
Synthesis, Spectroscopic Characterization, and Photophysical Studies of Heteroleptic Silver Complexes Bearing 2,9-Bis(styryl)-1,10-phenanthroline Ligands and Bis[(2-diphenylphosphino)phenyl] Ether

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
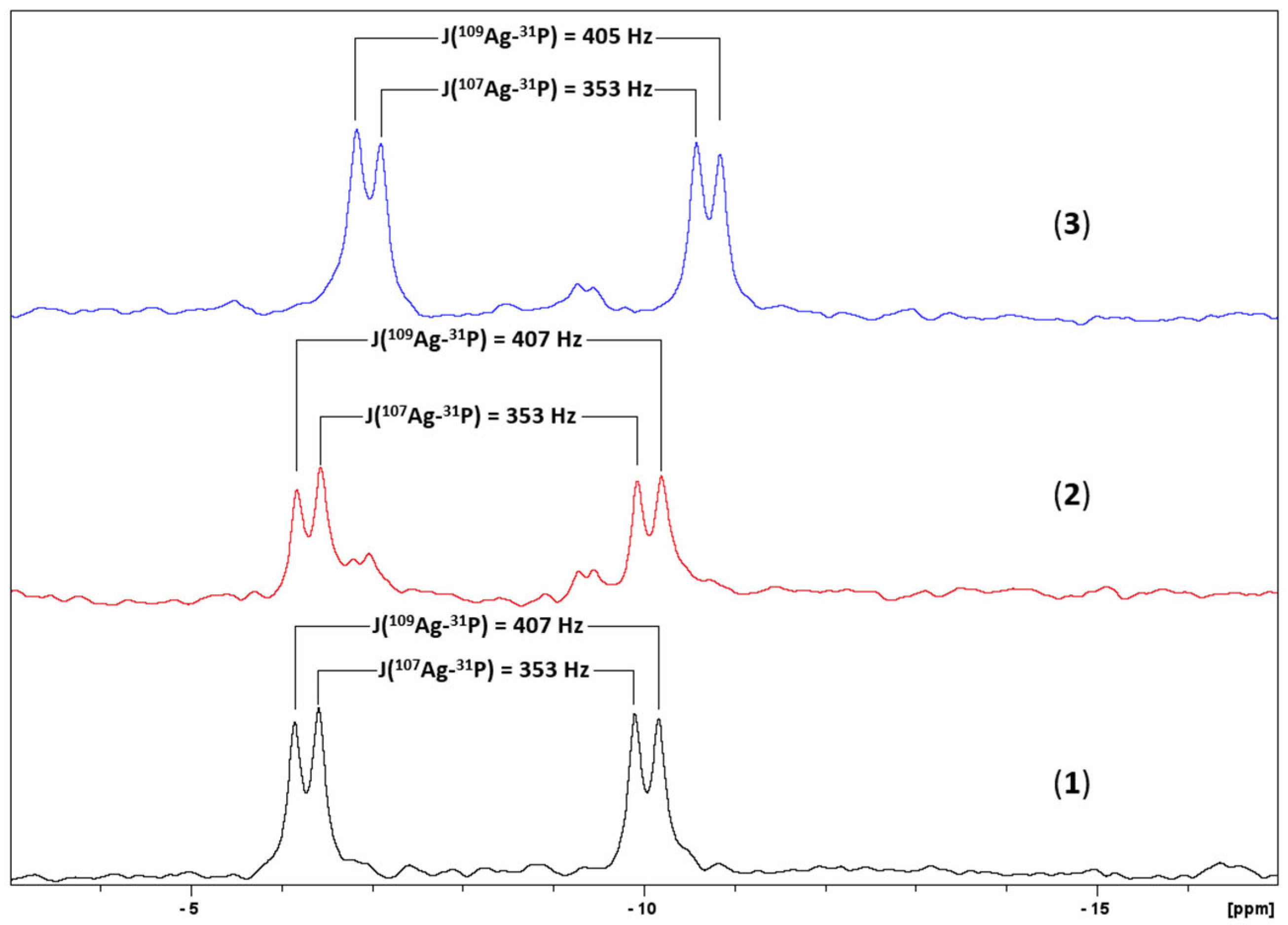
2.1. Synthesis and Characterization
2.2. Characterization in Solution
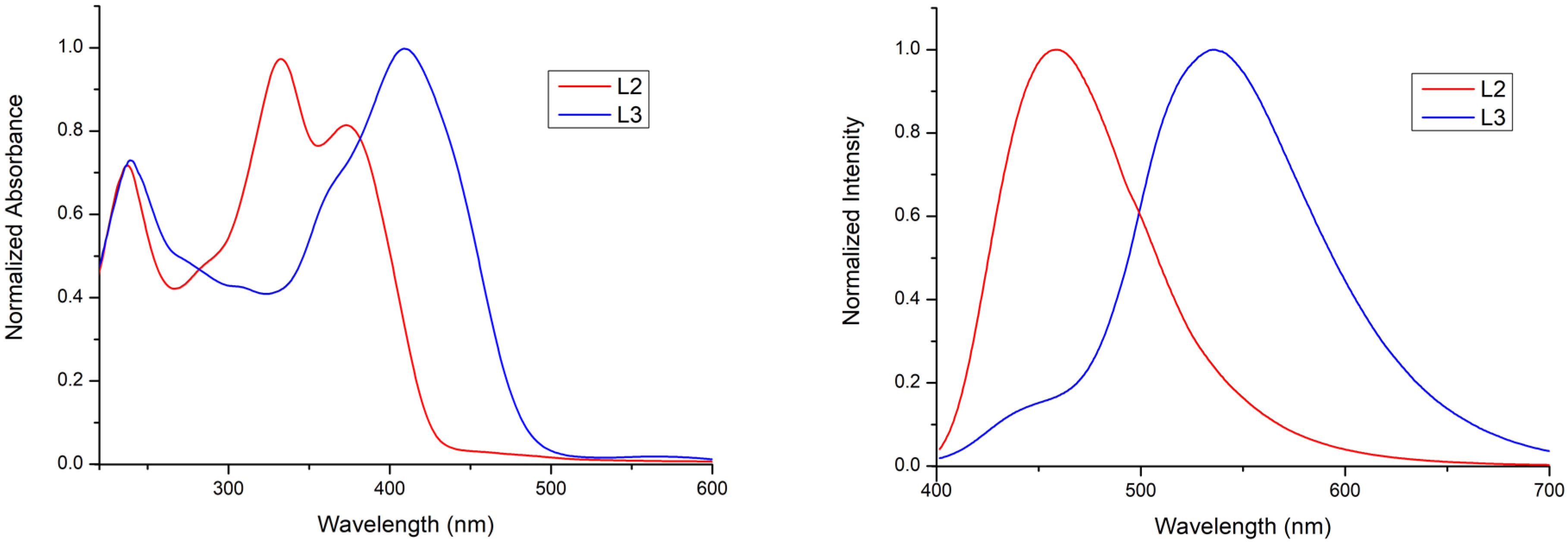
2.3. Optical-Photophysical Properties of Ligands in Solution
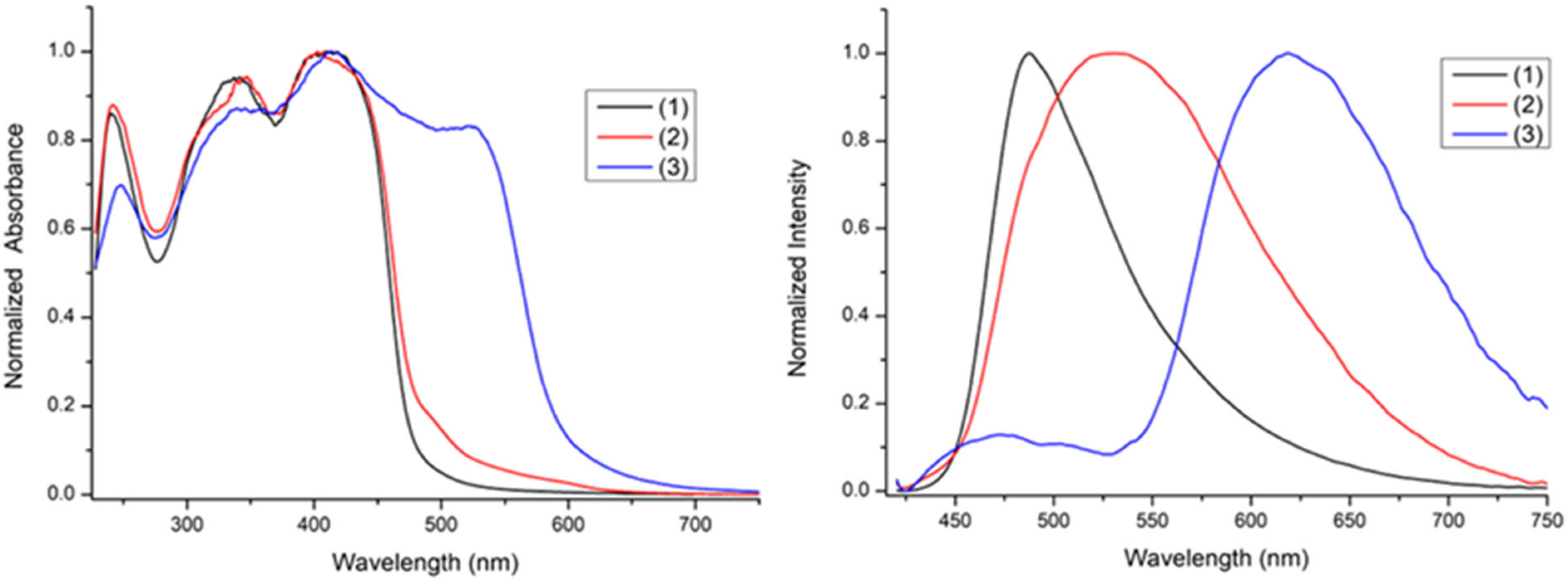
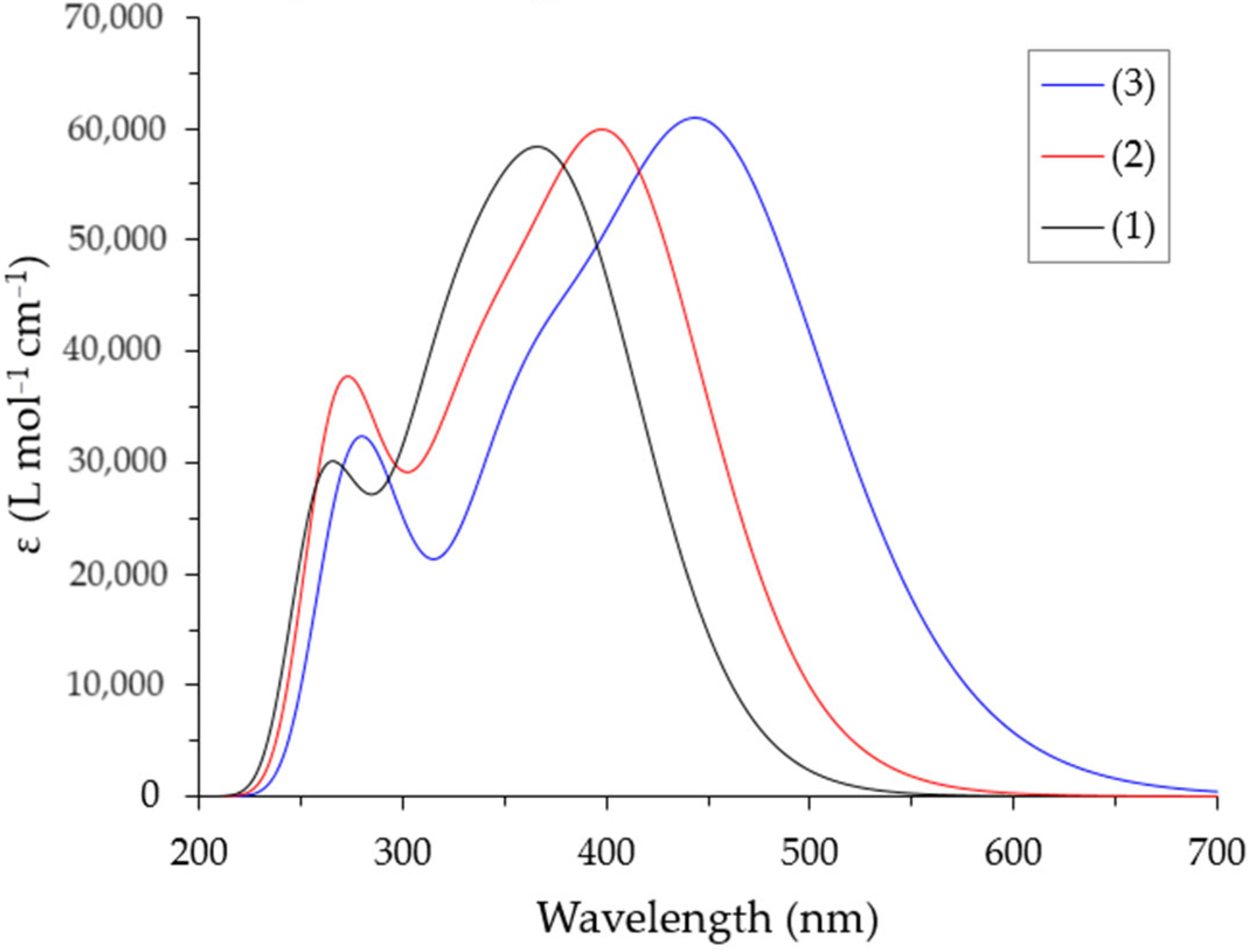
2.4. Optical Properties of Complexes in Solution
2.5. Luminescent Behavior in Solid State
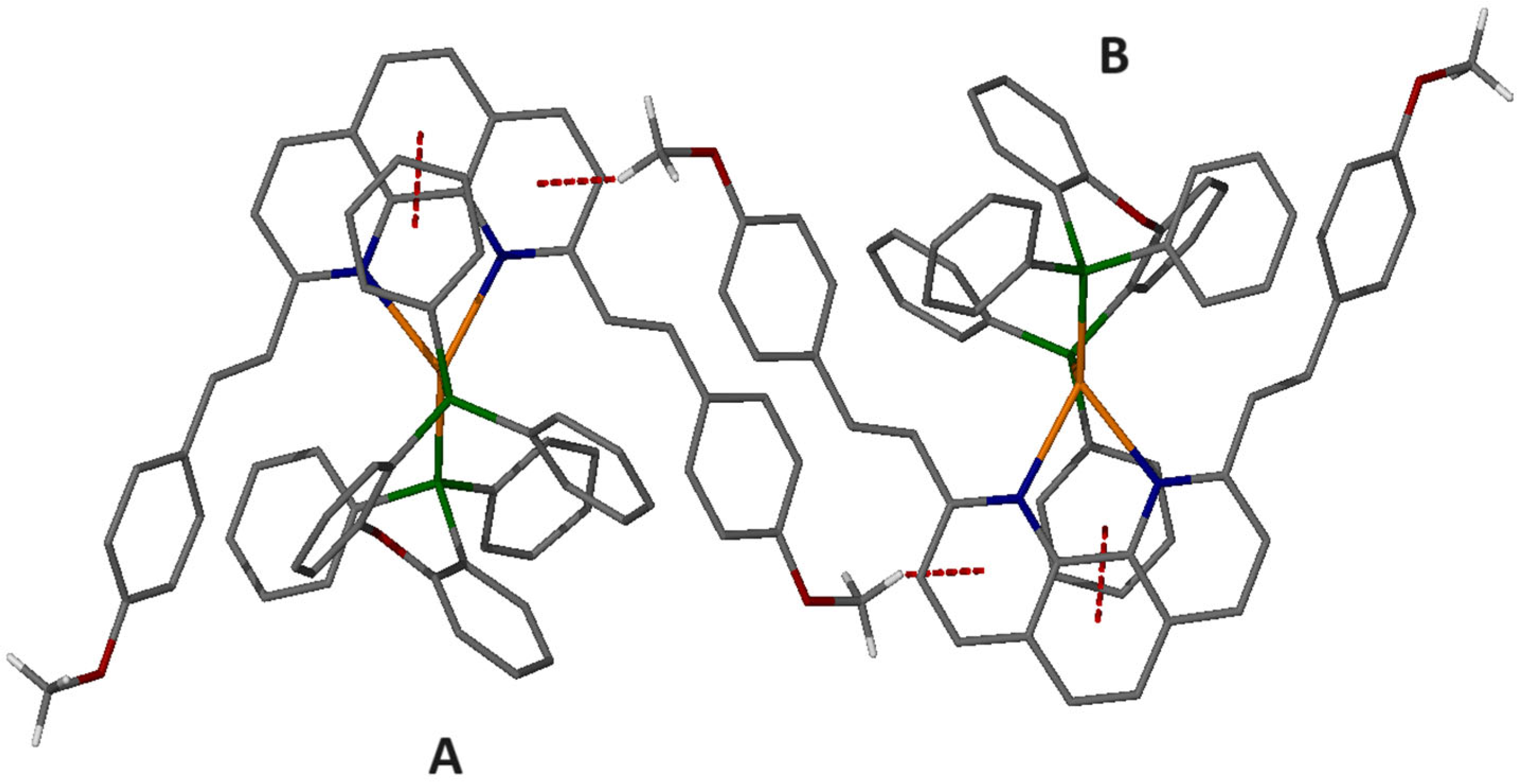
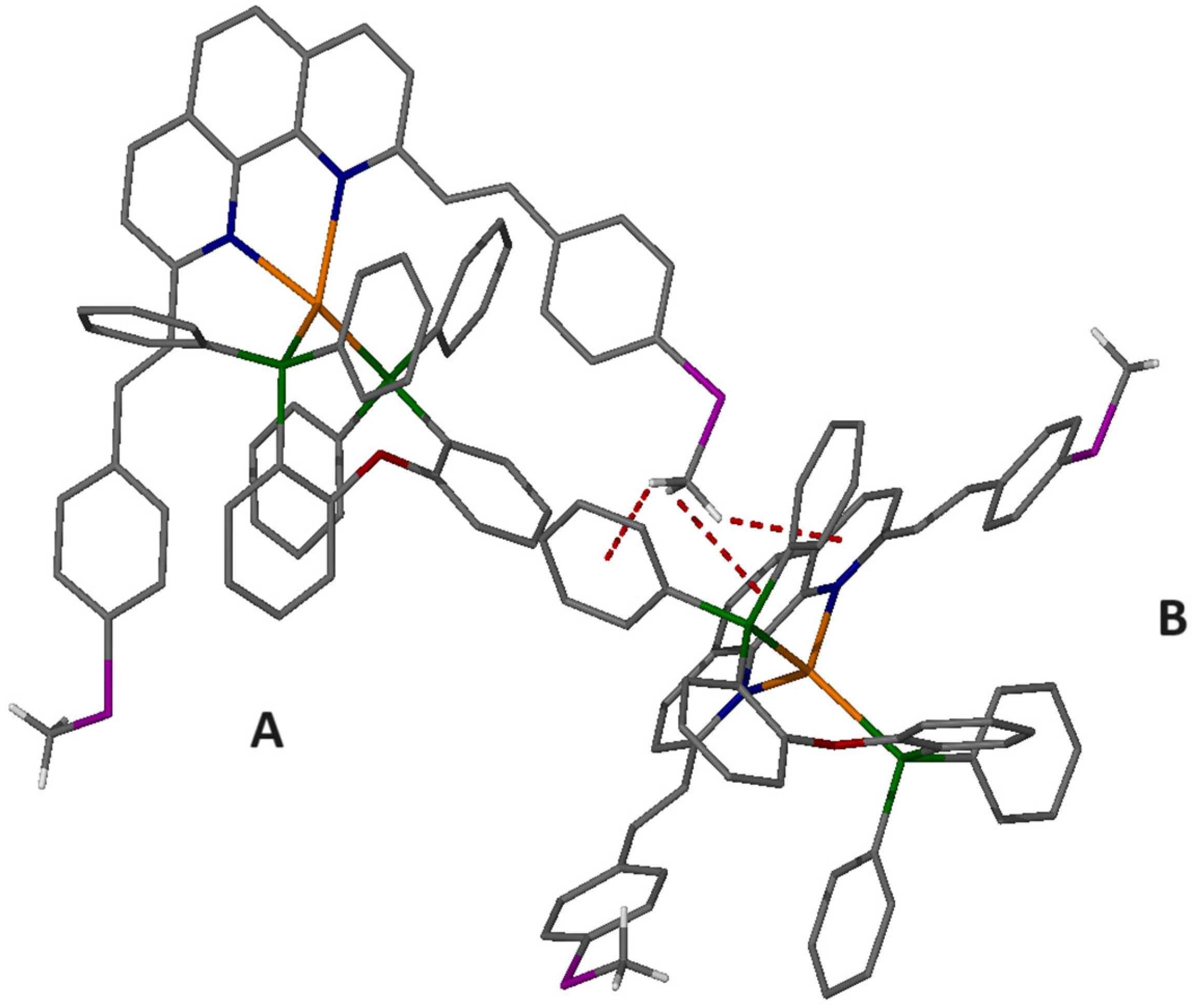
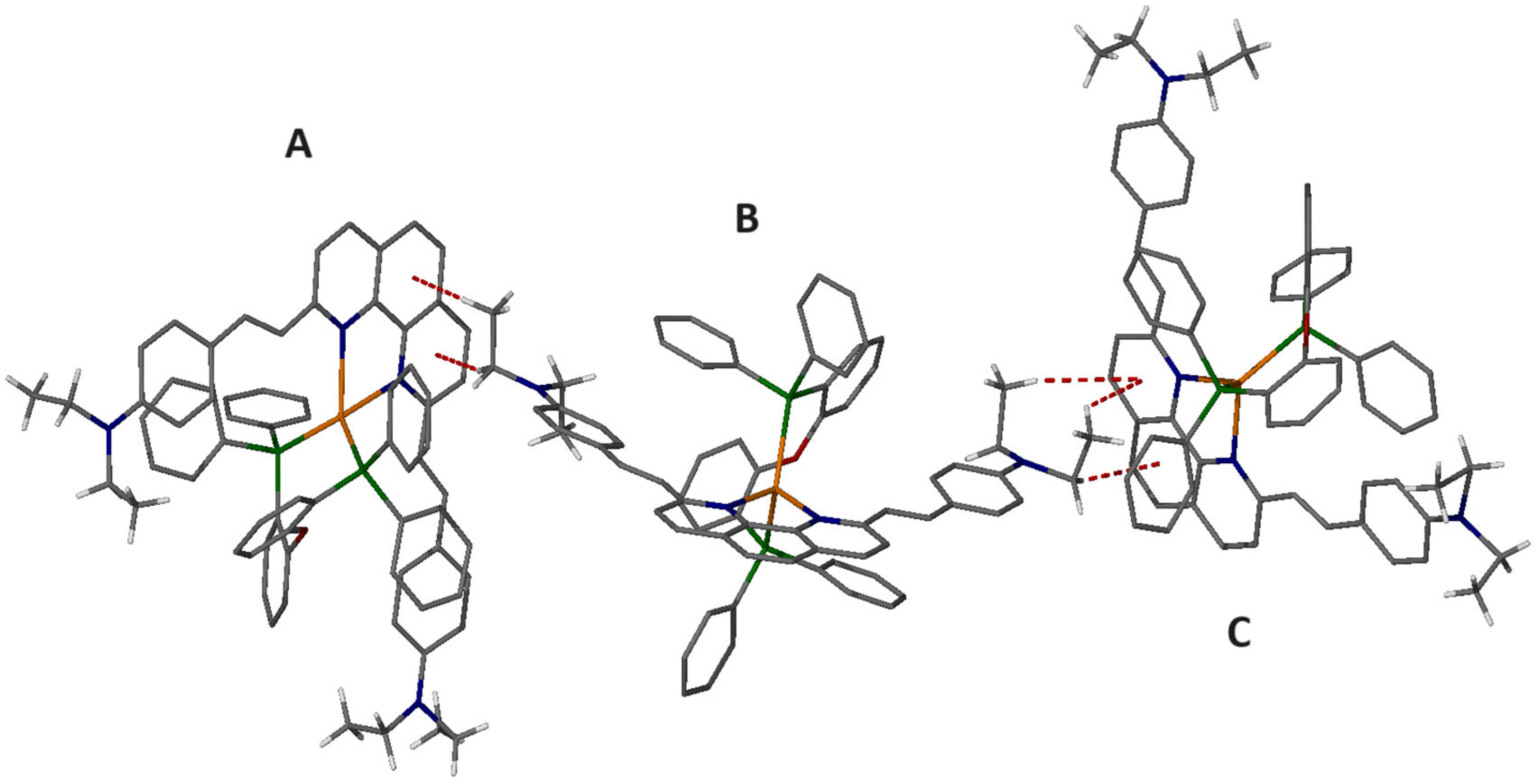
2.6. Single-Crystal Structure Analysis
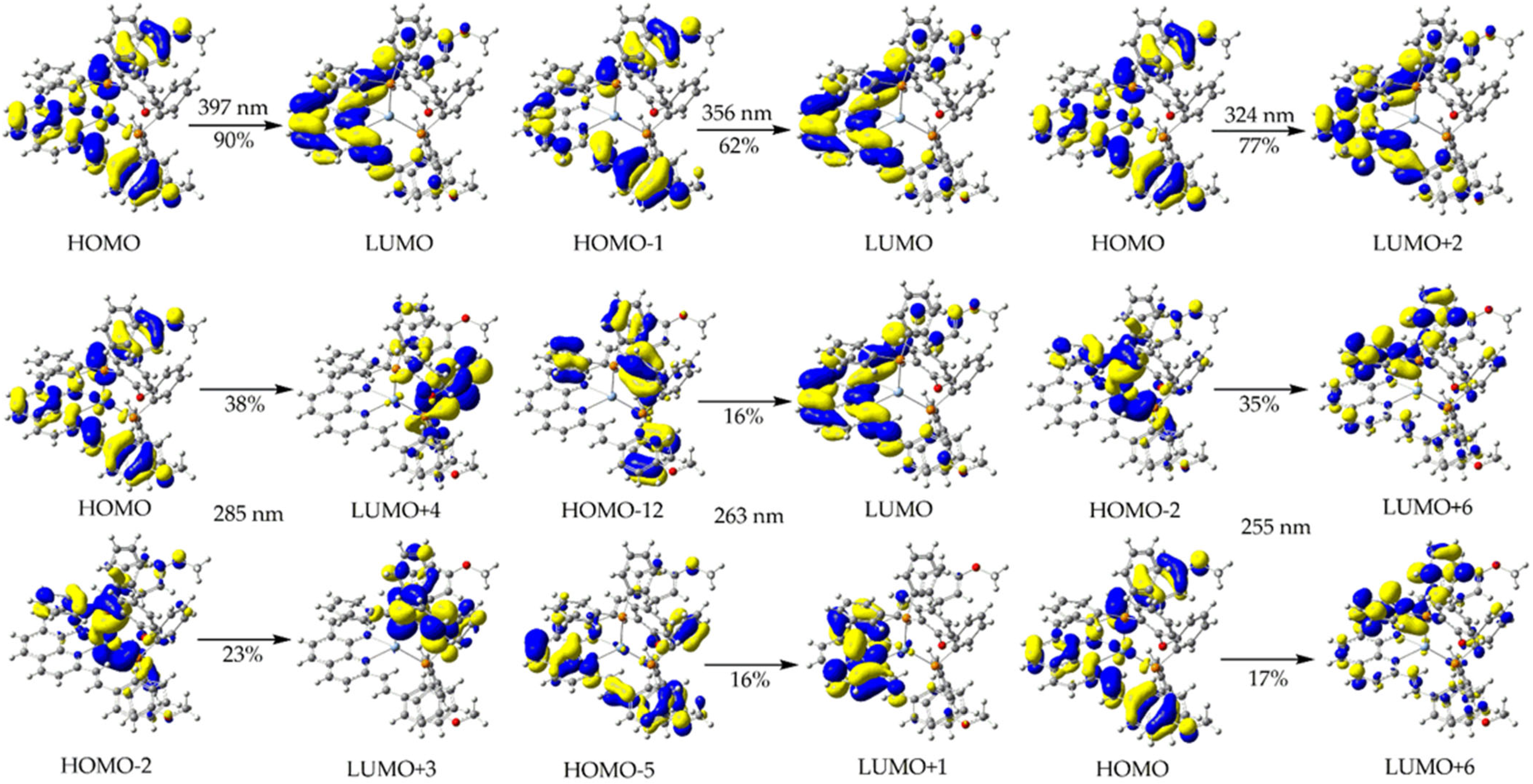
2.7. Computational Study
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Crystal Structure Determination
3.4. Computational Details
3.5. Synthesis of Complexes
- [Ag(L1)(POP)][BF4] (1)
- [Ag(L2)(POP)][BF4] (2)
- [Ag(L3)(POP)][BF4] (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernández-Moreira, V.; Thorp-Greenwood, F.L.; Coogan, M.P. Application of D6 Transition Metal Complexes in Fluorescence Cell Imaging. Chem. Commun. 2010, 46, 186–202. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Wong, K.M.C. Luminescent Metal Complexes of D6, D8 and d 10 Transition Metal Centres. Chem. Commun. 2011, 47, 11579–11592. [Google Scholar] [CrossRef]
- Lo, K.K.-W.; Choi, A.W.; Law, W.H.-T. Applications of Luminescent Inorganic and Organometallic Transition Metal Complexes as Biomolecular and Cellular Probes. Dalton Trans. 2012, 41, 6021. [Google Scholar] [CrossRef]
- Min, J.; Zhang, Q.; Sun, W.; Cheng, Y.; Wang, L. Neutral Copper(i) Phosphorescent Complexes from Their Ionic Counterparts with 2-(2′-Quinolyl)Benzimidazole and Phosphine Mixed Ligands. Dalton Trans. 2011, 40, 686–693. [Google Scholar] [CrossRef]
