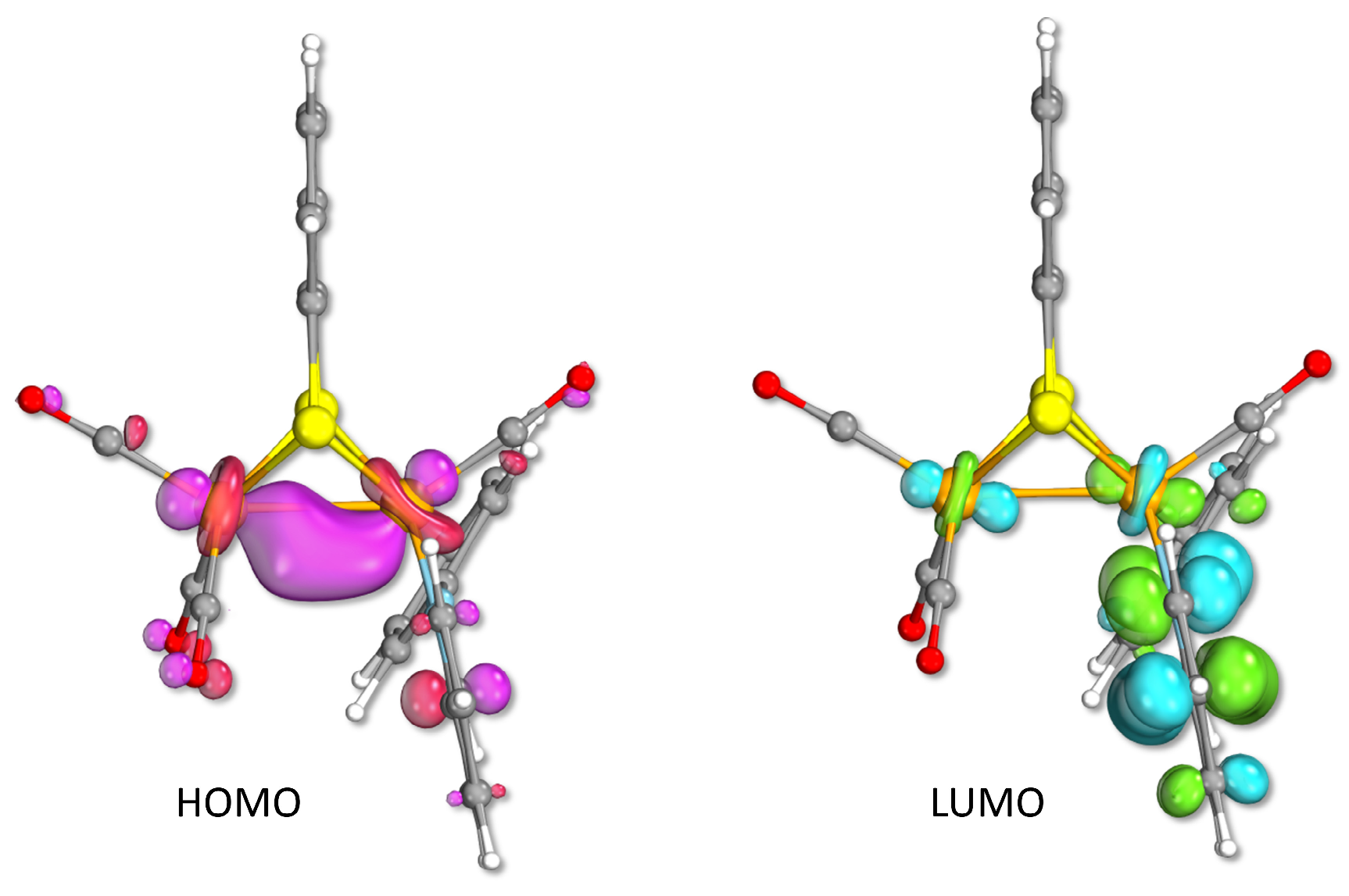
2.3. Density Functional Theory Investigation of Geometries and Electronic Structures of the Anionic Species Arising from the Reduction of Complexes 1 and 2
2.4. Density Functional Theory Investigation of the Mechanism of Proton Reduction for Complex 1
3. Materials and Methods
DFT Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- IR (CH2Cl2, cm−1): ν(CO) 2022 (vs), 1956(s), 1924(s).
