

Fluorescent Vitamin B12–Platinum(II) Derivatives as Potential Metallotheranostic Agents for the Treatment and Imaging of Tumors
 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Syntheses and Characterizations
2.1.1. Fluorophore-Functionalized Cyanocobalamin
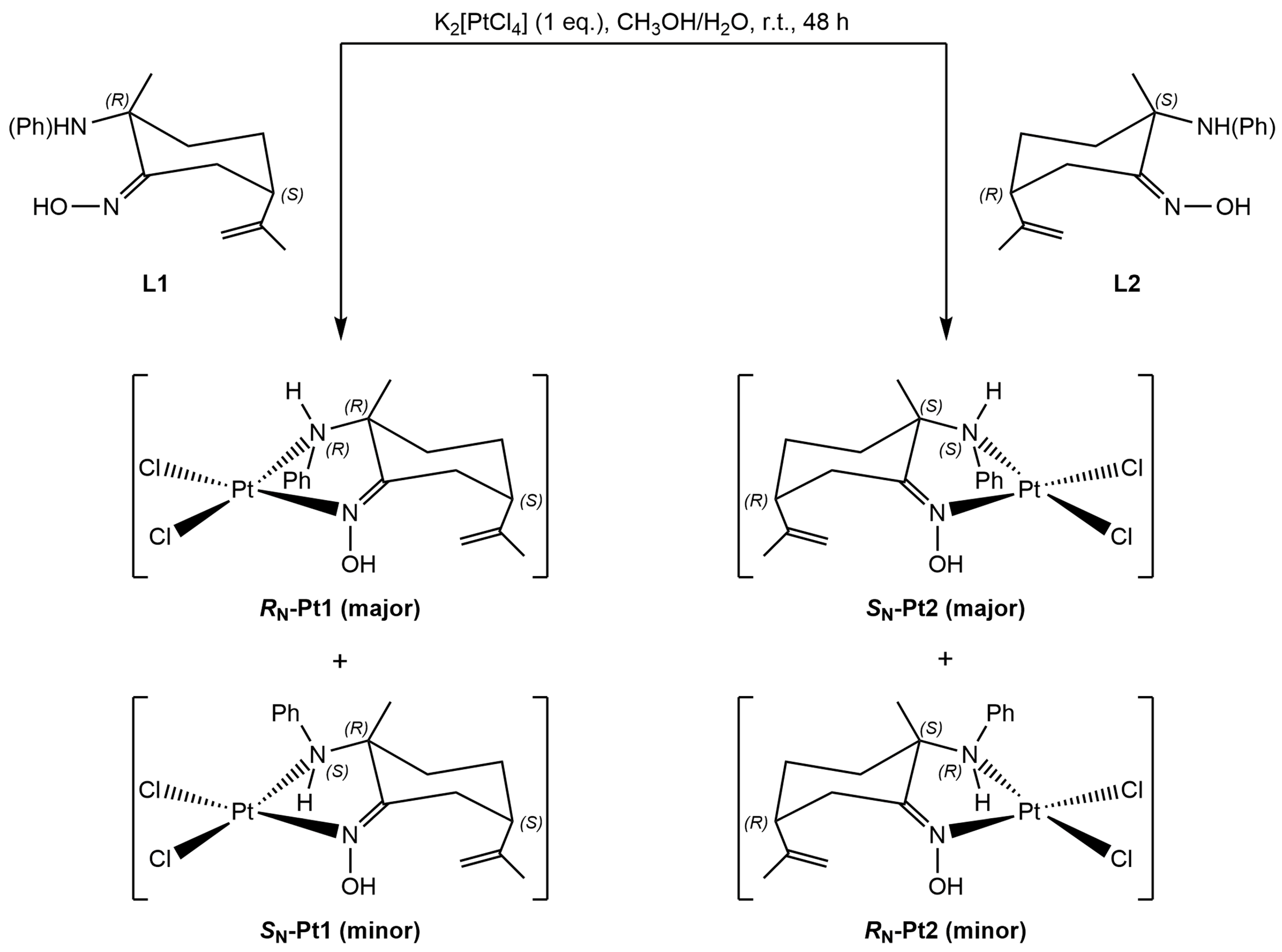
2.1.2. Amino-Oxime Platinum(II) Complexes
2.1.3. Vitamin B12–Platinum(II) Derivatives
2.1.4. Fluorescent Vitamin B12–Platinum(II) Derivatives
2.2. Fluorescence Measurements
2.2.1. Fluorescence Properties of Vitamin B12
2.2.2. Fluorescence Properties of Rhodamine 6G and its Vitamin B12 Conjugate
2.2.3. Fluorescence Properties of the Vitamin B12–Platinum(II) Derivatives
2.3. Lipophilicity Studies (log D7.4)
2.4. In Vitro Biological Studies
2.4.1. Confocal Microscopy
2.4.2. Antiproliferative Activity
2.4.3. Cell Cycle Assay
2.4.4. DNA Interaction
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2022, 10, 1367–1401. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Pettenuzzo, A.; Pigot, R.; Ronconi, L. Vitamin B12-metal conjugates for targeted chemotherapy and diagnosis: Current status and future prospects. Eur. J. Inorg. Chem. 2017, 12, 1625–1638. [Google Scholar] [CrossRef]
- Russell-Jones, G.J.; Arthur, L.; Walker, H. Vitamin B12-mediated transport of nanoparticles across Caco-2 cells. Int. J. Pharm. 1999, 179, 247–255. [Google Scholar] [CrossRef]
- Obeid, R.; Heil, S.G.; Verhoeven, M.M.A.; van den Heuvel, E.G.H.M.; de Groot, L.C.P.G.M.; Eussen, S.J.P.M. Vitamin B12 intake from animal foods, biomarkers, and health aspects. Front. Nutr. 2019, 6, 93. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Rasmussen, M.R.; Andersen, C.B.; Nexø, E.; Moestrup, S.K. Vitamin B12 transport from food to the body’s cells—A sophisticated, multistep pathway. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 345–354. [Google Scholar] [CrossRef]
- Gendron, L.N.; Zites, D.C.; LaRochelle, E.P.M.; Gunn, J.R.; Pogue, B.W.; Shell, T.A.; Shell, J.R. Tumor targeting vitamin B12 derivatives for X-ray induced treatment of pancreatic adenocarcinoma. Photodiagn. Photodyn. Ther. 2020, 30, 101637. [Google Scholar] [CrossRef] [PubMed]
- Mundwiler, S.; Spingler, B.; Kurz, P.; Kunze, S.; Alberto, R. Cyanide-bridged vitamin B12-cisplatin conjugates. Chem. Eur. J. 2005, 11, 4089–4095. [Google Scholar] [CrossRef]
- Ruiz-Sánchez, P.; Mundwiler, S.; Spingler, B.; Buan, N.R.; Escalante-Semerena, J.C.; Alberto, R. Syntheses and characterization of vitamin B12-Pt(II) conjugates and their adenosylation in an enzymatic assay. J. Biol. Inorg. Chem. 2008, 13, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sánchez, P.; König, C.; Ferrari, S.; Alberto, R. Vitamin B₁₂ as a carrier for targeted platinum delivery: In vitro cytotoxicity and mechanistic studies. J. Biol. Inorg. Chem. 2011, 16, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.Q.; Furger, E.; Alberto, R. Two-step activation prodrugs: Transplatin mediated binding of chemotherapeutic agents to vitamin