- Zhang, Q.; Ding, J.; Cheng, Y.; Wang, L.; Xie, Z.; Jing, X.; Wang, F. Novel Heteroleptic Cu1 Complexes with Tunable Emission Color for Efficient Phosphorescent Light-Emitting Diodes. Adv. Funct. Mater. 2007, 17, 2983–2990. [Google Scholar] [CrossRef]
- Takeda, H.; Kobayashi, A.; Tsuge, K. Recent Developments of Photoactive Cu(I) and Ag(I) Complexes with Diphosphine and Related Ligands. Coord. Chem. Rev. 2022, 470, 214700. [Google Scholar] [CrossRef]
- Shafikov, M.Z.; Czerwieniec, R.; Yersin, H. Ag(i) Complex Design Affording Intense Phosphorescence with a Landmark Lifetime of over 100 Milliseconds. Dalton Trans. 2019, 48, 2802–2806. [Google Scholar] [CrossRef]
- Shafikov, M.Z.; Suleymanova, A.F.; Czerwieniec, R.; Yersin, H. Thermally Activated Delayed Fluorescence from Ag(I) Complexes: A Route to 100% Quantum Yield at Unprecedentedly Short Decay Time. Inorg. Chem. 2017, 56, 13274–13285. [Google Scholar] [CrossRef]
- Zuideveld, M.A.; Swennenhuis, B.H.G.; Boele, M.D.K.; Guari, Y.; Van Strijdonck, G.P.F.; Reek, J.N.H.; Kamer, P.C.J.; Goubitz, K.; Fraanje, J.; Lutz, M.; et al. The Coordination Behaviour of Large Natural Bite Angle Diphosphine Ligands towards Methyl and 4-Cyanophenylpalladium(II) Complexes. J. Chem. Soc. Dalton Trans. 2002, 11, 2308–2317. [Google Scholar] [CrossRef]
- Freixa, Z.; Van Leeuwen, P.W.N.M. Bite Angle Effects in Diphosphine Metal Catalysts: Steric or Electronic? Dalton Trans. 2003, 10, 1890–1901. [Google Scholar] [CrossRef]
- Flecken, F.; Grell, T.; Hanf, S. Transition Metal Complexes of the PPO/POP Ligand: Variable Coordination Chemistry and Photo-Luminescence Properties. Dalton Trans. 2022, 51, 8975–8985. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Li, H.Y.; Young, D.J.; Cao, X.; Zhu, D.L.; Ren, Z.G.; Li, H.X. Heteroleptic Copper(i) Complexes [Cu(Dmp)(N^P)]BF4 for Photoinduced Atom-Transfer Radical Addition Reactions. Dalton Trans. 2023, 52, 8142–8154. [Google Scholar] [CrossRef]
- Beaudelot, J.; Oger, S.; Peruško, S.; Phan, T.A.; Teunens, T.; Moucheron, C.; Evano, G. Photoactive Copper Complexes: Properties and Applications. Chem. Rev. 2022, 122, 16365–16609. [Google Scholar] [CrossRef]