- 1H NMR (CD2Cl2, δ, ppm) (ill-resolved spectrum): δ 8.48 (br, 1H), 8.43 (br, 1H), 7.90 (br, 1H), 7.68 (br, 1H), 7.50 (br, 2H), 7.41, 7.37 (br, 3H), 7.13 (br, 2H), 6.92 (br, 1H), 6.54 (br, 2H): pma + bdt
- 13C-{1H} NMR (CD2Cl2, δ, ppm): δ 216.3 (CO), 211.6(3xCO), [157.2, 156.3, 152.5, 151.8, 151.2, 132.5, 128.4, 127.7, 127.6, 126.5, 125.1, 124.1, 121.6] (pma + bdt)
- Elemental Analysis for C22H14Fe2N2O4S2: 48.33% C, 2.56% H, 5.13% N
- Found 47.44% C, 2.50% H, 5.05% N
- IR (CH2Cl2, cm−1): ν(CO) 2014 (vs), 1946(s), 1904(s).
- 1H NMR (CD2Cl2, δ, ppm): δ [8.77 (d, J = 5 Hz, 1H), 8.56 (s, 1H), 7.84 (d, J = 10 Hz, 1H), 7.65 (m, 1H), 7.41 (m, 4H), 7.35 (m, 1H), 7.17 (m, 1H)] (pma); [2.20 (m, 1H), 2.02 (m, 1H), 1.91 (m, 1H), 1.74 (m, 1H), 1.50 (m, obscured, 1H) 1.12 ((m, 1H)] (pdt).
- 13C-{1H} NMR (CD2Cl2, δ, ppm): δ 215. 8 (CO), 212.7 (3xCO), [157.6, 156.6, 152.9, 150.8, 132.5, 128.8, 128.6, 127.6, 124.7, 121.1] (pma); [30.4, 24.7, 24.3] (pdt)
- Elemental Analysis calculated for C19H16Fe2N2O4S2: 44.52% C, 3.12% H, 5.47% N
- Found 45.46% C, 3.14% H, 5.26% N
References
- Peters, J.W.; Lanzilotta, W.N.; Lemon, B.J.; Seefeldt, L.C. X-ray crystal structure of the Fe-only hydrogenase (Cpl) from Clostridium pasteurianum to 1.8 Angstrom resolution. Science 1998, 282, 1853–1858. [Google Scholar] [CrossRef]
- Nicolet, Y.; Piras, C.; Legrand, P.; Hatchikian, C.E.; Fontecilla-Camps, J.C. Desulfovibrio desulfuricans iron hydrogenase: The structure shows unusual coordination to an active site Fe binuclear center. Structure 1999, 7, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Berggren, G.; Adamska, A.; Lambertz, C.; Simmons, T.R.; Esselborn, J.; Atta, M.; Gambarelli, S.; Mouesca, J.M.; Reijerse, E.; Lubitz, W.; et al. Biomimetic assembly and activation of [FeFe]-hydrogenases. Nature 2013, 4, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.-P. [FeFe]-Hydrogenases: Recent developments and future perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef]
- Laun, K.; Baranova, I.; Duan, J.; Kertess, L.; Wittkamp, F.; Apfel, U.-P.; Happe, T.; Senger, M.; Stripp, S.T. Site-selective protonation of the one-electron reduced cofactor in [FeFe]-hydrogenase. Dalton Trans. 2021, 50, 3641–3650. [Google Scholar] [CrossRef] [PubMed]
- Birrell, J.A.; Rodríguez-Maciá, P.; Reijerse, E.J.; Martini, M.A.; Lubitz, W. The catalytic cycle of [FeFe] hydrogenase: A tale of two sites. Coord. Chem. Rev. 2021, 449, 214191. [Google Scholar] [CrossRef]
- Caserta, G.; Zuccarello, L.; Barbosa, C.; Silveira, C.M.; Moe, E.; Katz, S.; Hildebrandt, P.; Zebger, I.; Todorovic, S. Unusual structures and unknown roles of FeS clusters in metalloenzymes seen from a resonance Raman spectroscopic perspective. Coord. Chem. Rev. 2022, 452, 214287. [Google Scholar] [CrossRef]
- Hogarth, G. An unexpected leading role for [Fe2(CO)6(μ-pdt)] in our understanding of [FeFe]-H2ases and the search for clean hydrogen production. Coord. Chem. Rev. 2023, 490, 215174. [Google Scholar] [CrossRef]