B12. Org. Biomol. Chem. 2013, 11, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.T.Q.; Stürup, S.; Lambert, I.H.; Gammelgaard, B.; Furger, E.; Alberto, R. Cellular uptake of metallated cobalamins. Metallomics 2016, 8, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Kunze, S.; Zobi, F.; Kurz, P.; Spingler, B.; Alberto, R. Vitamin B12 as a ligand for technetium and rhenium complexes. Angew. Chem. Int. Ed. 2004, 43, 5025–5029. [Google Scholar] [CrossRef] [PubMed]
- Rossier, J.; Hauser, D.; Kottelat, E.; Rothen-Rutishauserb, B.; Zobi, F. Organometallic cobalamin anticancer derivatives for targeted prodrug delivery via transcobalamin-mediated uptake. Dalton Trans. 2017, 46, 2159–2164. [Google Scholar] [CrossRef]
- Atoum, M.F.; Alzoughool, F.E.; Al-Mazaydeh, Z.A.; Rammaha, M.S.; Tahtamouni, L.H. Vitamin B12 enhances the antitumor activity of 1,25-dihydroxyvitamin D3 via activation of caspases and targeting actin cytoskeleton. Tumor Biol. 2022, 44, 17–35. [Google Scholar] [CrossRef]
- McGreevy, J.M.; Cannon, M.J.; Grissom, C.B. Minimally invasive lymphatic mapping using fluorescently labeled vitamin B12. J. Surg. Res. 2003, 111, 38–44. [Google Scholar] [CrossRef]
- Fedosov, S.N.; Grissom, C.B.; Fedosova, N.U.; Moestrup, S.K.; Nexø, E.; Petersen, T.E. Application of a fluorescent cobalamin analogue for analysis of the binding kinetics. A study employing recombinant human transcobalamin and intrinsic factor. FEBS J. 2006, 273, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Vortherms, A.R.; Kahkoska, A.R.; Rabideau, A.E.; Zubieta, J.; Andersen, L.L.; Madsen, M.; Doyle, R.P. A water soluble vitamin B12-Re(I) fluorescent conjugate for cell uptake screens: Use in the confirmation of cubilin in the lung cancer line A549. Chem. Commun. 2011, 47, 9792–9794. [Google Scholar] [CrossRef]
- Wuerges, J.; Garau, G.; Geremia, S.; Fedosov, S.N.; Petersen, T.E.; Randaccio, L. Structural basis for mammalian vitamin B12 transport by transcobalamin. Proc. Natl. Acad. Sci. USA 2006, 103, 4386–4391. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.K.; Vortherms, A.R.; Fairchild, T.J.; Doyle, R.P. Vitamin B12 as a carrier for the oral delivery of insulin. ChemMedChem 2007, 2, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, L.; Kotra, L.P. Cyanocobalamin (vitamin B12) conjugates with enhanced solubility. Bioorg. Med. Chem. 2007, 15, 1780–1787. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Chen, X.; Smyrniotis, C.; Xenakis, A.; Hu, T.; Bertozzi, C.R.; Wu, P. Metabolic labeling of sialic acids in living animals with alkynyl sugars. Angew. Chem. Int. Ed. 2009, 48, 4030–4033. [Google Scholar] [CrossRef]
- Clardy, S.M.; Allis, D.G.; Fairchild, T.J.; Doyle, R.P. Vitamin B12 in drug delivery: Breaking through the barriers to a B12 bioconjugate pharmaceutical. Expert Opin. Drug Deliv. 2011, 8, 127–140. [Google Scholar] [CrossRef]
- Lee, M.; Grissom, C.B. Design, synthesis, and characterization of fluorescent cobalamin analogues with high quantum efficiencies. Org. Lett. 2009, 11, 2499–2502. [Google Scholar] [CrossRef]