- Glykos, D.; Plakatouras, J.C.; Malandrinos, G. Bis(2-Phenylpyridinato,-C2′,N)[4,4′-Bis(4-Fluorophenyl)-6,6′-Dimethyl-2,2′-Bipyridine] Iridium(III) Hexafluorophosphate. Molbank 2023, 2023, 4–11. [Google Scholar] [CrossRef]
- Kouvatsis, P.; Glykos, D.; Plakatouras, J.C.; Malandrinos, G. [6-(Furan-2-Yl)-2,2′-Bipyridine]Bis(Triphenylphosphine) Copper(I) Tetrafluoroborate. Molbank 2023, 2023, M1724. [Google Scholar] [CrossRef]
- Glykos, D.; Plakatouras, J.C.; Malandrinos, G. [4,4′-Bis(4-Fluorophenyl)-6,6′-Dimethyl-2,2′-Bipyridine] [Bis (2-(Diphenylphosphino) Phenyl) Ether] Silver(I) Hexafluorophosphate. Molbank 2023, 2023, M1675. [Google Scholar] [CrossRef]
- Glykos, D.; Plakatouras, J.C.; Malandrinos, G. Solution-State Studies, X-Ray Structure Determination and Luminescence Properties of an Ag(I) Heteroleptic Complex Containing 2,9-Bis(Styryl)-1,10-Phenanthroline Derivative and Triphenylphosphine. Inorganics 2023, 11, 467. [Google Scholar] [CrossRef]
- Kouvatsis, P.; Glykos, D.; Plakatouras, J.C.; Malandrinos, G. [6-(Thiophen-2-Yl)-2,2′-Bipyridine]Bis(Triphenylphosphine) Copper(I) Tetrafluoroborate. Molbank 2023, 2023, M1605. [Google Scholar] [CrossRef]
- Bonaccorso, C.; Cesaretti, A.; Elisei, F.; Mencaroni, L.; Spalletti, A.; Fortuna, C.G. New Styryl Phenanthroline Derivatives as Model D−π−A−π−D Materials for Non-Linear Optics. ChemPhysChem 2018, 19, 1917–1929. [Google Scholar] [CrossRef]
- Sun, L.Z.; Kuang, X.N.; Lin, S.; Zhao, L.; Yu, X.; Li, Z.F.; Liu, M.; Xin, X.L.; Yang, Y.P.; Jin, Q.H. Nine Heteroleptic Copper(I)/Silver(I) Complexes Prepared from Phosphine and Diimine Ligands: Syntheses, Structures and Terahertz Spectra. Polyhedron 2020, 175, 114177. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Cui, Y.Z.; Jin, Q.H.; Yang, Y.P.; Liu, M.; Li, Z.F.; Bi, K.L.; Zhang, C.L. Syntheses, Structural Characterizations and Terahertz Spectra of Ag(I)/Cu(I) Complexes with Bis[2-(Diphenylphosphino)Phenyl]Ether and N^N Ligands. Polyhedron 2017, 122, 86–98. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, S.; Zhao, Z.; He, B.; Li, X.L. Photophysical Properties of Homobimetallic Cu(i)-Cu(i) and Heterobimetallic Cu(i)-Ag(i) Complexes of 2-(6-Bromo-2-Pyridyl)-1H-Imidazo[4,5-f][1,10]Phenanthroline. New J. Chem. 2022, 46, 8881–8891. [Google Scholar] [CrossRef]