- Tard, C.; Liu, X.; Ibrahim, S.K.; Bruschi, M.; De Gioia, L.; Davies, S.C.; Yang, X.; Wang, L.S.; Sawers, G.; Pickett, C.J. Synthesis of the H-cluster framework of iron-only hydrogenase. Nature 2005, 433, 610–613. [Google Scholar] [CrossRef]
- Jørgensen, C.K. Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one electron energy. Coord. Chem. Rev. 1966, 1, 164–178. [Google Scholar] [CrossRef]
- Elleouet, C.; Pétillon, F.Y.; Schollhammer, P. Role of a redox-active ligand close to a dinuclear activating framework. In Modes of Cooperative Effects in Dinuclear Complexes; Kalck, P., Ed.; Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2023; Volume 70, pp. 99–156. [Google Scholar] [CrossRef]
- Becker, R.; Amirjalayer, S.; Li, P.; Woutersen, S.; Reek, J.N.H. An iron-iron hydrogenase mimic with appended electron reservoir for efficient proton reduction in aqueous media. Sci. Adv. 2016, 2, e1501014. [Google Scholar] [CrossRef] [PubMed]
- Camara, J.M.; Rauchfuss, T.B. Combining acid–base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat. Chem. 2011, 4, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Laureanti, J.A.; Groy, T.L.; Jones, A.K. Synthesis and electrocatalytic activity of [FeFe]-hydrogenase model complexes with non-innocent chelating nitrogen-donor ligands. Eur. J. Inorg. Chem. 2017, 2017, 2942–2950. [Google Scholar] [CrossRef]
- Roy, S.; Groy, T.L.; Jones, A.K. Biomimetic model for [FeFe]-hydrogenase: Asymmetrically disubstituted diiron complex with a redox-active 2,2′-bipyridyl ligand. Dalton Trans. 2013, 42, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Rahaman, A.; Holt, K.B.; Nordlander, E.; Richmond, M.G.; Kabir, S.E.; Hogarth, G. Hydrogenase biomimetics with redox-active ligands: Electrocatalytic proton reduction by [Fe2(CO)4(κ2-diamine)(μ-edt)] (diamine = 2,2′-bipy, 1,10-phen). Polyhedron 2016, 116, 127–135. [Google Scholar] [CrossRef]
- Arrigoni, F.; Elleouet, C.; Mele, A.; Pétillon, F.Y.; De Gioia, L.; Schollhammer, P.; Zampella, G. Insights into the two-electron reductive process of [FeFe]H2ase biomimetics: Cyclic voltammetry and DFT investigation on chelate control of redox properties of [Fe2(CO)4(κ2-chelate)(μ-dithiolate)]. Chem. Eur. J. 2020, 26, 17536–17545. [Google Scholar] [CrossRef]
- Marx, M.; Mele, A.; Spannenberg, A.; Steinlechner, C.; Junge, H.; Schollhammer, P.; Beller, M. Addressing the reproducibility of photocatalytic carbon dioxide reduction. ChemCatChem 2020, 12, 1603–1608. [Google Scholar] [CrossRef]
- Orain, P.-Y.; Capon, J.-F.; Kervarec, N.; Gloaguen, F.; Pétillon, F.; Pichon, R.; Schollhammer, P.; Talarmin, J. Use of 1,10-phenanthroline in diiron dithiolate derivatives related to the [Fe–Fe] hydrogenase active site. Dalton Trans. 2007, 3754–3756. [Google Scholar] [CrossRef]