- Mchedlov-Petrosyan, N.O.; Fedorov, L.A.; Sokolovskii, S.A.; Surov, Y.N.; Salinas Maiorga, R. Structural conversions of rhodamines in solution. Russ. Chem. Bull. 1992, 41, 403–409. [Google Scholar] [CrossRef]
- Bhanja, A.K.; Mishra, S.; Naskar, K.; Maity, S.; Das Saha, K.; Sinha, C. Specific recognition of Cr3+ under physiological conditions by allyl substituted appendage rhodamine and its cell-imaging studies. Dalton Trans. 2017, 46, 16516–16524. [Google Scholar] [CrossRef]
- Lu, D.; Yang, T.; Tang, N.; Li, C.; Song, Y.; Wang, L.; Wong, W.Y.; Yin, S.F.; Xing, Y.; Kambe, N.; et al. A pH-dependent rhodamine fluorophore with antiproliferative activity of bladder cancer in vitro/vivo and apoptosis mechanism. Eur. J. Med. Chem. 2022, 236, 114293. [Google Scholar] [CrossRef] [PubMed]
- Majoube, M.; Henry, M. Fourier transform Raman and infrared and surface-enhanced Raman spectra for rhodamine 6G. Spectrochim. Acta A 1991, 47, 1459–1466. [Google Scholar] [CrossRef]
- Alkadi, H.; Jbeily, R. Role of chirality in drugs: An overview. Infect. Disord. Drug Targets 2018, 18, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Pannunzio, A.; Coluccia, M.; Natile, G. Effect of chirality in platinum drugs. Coord. Chem. Rev. 2015, 284, 286–297. [Google Scholar] [CrossRef]
- Pacuła-Miszewska, A.J.; Obieziurska-Fabisiak, M.; Ścianowski, J. Monoterpenes as chiral building blocks. In Chiral Building Blocks in Asymmetric Synthesis: Synthesis and Applications; Wojaczyńska, E., Wojaczyński, J., Eds.; Wiley-VCH: Weinheim, Germany, 2022; pp. 235–266. [Google Scholar]
- de la Cueva-Alique, I.; Sierra, S.; Muñoz-Moreno, L.; Pérez-Redondo, A.; Bajo, A.M.; Marzo, I.; Gude, L.; Cuenca, T.; Royo, E. Biological evaluation of water soluble arene Ru(II) enantiomers with amino-oxime ligands. J. Inorg. Biochem. 2018, 183, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Benabdelouahab, Y.; Muñoz-Moreno, L.; Frik, M.; de la Cueva-Alique, I.; El Amrani, M.A.; Contel, M.; Bajo, A.M.; Cuenca, T.; Royo, E. Hydrogen bonding and anticancer properties of water-soluble chiral p-cymene Ru(II) compounds with amino-oxime ligands. Eur. J. Inorg. Chem. 2015, 2015, 2295–2307. [Google Scholar] [CrossRef]
- de la Cueva-Alique, I.; Muñoz-Moreno, L.; Benabdelouahab, Y.; Elie, B.T.; El Amrani, M.A.; Mosquera, M.E.; Contel, M.; Bajo, A.M.; Cuenca, T.; Royo, E. Novel enantiopure cyclopentadienyl Ti(IV) oximato compounds as potential anticancer agents. J. Inorg. Biochem. 2016, 156, 22–34. [Google Scholar] [CrossRef]
- de la Cueva-Alique, I.; Sierra, S.; Pérez-Redondo, A.; Marzo, I.; Gude, L.; Cuenca, T.; Royo, E. Study of the anticancer properties of optically active titanocene oximato compounds. J. Organomet. Chem 2019, 881, 150–158. [Google Scholar] [CrossRef]