- Kaeser, A.; Delavaux-Nicot, B.; Duhayon, C.; Coppel, Y.; Nierengarten, J.F. Heteroleptic Silver(I) Complexes Prepared from Phenanthroline and Bis-Phosphine Ligands. Inorg. Chem. 2013, 52, 14343–14354. [Google Scholar] [CrossRef]
- Moudam, O.; Tsipis, A.C.; Kommanaboyina, S.; Horton, P.N.; Coles, S.J. First Light-Emitting Electrochemical Cell with [Ag(i)(N^N)(P^P)] Type Complex. RSC Adv. 2015, 5, 95047–95053. [Google Scholar] [CrossRef]
- Wang, Y.; Kuang, X.N.; Cui, Y.Z.; Xin, X.L.; Han, H.L.; Liu, M.; Yang, Y.P.; Jin, Q.H. Synthesis, Structure, Luminescent Properties, and Photocatalytic Behavior of 0D–3D Silver(I) Complexes Bearing Both Diphosphine Ligands and 1,10-Phenanthroline Derivatives. Polyhedron 2018, 155, 135–143. [Google Scholar] [CrossRef]
- Lipinski, S.; Cavinato, L.M.; Pickl, T.; Biffi, G.; Pöthig, A.; Coto, P.B.; Fernández-Cestau, J.; Costa, R.D. Dual-Phosphorescent Heteroleptic Silver(I) Complex in Long-Lasting Red Light-Emitting Electrochemical Cells. Adv. Opt. Mater. 2023, 11, 2203145. [Google Scholar] [CrossRef]
- Teng, T.; Li, K.; Cheng, G.; Wang, Y.; Wang, J.; Li, J.; Zhou, C.; Liu, H.; Zou, T.; Xiong, J.; et al. Lighting Silver(I) Complexes for Solution-Processed Organic Light-Emitting Diodes and Biological Applications via Thermally Activated Delayed Fluorescence. Inorg. Chem. 2020, 59, 12122–12131. [Google Scholar] [CrossRef]
- Nemati Bideh, B.; Shahroosvand, H.; Nazeeruddin, M.K. High-Efficiency Deep-Red Light-Emitting Electrochemical Cell Based on a Trinuclear Ruthenium(II)–Silver(I) Complex. Inorg. Chem. 2021, 60, 11915–11922. [Google Scholar] [CrossRef]
- Beliaeva, M.; Belyaev, A.; Grachova, E.V.; Steffen, A.; Koshevoy, I.O. Ditopic Phosphide Oxide Group: A Rigidifying Lewis Base to Switch Luminescence and Reactivity of a Disilver Complex. J. Am. Chem. Soc. 2021, 143, 15045–15055. [Google Scholar] [CrossRef]