- Ezzaher, S.; Orain, P.-Y.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Roisnel, T.; Schollhammer, P.; Talarmin, J. First insights into the protonation of dissymetrically disubstituted di-iron azadithiolate models of the [FeFe]H2ases active site. Chem. Commun. 2008, 2547–2549. [Google Scholar] [CrossRef]
- Seidel, R.A.; Hall, M.B.; Swenson, M.T.; Nichol, G.S.; Lichtenberger, D.L.; Evans, D.H.; Glass, R.S. Synthesis and characterization of [FeFe]-hydrogenase mimics appended with a 2-phenylazopyridine ligand. J. Sulfur Chem. 2013, 34, 566–579. [Google Scholar] [CrossRef]
- Si, Y.; Charreteur, K.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Non-innocent bma ligand in a dissymetrically disubstituted diiron dithiolate related to the active site of the [FeFe] hydrogenases. J. Inorg. Biochem. 2010, 104, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.; De Gioia, L. A theoretical study on the enhancement of functionally relevant electron transfers in biomimetic models of [FeFe]-hydrogenases. Inorg. Chem. 2011, 50, 6987–6995. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Hollingworth, N.; Warren, M.; Hvorat, D.A.; Richmond, M.G.; Hogarth, G. Hydrogenase biomimics containing redox-active ligands: Fe2(CO)4(μ-edt)(κ2-bpcd) with electron-acceptor 4,5-bis(diphenylphosphino)-4-cyclopenten-1,3-dione (bpcd) as a potential [Fe4–S4]H surrogate. Dalton Trans. 2019, 48, 6051–6060. [Google Scholar] [CrossRef]
- Morvan, D.; Capon, J.-F.; Gloaguen, F.; Le Goff, A.; Marchivie, M.; Michaud, F.; Schollhammer, F.; Talarmin, J.; Yaouanc, J.-J.; Pichon, R.; et al. N-heterocyclic carbene ligands in nonsymmetric diiron models of hydrogenase active sites. Organometallics 2007, 26, 2042–2052. [Google Scholar] [CrossRef]
- Chouffai, D.; Zampella, G.; Capon, J.-F.; De Gioia, L.; Le Goff, A.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Electrochemical and theoretical studies of the impact of the chelating ligand on the reactivity of [Fe2(CO)4(κ2-LL)(μ-pdt)]+ complexes with different substrates (LL = IMe-CH2-IMe, dppe; IMe = 1-methylimidazol-2-ylidene). Organometallics 2012, 31, 1082–1091. [Google Scholar] [CrossRef]
- Arrigoni, F.; De Gioia, L.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Zampella, G. Normal vs. inverted ordering of reduction potentials in [FeFe]-hydrogenases biomimetics: Effect of the dithiolate bulk. Chem. Eur. J. 2023, 29, e202300569. [Google Scholar] [CrossRef]
- Spall, S.J.P.; Keane, T.; Tory, J.; Cocker, D.C.; Adams, H.; Fowler, H.; Meijer, A.J.H.M.; Hartl, F.; Weinstein, J.A. Manganese tricarbonyl complexes with asymmetric 2-iminopyridine ligands: Toward decoupling steric and electronic factors in electrocatalytic CO2 reduction. Inorg. Chem. 2016, 55, 12568–12582. [Google Scholar] [CrossRef]
- Lu, C.C.; Weyhermüller, T.; Bil, E.; Wieghardt, K. Accessing the different redox states of α-iminopyridines within cobalt complexes. Inorg. Chem. 2009, 48, 6055–6064. [Google Scholar] [CrossRef]
