
Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand


Institute of Inorganic Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(2), 28; https://doi.org/10.3390/inorganics5020028
Submission received: 30 March 2017
/
Revised: 7 April 2017
/
Accepted: 14 April 2017
/
Published: 20 April 2017
(This article belongs to the Special Issue s-Block Metal Complexes)
Abstract
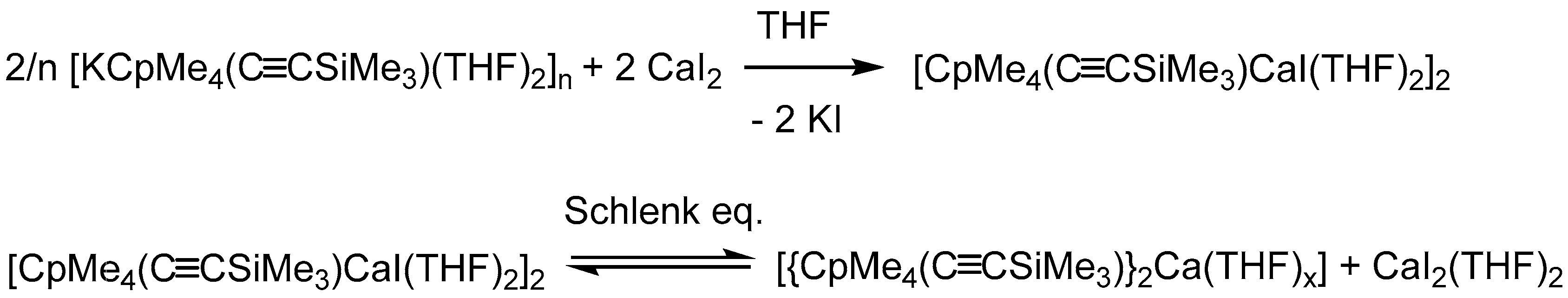
:Sodium, potassium, and calcium compounds of trimethyl((2,3,4,5-tetramethylcyclopentadien-1-yl)ethynyl)silane (CpMe4(C≡CSiMe3)) were synthesized and characterized by X-ray diffraction and standard analytical methods. The sodium derivative was obtained by deprotonation of CpMe4(C≡CSiMe3)H with Na{N(SiMe3)2} to give a monomeric complex [NaCpMe4(C≡CSiMe3)(THF)3]. In a similar reaction, starting from K{N(SiMe3)2} the corresponding potassium compound [KCpMe4(C≡CSiMe3)(THF)2]n, which forms a polymeric super sandwich structure in the solid state, was obtained. Subsequently, salt metathesis reactions were conducted in order to investigate the versatility of the CpMe4(C≡CSiMe3)− ligand in alkaline earth chemistry. The reaction of [KCpMe4(C≡CSiMe3)(THF)2]n with CaI2 afforded the dimeric complex [CaCpMe4(C≡CSiMe3)I(THF)2]2, in which both CpMe4(C≡CSiMe3)Ca units are bridged by iodide in a μ2 fashion. In-depth NMR investigation indicates that [CaCpMe4(C≡CSiMe3)I(THF)2]2 is in a Schlenk equilibrium with [{CpMe4(C≡CSiMe3)}2Ca(THF)x] and CaI2(THF)2, as is already known for [CaCp*I(THF)2].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Cyclopentadienyl salts of the alkali metals are probably one of the most versatile reagents in organometallic chemistry. They have been used for the synthesis of countless cyclopentadienyl complexes. Potassium cyclopentadienyl (KCp) was first reported by J. Thiele, who reacted