- Wang, C.; Li, Z. Molecular Conformation and Packing: Their Critical Roles in the Emission Performance of Mechanochromic Fluorescence Materials. Mater. Chem. Front. 2017, 1, 2174–2194. [Google Scholar] [CrossRef]
- Yuan, M.-S.; Du, X.; Xu, F.; Wang, D.-E.; Wang, W.-J.; Li, T.-B.; Tu, Q.; Zhang, Y.; Du, Z.; Wang, J. Aggregation-Induced Bathochromic Fluorescent Enhancement for Fluorenone Dyes. Dye Pigment 2015, 123, 355–362. [Google Scholar] [CrossRef]
- Jia, J.H.; Liang, D.; Yu, R.; Chen, X.L.; Meng, L.; Chang, J.F.; Liao, J.Z.; Yang, M.; Li, X.N.; Lu, C.Z. Coordination-Induced Thermally Activated Delayed Fluorescence: From Non-TADF Donor-Acceptor-Type Ligand to TADF-Active Ag-Based Complexes. Chem. Mater. 2020, 32, 620–629. [Google Scholar] [CrossRef]
- Balakrishna, M.S.; Venkateswaran, R.; Mobin, S.M. Mixed-Ligand Silver(I) Complexes Containing Bis[2-(Diphenylphosphino)Phenyl]Ether and Pyridyl Ligands. Inorganica Chim. Acta 2009, 362, 271–276. [Google Scholar] [CrossRef]
- Cui, Y.Z.; Yuan, Y.; Han, H.L.; Li, Z.F.; Liu, M.; Jin, Q.H.; Yang, Y.P.; Zhang, Z.W. Synthesis, Characterization, and Luminescent Properties of Silver(I) Complexes Based on Diphosphine Ligands and 2,9-Dimethyl-1,10-Phenanthroline. Z. Anorg. Allg. Chem. 2016, 642, 953–959. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, Y.Z.; Li, Z.F.; Liu, M.; Yang, Y.P.; Zhang, Z.W.; Xin, X.L.; Jin, Q.H. Synthesis, Characterization, and Luminescent Properties of Silver(I) Complexes Based on Diphosphine Ligands and 6,7-Dicyanodipyridoquinoxaline. Z. Anorg. Allg. Chemie 2017, 643, 1253–1261. [Google Scholar] [CrossRef]
- Ishida, H.; Tobita, S.; Hasegawa, Y.; Katoh, R.; Nozaki, K. Recent Advances in Instrumentation for Absolute Emission Quantum Yield Measurements. Coord. Chem. Rev. 2010, 254, 2449–2458. [Google Scholar] [CrossRef]
- APEX 3; SAINT, SHELXT 2016; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. SADABS 1996; University of Göttingen: Göttingen, Germany.