- de la Cueva-Alique, I.; de la Torre-Rubio, E.; Muñoz-Moreno, L.; Calvo-Jareño, A.; Perez-Redondo, A.; Gude, L.; Cuenca, T.; Royo, E. Stereoselective synthesis of oxime containing Pd(II) compounds: Highly effective, selective and stereoregulated cytotoxicity against carcinogenic PC-3 cells. Dalton Trans. 2022, 51, 12812–14828. [Google Scholar] [CrossRef]
- de la Cueva-Alique, I.; Muñoz-Moreno, L.; de la Torre-Rubio, E.; Bajo, A.M.; Gude, L.; Cuenca, T.; Royo, E. Water soluble, optically active monofunctional Pd(II) and Pt(II) compounds: Promising adhesive and antimigratory effects on human prostate PC-3 cancer cells. Dalton Trans. 2019, 48, 14279–14293. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Oxime and oximate metal complexes: Unconventional synthesis and reactivity. Coord. Chem. Rev. 1999, 181, 147–175. [Google Scholar] [CrossRef]
- Motaleb, M.A.; Selim, A.A. Dioximes: Synthesis and biomedical applications. Bioorg. Chem. 2019, 82, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Plotnikov, M.B.; Khlebnikov, A.I.; Plotnikova, T.M.; Quinn, M.T. Oximes: Novel therapeutics with anticancer and anti-inflammatory potential. Biomolecules 2021, 11, 777. [Google Scholar] [CrossRef] [PubMed]
- Brecknell, D.J.; Carman, R.M.; Singaram, B.; Verghese, J. Silvestrene nitrosochlorides and derived amino-oximes. Aust. J. Chem. 1977, 30, 195–203. [Google Scholar] [CrossRef]
- Faller, J.W.; Patel, B.P.; Albrizzio, M.A.; Curtis, M. Diastereoselectivity in chiral ruthenium complexes of bidentate bisphosphine monoxide ligands: controlling epimerization in aldehyde complexes and 16-electron intermediates. Organometallics 1999, 18, 3096–3104. [Google Scholar] [CrossRef]
- Faller, J.W.; Sarantopoulos, N. Novel binding modes and hemilability in atropisomeric phosphino−amino palladium complexes. Organometallics 2004, 23, 2008–2014. [Google Scholar] [CrossRef]
- Bigotto, A.; Costa, G.; Galasso, V.; De Alti, G. Infra-red spectra and normal vibrations of bis-dimethylglyoximates of transition metals. Spectrochim. Acta A 1970, 26, 1939–1949. [Google Scholar] [CrossRef]
- Thorton, D.A. Metal complexes of aniline: Infrared and raman spectra. J. Coord. Chem. 1991, 24, 261–289. [Google Scholar] [CrossRef]
- Nakamoto, K. Halogeno Complexes. In Infrared and Raman Spectra of Inorganic and Coordination Compounds—Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 193–196. [Google Scholar]
- Bandyopadhyay, N.; Zhu, M.; Lu, L.; Mitra, D.; Das, M.; Das, P.; Samanta, A.; Naskar, J.P. Synthesis, structure, spectral characterization, electrochemistry and evaluation of antibacterial potentiality of a novel oxime-based palladium(II) compound. Eur. J. Med. Chem. 2015, 89, 59–66. [Google Scholar] [CrossRef]
- Salih, B.M.M.; Satyanarayana, S. Vitamin B12 models: Synthesis and characterization of cyano bridged dicobaloximes and antimicrobial activity. Afr. J. Pure Appl. Chem. 2009, 3, 170–176. [Google Scholar]
- Lippolis, V.; Blake, A.J.; Cooke, P.A.; Isaia, F.; Li, W.-S.; Schröder, M. Synthesis and full characterisation of the first discrete binuclear complex featuring a two-electron (σ) μ2-κC:κC bridging cyanide. Chem. Eur. J. 1999, 5, 1987–1991. [Google Scholar] [CrossRef]