- Shejwalkar, P.; Rath, N.P.; Bauer, E.B. New iron(II) a-iminopyridine complexes and their catalytic activity in the oxidation of activated methylene groups and secondary alcohols to ketones. Dalton Trans. 2011, 40, 7617–7631. [Google Scholar] [CrossRef]
- Sieh, D.; Lacy, D.C.; Peters, J.C.; Kubiak, C.P. Reduction of CO2 by pyridine monoimine molybdenum carbonyl complexes: Cooperative metal–ligand binding of CO2. Chem. Eur. J. 2015, 21, 8497–8503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Orain, P.-Y.; Capon, J.-F.; Gloaguen, F.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; Zampella, G.; De Gioia, L.; Roisnel, T. Investigation on the protonation of a trisubstituted [Fe2(CO)3(PPh3)(κ2-phen)(μ-pdt)] complex: Rotated versus unrotated Intermediate pathways. Inorg. Chem. 2010, 49, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Electrochemical proton reduction by thiolate-bridged hexacarbonyldiiron clusters. J. Electroanal. Chem. 2004, 566, 241–247. [Google Scholar] [CrossRef]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Activation of proton by the two-electron reduction of a di-iron organometallic complex. J. Electroanal. Chem. 2006, 595, 47–52. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Vannucci, A.K.; Chen, J.; Tori Lockett, L.; Okumura, N.; Petro, B.J.; Zakai, U.I.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Hydrogen generation from weak acids: Electrochemical and computational studies of a diiron hydrogenase mimic. J. Am. Chem. Soc. 2007, 129, 12521–12530. [Google Scholar] [CrossRef] [PubMed]
- Windhager, J.; Rudolph, M.; Braütigam, S.; Görls, H.; Weigand, W. Reactions of 1,2,4-trithiolane, 1,2,5-trithiepane, 1,2,5-trithiocane and 1,2,6-trithionane with nonacarbonyldiiron: Structural determination and electrochemical investigation. Eur. J. Inorg. Chem. 2007, 2007, 2748–2760. [Google Scholar] [CrossRef]
- Almazahreh, L.R.; Arrigoni, F.; Abul-Futouh, H.; El-Khateeb, M.; Görls, H.; Elleouet, C.; Schollhammer, P.; Bertini, L.; De Gioia, L.; Rudolph, M.; et al. Proton shuttle mediated by (SCH2)2P=O moiety in [FeFe]-hydrogenase mimics: Electrochemical and DFT studies. ACS Catal. 2021, 11, 7080–7098. [Google Scholar] [CrossRef]
- Arrigoni, F.; Rizza, F.; Vertemara, J.; Breglia, R.; Greco, C.; Bertini, L.; Zampella, G.; De Gioia, L. Rational Design of Fe2(μ-PR2)2(L)6 coordination compounds featuring tailored potential inversion. ChemPhysChem 2020, 21, 2279–2292. [Google Scholar] [CrossRef]
- Wright, R.J.; Zhang, W.; Yang, X.; Fasuloa, M.; Tilley, T.D. Isolation, observation, and computational modeling of proposed intermediates in catalytic proton reductions with the hydrogenase mimic Fe2(CO)6S2C6H4. Dalton Trans. 2012, 41, 73–82. [Google Scholar] [CrossRef]