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Barbour, L.J. X-Seed—A Software Tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16W, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Vetere, V.; Adamo, C.; Maldivi, P. Performance of the ‘parameter free’ PBE0 functional for the modeling of molecular properties of heavy metals. Chem. Phys. Lett. 2000, 325, 99–105. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Inexpensive and accurate predictions of optical excitations in transition-metal complexes: The TDDFT/PBE0 route. Theor. Chem. Acc. 2000, 105, 169–172. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Scuseria, G.E.; Barone, V. Accurate excitation energies from time-dependent density functional theory: Assessing the PBE0 model. J. Chem. Phys. 1999, 111, 2889–2899. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett. 1997, 274, 242–250. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H Atoms | 1 | 2 | 3 | Δδ (1) * | Δδ (2) * | Δδ (3) * |
|---|---|---|---|---|---|---|
| H(3)/H(8) | 8.24 | 8.28 | 8.14 | 0.31 | 0.34 | 0.24 |
| H(4)/H(7) | 8.44 | 8.48 | 8.32 | 0.22 | 0.25 | 0.16 |
| H(5)/H(6) | 7.83 | 7.86 | 7.77 | 0.09 | 0.11 | 0.09 |
| H(a) | 7.48 | 7.51 | 7.39 | −0.29 | −0.26 | −0.33 |
| H(b) | 7.42 | 7.48 | 7.30 | −0.2 | −0.21 | −0.24 |
| A, H(2,6) | 6.85 | 6.95 | 6.70 | −0.83 | −0.69 | −0.9 |
| A, H(3,5) | 6.65 | 6.81 | 6.33 | −0.34 | −0.51 | −0.4 |
| (-OMe) (-CH3) | 3.88 | - | - | −0.02 | - | - |
| (-SMe) (-CH3) | - | 2.56 | - | - | 0 | - |
| (-NEt2) (-CH2) | - | - | 3.42 | - | - | −0.03 |
| (-NEt2) (-CH3) | - | - | 1.25 | - | - | −0.01 |
| POP(C ring) H3 | 6.73 | 6.74 | 6.85 | 0.01 | 0.02 | 0.13 |
| POP(C ring) H4 | 7.19 | 7.21 | 7.02 | −0.03 | −0.01 | −0.20 |
| POP(C ring) H5 | 7.12 | 7.12 | 7.15 | 0.13 | 0.13 | 0.16 |
| POP(C ring) H6 | 6.91 | 6.92 | 6.93 | 0.07 | 0.08 | 0.09 |
| Compound | λabs/nm (ε/Μ−1cm−1) (CH2Cl2) | λem/nm (CH2Cl2) | Φem (CH2Cl2) | λem/nm (Solid) | Φem (Solid) |
|---|---|---|---|---|---|
| L2 | 235 (44,000), 332 (60,000), 377 (48,000) | 460 | 10% | - | - |