- Laidlaw, W.M.; Denning, R.G. Synthesis and characterisation of cyanide-bridged heterobinuclear mixed-valence compounds based on cyclopentadienylorganophosphine-ruthenium(II) and -osmium(II) cyano and pentaammine-ruthenium(III) and -osmium(III) moieties. J. Chem. Soc. Dalton Trans. 1994, 1987–1994. [Google Scholar] [CrossRef]
- Kettle, S.F.A.; Aschero, G.L.; Diana, E.; Rossetti, R.; Stanghellini, P.L. The vibrational spectra of the cyanide ligand revisited: terminal cyanides. Inorg. Chem. 2006, 45, 4928–4937. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K. Cyano and Nitrile Complexes. In Infrared and Raman Spectra of Inorganic and Coordination Compounds—Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 116–117. [Google Scholar]
- Karadag, A.; Pasaoglu, H.; Kastas, G.; Büyükgüngör, O. Synthesis, IR spectrum, thermal behaviour and crystal structure of a novel one-dimensional cyano-bridged zinc(II)/nickel(II) complex. Z. Kristallogr. Cryst. Mater. 2005, 220, 74–78. [Google Scholar] [CrossRef]
- Hanusa, T.P. Cyanide complexes of the transition metals. In Encyclopedia of Inorganic Chemistry, 2nd ed.; King, R.B., Crabtree, R.H., Lukehart, C.M., Atwood, D.A., Scott, R.A., Eds.; John Wiley & Sons: Chichester, UK, 2006. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds—Part A: Theory and Applications in Inorganic Chemistry, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 180–184. [Google Scholar]
- v. Ahsen, B.; Bley, B.; Proemmel, S.; Wartchow, R.; Willner, H.; Aubke, F. Synthesen und schwingungsspektren der homoleptischen acetonitrilkomplex-kationen [Au(NCCH3)2]+, [Pd(NCCH3)4]2+, [Pt(NCCH3)4]2+ und des adduktes CH3CN⋅SbF5. Kristallstrukturen der salze [M(NCCH3)4][SbF6]2⋅CH3CN, M = Pd, Pt. Z. Anorg. Allg. Chem. 1998, 624, 1225–1234. [Google Scholar] [CrossRef]
- Tsiminis, G.; Schartner, E.P.; Brooks, J.L.; Hutchinson, M.R. Measuring and tracking vitamin B12: A review of current methods with a focus on optical spectroscopy. Appl. Spectrosc. Rev. 2017, 52, 439–455. [Google Scholar] [CrossRef]
- Bao-Sheng, L.; Jing, G.; Geng-Liang, Y. Determination of vitamin B12 concentration by fluorescence quenching with Acridine Orange-Rhodamine 6G energy transfer system. Spect. Spectral Anal. 2005, 25, 1080–1082. [Google Scholar]
- Shang, Z.B.; Wen, Y.J.; Yan, X.Q.; Sun, H.H.; Wang, Y.; Jin, W.J. Synthesis of a novel fluorescent probe based on 7-nitrobenzo-2-oxa-1,3-diazole skeleton for the rapid determination of vitamin B12 in pharmaceuticals. Luminescence 2014, 29, 598–602. [Google Scholar] [CrossRef]
- Song, P.-S. Spectroscopic analysis of vitamin B12 derivatives. Meth. Enzymol. 1980, 678, 5–11. [Google Scholar]
- Lodowski, P.; Toda, M.J.; Ciura, K.; Jaworska, M.; Kozlowski, P.M. Photolytic properties of Antivitamins B12. Inorg. Chem. 2018, 57, 7838–7850. [Google Scholar] [CrossRef]
- Jones, A.R. The photochemistry and photobiology of vitamin B12. Photochem. Photobiol. Sci. 2017, 16, 820–834. [Google Scholar] [CrossRef]