- Mirmohades, M.; Pullen, S.; Stein, M.; Maji, S.; Ott, S.; Hammarström, L.; Lomoth, R. Direct observation of key catalytic intermediates in a photoinduced proton reduction cycle with a diiron carbonyl complex. J. Am. Chem. Soc. 2014, 136, 17366–17369. [Google Scholar] [CrossRef] [PubMed]
- Etinski, M.; Stanković, I.M.; Rakesh, C.; Puthenkalathil, R.C.; Ensing, B. A DFT study of structure and electrochemical properties of diiron-hydrogenase models with benzenedithiolato and benzenediselenato ligands. New J. Chem. 2020, 44, 932–941. [Google Scholar] [CrossRef]
- Gao, S.; Liang, Q.; Duan, Q.; Jiang, D.; Zhao, J. Electrochemical proton reductions in varying acidic media by a simple synthetic hydrogenase mimic. Int. J. Hydrogen Energy 2018, 43, 7245–7256. [Google Scholar] [CrossRef]
- Winter, A.; Zsolnai, L.; Huttner, G. Dinuclear and trinuclear carbonyliron complexes containing 1,2- and 1,3-dithiolato bridging ligands. Z. Naturforsch 1982, 37b, 1430–1436. [Google Scholar] [CrossRef]
- Lyon, E.J.; Georgakaki, I.P.; Reibenspies, J.H.; Darensbourg, M.Y. Coordination sphere flexibility of active-site models for Fe-only hydrogenase: Studies in intra- and intermolecular diatomic ligand exchange. J. Am. Chem. Soc. 2001, 123, 3268–3278. [Google Scholar] [CrossRef]
- Cabeza, J.A.; Martinez-Garcia, M.A.; Riera, V.; Ardura, D.; Garcia-Granda, S. Binuclear iron(I), ruthenium(I), and osmium(I) hexacarbonyl complexes containing a bridging benzene-1,2-dithiolate ligand. Synthesis, X-ray structures, protonation reactions, and EHMO calculations. Organometallics 1998, 17, 1471–1477. [Google Scholar] [CrossRef]
- Arrigoni, F.; Mohamed Bouh, S.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; De Gioia, L.; Zampella, G. Electrochemical and theoretical investigations of the oxidatively induced reactivity of the complex [Fe2(CO)4(κ2-dmpe)(µ-adtBn)] related to the active site of [FeFe] hydrogenases. Chem. Eur. J. 2018, 24, 15036–15051. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A At. Mol. Opt. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Chatelain, L.; Breton, J.-B.; Arrigoni, F.; Shollhammer, P.; Zampella, G. Geometrical influence on the non-biomimetic heterolytic splitting of H2 by bio-inspired [FeFe]-hydrogenase complexes: A rare example of inverted frustrated Lewis pair based reactivity. Chem. Sci. 2022, 13, 4863–4873. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, F.; Bertini, L.; Breglia, R.; Greco, C.; De Gioia, L.; Zampella, G. Catalytic H2 evolution/oxidation in [FeFe]-hydrogenase biomimetics: Account from DFT on the interplay of related issues and proposed solutions. New J. Chem. 2020, 44, 17596–17615. [Google Scholar] [CrossRef]
- Chambers, J.M.; Rauchfuss, T.B.; Arrigoni, F.; Zampella, G. Effect of pyramidalization of the M2(SR)2 center: The case of (C5H5)2Ni2(SR)2. Organometallics 2016, 35, 836–846. [Google Scholar] [CrossRef]