| L3 | 239 (45,454), 360 (40,184), 407 (62,582) | 538 | 6% | - | - |
| Compound | λabs/nm (ε/Μ−1cm−1) (CH2Cl2) | λem/nm (CH2Cl2) | Φem (CH2Cl2) | λem/nm (Solid) | Φem (Solid) |
|---|---|---|---|---|---|
| 1 | 230 (96,153), 335 (62,500), 385 (65,384) | 445 | 11% | 487 | 9.5% |
| 2 | 235 (63,532), 350 (55,908), 390 (60,991) | 465 | 20% | 534 | 5.6% |
| 3 | 245 (56,112), 445 (80,160) | 550 | 23% | 625 | 2.5% |
| Bond Distances | (Å) | Bond Angles | (°) |
|---|---|---|---|
| Ag(1)-N(1) | 2.350(3) | N(1)-Ag(1)-N(2) | 72.11(12) |
| Ag(1)-N(2) | 2.356(4) | N(1)-Ag(1)-P(2) | 127.15(9) |
| Ag(1)-P(2) | 2.4104(12) | N(2)-Ag(1)-P(2) | 133.36(9) |
| Ag(1)-P(1) | 2.5661(12) | N(1)-Ag(1)-P(1) | 97.05(9) |
| N(2)-Ag(1)-P(1) | 96.20(9) | ||
| P(2)-Ag(1)-P(1) | 118.64(4) |
| Bond Distances | (Å) | Bond Angles | (°) |
|---|---|---|---|
| Ag(1)-N(1) | 2.385(3) | N(1)-Ag(1)-N(2) | 70.43(11) |
| Ag(1)-N(2) | 2.392(3) | N(1)-Ag(1)-P(2) | 114.60(9) |
| Ag(1)-P(2) | 2.5240(12) | N(2)-Ag(1)-P(2) | 118.77(8) |
| Ag(1)-P(1) | 2.5245(12) | N(1)-Ag(1)-P(1) | 117.89(8) |
| N(2)-Ag(1)-P(1) | 110.36(9) | ||
| P(2)-Ag(1)-P(1) | 116.67(4) |
| Bond Distances | (Å) | Bond Angles | (°) |
|---|---|---|---|
| Ag(1)-N(1) | 2.362(4) | N(1)-Ag(1)-N(2) | 71.13(12) |
| Ag(1)-N(2) | 2.377(4) | N(1)-Ag(1)-P(2) | 114.57(9) |
| Ag(1)-P(2) | 2.5117(14) | N(2)-Ag(1)-P(2) | 118.83(9) |
| Ag(1)-P(1) | 2.5144(13) | N(1)-Ag(1)-P(1) | 116.75(9) |
| N(2)-Ag(1)-P(1) | 113.40(9) | ||
| P(2)-Ag(1)-P(1) | 115.11(5) |
| Excitation (% Composition) | λ (nm) | f | Assignment |
|---|---|---|---|
| 1 | |||
| H → L (90%) | 397 | 0.723 | MLCT/IL/LL’CT |
| H-1 → L (62%), H → L + 1 (27%), H-2 → L + 1 (8%) | 356 | 0.352 | IL/LL’CT |
| H → L + 2 (77%), H-2 → L + 1 (5%), H-2 → L + 2 (5%) | 324 | 0.482 | MLCT/IL/LL’CT |
| H → L + 4 (38%), H-2 → L + 3 (23%), H → L + 3 (11%), H-2 → L + 4 (10%) | 285 | 0.056 | IL/LL’CT |
| H-12 → L (16%), H-5 → L + 1 (16%), H-9 → L + 1 (13%) | 263 | 0.058 | IL/LL’CT |
| H-2 → L + 6 (35%), H → L + 6 (17%), H → L + 9 (10%) | 255 | 0.087 | IL/LL’CT |
| 2 | |||