- Lawrence, A.D.; Nemoto-Smith, E.; Deery, E.; Baker, J.A.; Schroeder, S.; Brown, D.G.; Tullet, J.M.A.; Howard, M.J.; Brown, I.R.; Smith, A.G.; et al. Construction of fluorescent analogs to follow the uptake and distribution of cobalamin (vitamin B12) in bacteria, worms, and plants. Cell Chem. Biol. 2018, 25, 941–951. [Google Scholar] [CrossRef]
- Sugiarto, T.; Isnaeni, I.; Putri, K.J. Analysis of dual peak emission from Rhodamine 6G organic dyes using photoluminescence. J. Phys. Conf. Ser. 2017, 817, 012047. [Google Scholar] [CrossRef]
- Mahasin, F.; Al-Kadhemy, H.; Abbas, K.N.; Abdalmuhdi, W.B. Physical properties of Rhodamine 6G laser dye combined in polyvinyl alcohol films as heat sensor. IOP Conf. Ser. Mater. Sci. Eng. 2020, 928, 072126. [Google Scholar]
- Karolin, J.; Bogen, S.T.; Johansson, L.B.Å. Polarized fluorescence and absorption spectroscopy of 1,32-dihydroxy-dotriacontane-bis-rhodamine 101 ester. A new and lipid bilayer-spanning probe. J. Fluoresc. 1995, 5, 279–284. [Google Scholar] [CrossRef]
- Geoghegan, K.F.; Emery, M.J.; Martin, W.H.; McColl, A.S.; Daumy, G.O. Site-directed double fluorescent tagging of human renin and collagenase (MMP-1) substrate peptides using the periodate oxidation of N-terminal serine. An apparently general strategy for provision of energy-transfer substrates for proteases. Bioconjug. Chem. 1993, 4, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Rajasekar, M. Recent Trends in Rhodamine derivatives as fluorescent probes for biomaterial applications. J. Mol. Struct. 2021, 1235, 130232. [Google Scholar] [CrossRef]
- Tian, M.; Peng, X.; Fan, J.; Wang, J.; Sun, S. A fluorescent sensor for pH based on rhodamine fluorophore. Dye. Pigment. 2012, 95, 112–115. [Google Scholar] [CrossRef]
- Zehentbauer, F.M.; Moretto, C.; Stephen, R.; Thevar, T.; Gilchrist, J.R.; Pokrajac, D.; Richard, K.L.; Kiefer, J. Fluorescence spectroscopy of Rhodamine 6G: Concentration and solvent effects. Spectrochim. Acta A 2014, 121, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Beija, M.; Afonso, C.A.; Martinho, J.M. Synthesis and applications of Rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Andrés, A.; Rosés, M.; Ràfols, C.; Bosch, E.; Espinosa, S.; Segarra, V.; Huerta, J.M. Setup and validation of shake-flask procedures for the determination of partition coefficients (logD) from low drug amounts. Eur. J. Pharm. Sci. 2015, 76, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Rossier, J.; Delasoie, J.; Haeni, L.; Hauser, D.; Rothen-Rutishauser, B.; Zobi, F. Cytotoxicity of Mn-based photoCORMs of ethynyl-α-diimine ligands against different cancer cell lines: The key role of CO-depleted metal fragments. J. Inorg. Biochem. 2020, 209, 111122. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tavassoli, M.; Jacobsen, D.W. Receptor binding and internalization of immobilized transcobalamin II by mouse leukaemia cells. Nature 1980, 288, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Kutushov, M.; Gorelik, O. Low concentrations of Rhodamine-6G selectively destroy tumor cells and improve survival of melanoma transplanted mice. Neoplasma 2013, 60, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Magut, P.K.S.; Das, S.; Fernand, V.E.; Losso, J.; McDonough, K.; Naylor, B.M.; Aggarwal, S.; Warner, I.M. Tunable cytotoxicity of rhodamine 6G via anion variations. J. Am. Chem. Soc. 2013, 135, 15873–15879. [Google Scholar] [CrossRef]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9847/ (accessed on 15 March 2024).
- Stewart, Z.A.; Westfall, M.D.; Pietenpol, J.A. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol. Sci. 2003, 24, 139–145. [Google Scholar] [CrossRef]
- Jäckel, M.; Köpf-Maier, P. Influence of cisplatin on cell-cycle progression in xenografted human head and neck carcinomas. Cancer Chemother. Pharmacol. 1991, 27, 464–471. [Google Scholar] [CrossRef]
- Gear, A.R. Rhodamine 6G. A potent inhibitor of mitochondrial oxidative phosphorylation. J. Biol. Chem. 1974, 249, 3628–3637. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef] [PubMed]
- Chaires, J.B. Structural selectivity of drug-nucleic acid interactions probed by competition dialysis. In Topics in Current Chemistry—DNA Binders and Related Subjects; Waring, M.J., Chaires, J.B., Eds.; Springer: Berlin-Heidelberg, Germany, 2005; Volume 253, pp. 33–53. [Google Scholar]
- Oxford Diffraction. CrysAlis CCD; Oxford Diffraction Ltd.: Abingdon, UK, 2006. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- McArdle, P.; Gilligan, K.; Cunningham, D.; Dark, R.; Mahon, M. A method for the prediction of the crystal structure of ionic organic compounds—The crystal structures of o-toluidinium chloride and bromide and polymorphism of bicifadine hydrochloride. CrystEngComm 2004, 6, 303–309. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Taraszka, K.S.; Chen, E.; Metzger, T.; Chance, M.R. Identification of structural markers for vitamin B12 and other corrinoid derivatives in solution using FTIR spectroscopy. Biochemistry 1991, 30, 1222–1227. [Google Scholar] [CrossRef]
- Zhou, K.; Zelder, F. Vitamin B12 mimics having a peptide backbone and tuneable coordination and redox properties. Angew. Chem. Int. Ed. 2010, 49, 5178–5180. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Marzilli, L.G. Investigations of B12 derivatives with inorganic ligands using 2D NMR spectroscopy. Ligand-responsive shifts suggest that the deoxyadenosyl moiety in Coenzyme B12 has a steric trans influence. J. Am. Chem. Soc. 1993, 115, 9182–9190. [Google Scholar] [CrossRef]
- Horton, R.A.; Bagnato, J.D.; Grissom, C.B. Structural determination of 5’-OH alpha-ribofuranoside modified cobalamins via 13C and DEPT NMR. J. Org. Chem. 2003, 68, 7108–7111. [Google Scholar] [CrossRef]
- Brown, K.L.; Evans, D.R. Heteronuclear NMR studies of cobalt corrinoids. 14. Amide proton and nitrogen-15 NMR studies of base-on cobalamins. Inorg. Chem. 1993, 32, 2544–2548. [Google Scholar] [CrossRef]