- Arrigoni, F.; Mohamed Bouh, S.; De Gioia, L.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; Zampella, G. Influence of the dithiolate bridge on the oxidative processes of diiron models related to the active site of [FeFe] hydrogenases. Chem.-Eur. J. 2017, 23, 4364–4372. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154122. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A. Calculation of UV/Vis spectra in solution. J. Phys. Chem. 1996, 100, 3349–3353. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |
|---|---|---|
| Fe(1)-Fe(2) | 2.505 (2) | 2.5355 (5) |
| Fe(1)-S(1) | 2.241 (4) | 2.2183 (8) |
| Fe(2)-S(1) | 2.270 (4) | 2.2669 (8) |
| Fe(1)-S(2) | 2.254 (3) | 2.2179 (7) |
| Fe(1)-S(2) | 2.281 (4) | 2.2700 (8) |
| Fe(1)-N (Ph)) | 1.962 (11) | 1.962 (2) |
| Fe(1)-N (pyr) | 1.942 (11) | 1.974 (2) |
| C(11)-C(12) | 1.44 (2) | 1.426 (4) |
| C(5)-C(6) | 1.381 (4) | |
| C-N (C=N imine) | 1.277 (18) | 1.304 (3) |
| N-C (ispo-Ph) | 1.436 (16) | 1.432 (3) |
| C-N (pyr) | 1.365 (18) | 1.363 (3) |
| C16-N (pyr) | 1.388 (18) | 1.356 (3) |
| C(15)-C(16) | 1.378 (4) | 1.393 (19) |
| C(1)-O(1) | 1.139 (16) | 1.159 (3) |
| C(1)-Fe(1) | 1.785 (14) | 1.757 (3) |
| C-O (apical-Fe(2)) | 1.145 (17) | 1.144 (3) |
| C-Fe(2) (apical) | 1.813 (15) | 1.809 (3) |
| C(3)-O(3) (basal-Fe(2)) | 1.223 (18) | 1.154 (3) |
| Fe(2)-C(3) | 1.717 (16) | 1.779 (3) |
| C-O (basal-Fe(2)) | 1.167 (18) | 1.149 (3) |
| Fe(2)C (basal) | 1.748 (16) | 1.778 (3) |
| Fe(1)-S(1)-Fe(2) | 67.46 (11) | 64.84 (2) |
| Fe(1)-S(2)-Fe(2) | 67.07 (11) | 68.79 (2) |
| Fe(1)-C(1)-O(1) | 176.8 (14) | 172.9 (3) |
| Fe(2)-C-O (apical) | 177.0 (14) | 176.5 (2) |
| Fe(2)-C(3)-O(3) | 177.4 (15) | 176.9 (3) |
| Fe(2)-C-O (basal) | 178.6 (15) | 178.3 (3) |
| N(1)-Fe(1)-N(2) | 81.4 (5) | 80.47 (9) |
| Compound | Epc,red/V | (“E1/2red”/V) | Epa,ox/V |
|---|---|---|---|
| [Fe2(CO)4(κ2-pma)(µ-bdt)] (1) | −1.76 | (−1.66) | −0.04 |
| [Fe2(CO)4(κ2-pma)(µ-pdt)] (2) | −1.95 | (−1.89) | −0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mele, A.; Arrigoni, F.; De Gioia, L.; Elleouet, C.; Pétillon, F.Y.; Schollhammer, P.; Zampella, G. Use of the Asymmetrical Chelating N-Donor 2-Imino-Pyridine as a Redox [Fe4S4] Cubane Surrogate at a Di-Iron Site Related to [FeFe]-Hydrogenases. Inorganics 2023, 11, 463. https://doi.org/10.3390/inorganics11120463
Mele A, Arrigoni F, De Gioia L, Elleouet C, Pétillon FY, Schollhammer P, Zampella G. Use of the Asymmetrical Chelating N-Donor 2-Imino-Pyridine as a Redox [Fe4S4] Cubane Surrogate at a Di-Iron Site Related to [FeFe]-Hydrogenases. Inorganics. 2023; 11(12):463. https://doi.org/10.3390/inorganics11120463
Chicago/Turabian StyleMele, Andrea, Federica Arrigoni, Luca De Gioia, Catherine Elleouet, François Y. Pétillon, Philippe Schollhammer, and Giuseppe Zampella. 2023. "Use of the Asymmetrical Chelating N-Donor 2-Imino-Pyridine as a Redox [Fe4S4] Cubane Surrogate at a Di-Iron Site Related to [FeFe]-Hydrogenases" Inorganics 11, no. 12: 463. https://doi.org/10.3390/inorganics11120463