| H → L (94%) | 420 | 0.846 | IL |
| H-1 → L (90%), H → L + 1 (6%) | 400 | 0.575 | IL |
| H-1 → L + 1 (92%) | 358 | 0.136 | IL |
| H-6 → L (55%), H-4 → L + 1 (16%) | 299 | 0.093 | IL |
| H-6 → L + 1 (38%), H-8 → L (9%), H-3 → L + 2 (9%) | 278 | 0.078 | IL |
| H-4 → L + 2 (34%), H-12 → L (17%) | 266 | 0.089 | IL |
| 3 | |||
| H → L (90%), H-1 → L (6%) | 475 | 0.827 | IL |
| H-1 → L (90%), H → L (7%) | 431 | 0.658 | IL |
| H → L + 2 (95%) | 372 | 0.540 | IL |
| H-4 → L (83%) | 306 | 0.078 | IL |
| H → L + 9 (48%), H → L + 11 (12%) | 285 | 0.160 | IL |
| H-11 → L (28%), H-2 → L + 6 (26%), H-1 → L + 11 (19%) | 264 | 0.098 | IL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glykos, D.; Tsipis, A.C.; Plakatouras, J.C.; Malandrinos, G. Synthesis, Spectroscopic Characterization, and Photophysical Studies of Heteroleptic Silver Complexes Bearing 2,9-Bis(styryl)-1,10-phenanthroline Ligands and Bis[(2-diphenylphosphino)phenyl] Ether. Inorganics 2024, 12, 131. https://doi.org/10.3390/inorganics12050131
Glykos D, Tsipis AC, Plakatouras JC, Malandrinos G. Synthesis, Spectroscopic Characterization, and Photophysical Studies of Heteroleptic Silver Complexes Bearing 2,9-Bis(styryl)-1,10-phenanthroline Ligands and Bis[(2-diphenylphosphino)phenyl] Ether. Inorganics. 2024; 12(5):131. https://doi.org/10.3390/inorganics12050131
Chicago/Turabian StyleGlykos, Dimitrios, Athanassios C. Tsipis, John C. Plakatouras, and Gerasimos Malandrinos. 2024. "Synthesis, Spectroscopic Characterization, and Photophysical Studies of Heteroleptic Silver Complexes Bearing 2,9-Bis(styryl)-1,10-phenanthroline Ligands and Bis[(2-diphenylphosphino)phenyl] Ether" Inorganics 12, no. 5: 131. https://doi.org/10.3390/inorganics12050131
APA StyleGlykos, D., Tsipis, A. C., Plakatouras, J. C., & Malandrinos, G. (2024). Synthesis, Spectroscopic Characterization, and Photophysical Studies of Heteroleptic Silver Complexes Bearing 2,9-Bis(styryl)-1,10-phenanthroline Ligands and Bis[(2-diphenylphosphino)phenyl] Ether. Inorganics, 12(5), 131. https://doi.org/10.3390/inorganics12050131