- Kurumaya, K.; Kajiwara, M. Proton nuclear magnetic resonance (1H-NMR) signal assignment of vitamin B12 based on normal two-dimensional NMR and feeding experiments. Chem. Pharm. Bull. 1989, 1, 9–12. [Google Scholar] [CrossRef]
- Ramos, S.S.; Vilhena, A.F.; Santos, L.; Almeida, P. 1H and 13C NMR spectra of commercial rhodamine ester derivatives. Magn. Reson. Chem. 2000, 38, 475–478. [Google Scholar] [CrossRef]
- Akalin, E.; Akyüz, S. Force field and IR intensity calculations of aniline and transition metal(II) aniline complexes. J. Mol. Struct. 1999, 482–483, 175–181. [Google Scholar] [CrossRef]
- Ibn El Alami, M.S.; El Amrani, M.A.; Dahdouh, A.; Roussel, P.; Suisse, I.; Mortreux, A. α-Amino-oximes based on optically pure limonene: A new ligands family for ruthenium-catalyzed asymmetric transfer hydrogenation. Chirality 2012, 24, 675–682. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | (nm) | (nm) | Stokes Shift (nm) |
|---|---|---|---|
| vitamin B12 | 388 | 433 | 45 |
| B12-Pt1 | 367 | 416 | 49 |
| B12-Pt2 | 368 | 413 | 45 |
| R6G | 543 | 567 | 24 |
| R6G* | 326 | 563 | 237 |
| R6G*-B12 | 306 | 420 | 114 |
| R6G*-B12-Pt1 | 307 | 412 | 105 |
| R6G*-B12-Pt2 | 304 | 414 | 110 |
| Compound | Log D7.4 ± S.D. |
|---|---|
| B12-Pt1 | −0.81 ± 0.04 |
| B12-Pt2 | −0.78 ± 0.05 |
| R6G*-B12-Pt1 | 0.52 ± 0.01 |
| R6G*-B12-Pt2 | 0.50 ± 0.02 |
| Compound | IC50 ± S.D. (μM) | ||
|---|---|---|---|
| PC-3 | HeLa | MCF-7 | |
| vitamin B12 | no convergence | no convergence | no convergence |
| R6G | 1.11 ± 1.09 | 1.95 ± 1.09 | 1.62 ± 1.22 |
| Pt1 | 85.9 ± 19.3 | >100 | >100 |
| Pt2 | 86.8 ± 4.5 | >100 | >100 |
| B12-Pt1 | >100 | >100 | >100 |
| B12-Pt2 | >100 | >100 | >100 |
| R6G*-B12-Pt1 | 47.9 ± 1.0 | 77.2 ± 1.0 | 70.3 ± 1.0 |
| R6G*-B12-Pt2 | 42.7 ± 1.0 | 73.1 ± 1.0 | 58.1 ± 1.0 |
| B12-Pt1 + R6G | 2.58 ± 1.15 | 4.85 ± 1.10 | 0.80 ± 1.00 |
| cisplatin | 16.5 ± 1.1 | 14.5 ± 2.5 | 9.80 ± 0.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehder, R.; de la Torre-Rubio, E.; de la Cueva-Alique, I.; O’Malley, C.; Pérez-Redondo, A.; Gude, L.; Royo, E.; Ronconi, L. Fluorescent Vitamin B12–Platinum(II) Derivatives as Potential Metallotheranostic Agents for the Treatment and Imaging of Tumors. Inorganics 2024, 12, 91. https://doi.org/10.3390/inorganics12030091
Mehder R, de la Torre-Rubio E, de la Cueva-Alique I, O’Malley C, Pérez-Redondo A, Gude L, Royo E, Ronconi L. Fluorescent Vitamin B12–Platinum(II) Derivatives as Potential Metallotheranostic Agents for the Treatment and Imaging of Tumors. Inorganics. 2024; 12(3):91. https://doi.org/10.3390/inorganics12030091
Chicago/Turabian StyleMehder, Rozan, Elena de la Torre-Rubio, Isabel de la Cueva-Alique, Ciaran O’Malley, Adrián Pérez-Redondo, Lourdes Gude, Eva Royo, and Luca Ronconi. 2024. "Fluorescent Vitamin B12–Platinum(II) Derivatives as Potential Metallotheranostic Agents for the Treatment and Imaging of Tumors" Inorganics 12, no. 3: 91. https://doi.org/10.3390/inorganics12030091