potassium and cyclopentadiene in benzene [1], while the analogous sodium cyclopentadienyl (NaCp) was discovered approximately 50 years later by the groups of E. O. Fischer [2,3] and K. Ziegler [4]. The alkali metal cyclopentadienyls are generally available either by deprotonation of cyclopentadiene with an alkali metal base such as M{N(SiMe3)2}, MH, MOtBu, MOH or the alkali metal itself [5,6]. Some years ago, we showed that sodium and potassium cyclopentadienyl is most conveniently prepared in a one-pot synthesis directly from alkali metals with neat dicyclopentadiene at elevated temperature [7,8]. Especially in the chemistry of electron poor metals, cyclopentadienyl is often used in the form of its permethylated derivative pentamethylcyclopentadienyl η5-CpMe5 (Cp*) [8,9], because of the higher solubility of the corresponding metal complexes and the enlarged steric demand of the ligand, which prevents polymerization. Furthermore, other derivatives of cyclopentadienyl are easily accessible and increase the versatility of the cyclopentadienyl ligand [10,11,12]. For this reason, we became aware of the ligand trimethysilylethynyltetramethylcyclopentadiene CpMe4(C≡CSiMe3)H. CpMe4(C≡CSiMe3)− has been used before in group 8 chemistry. The postmodification of η5-CpMe4(C≡CSiMe3)− metal complexes may include access to metal acetylides [13,14], metal alkyne complexes [15,16], Sonogashira couplings [17,18,19], click reactions [20,21] and cyclizations [22,23,24]. [LiCpMe4(C≡CSiMe3)] has been generated in situ but, to the best of our knowledge, s-block compounds have not been isolated.
2. Results and Discussion
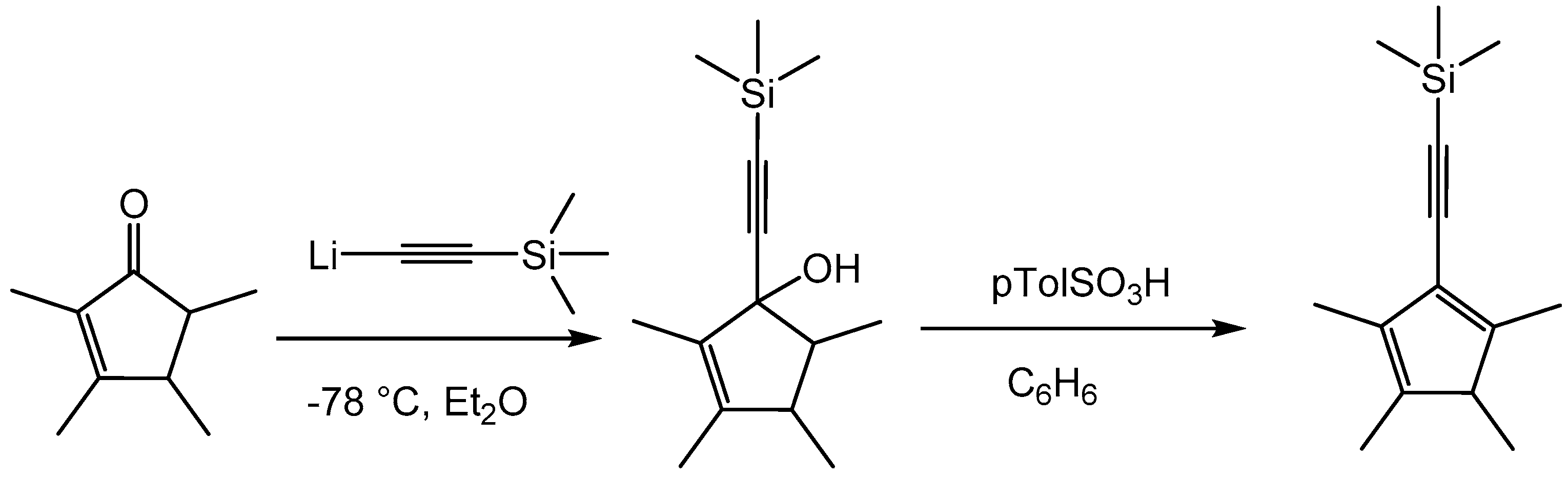
CpMe4(C≡CSiMe3)H was prepared in a modified procedure published by Pudelski et al. [23] (Scheme 1). CpMe4(C≡CSiMe3)H was obtained in an overall yield of 55% as a light yellow oil.
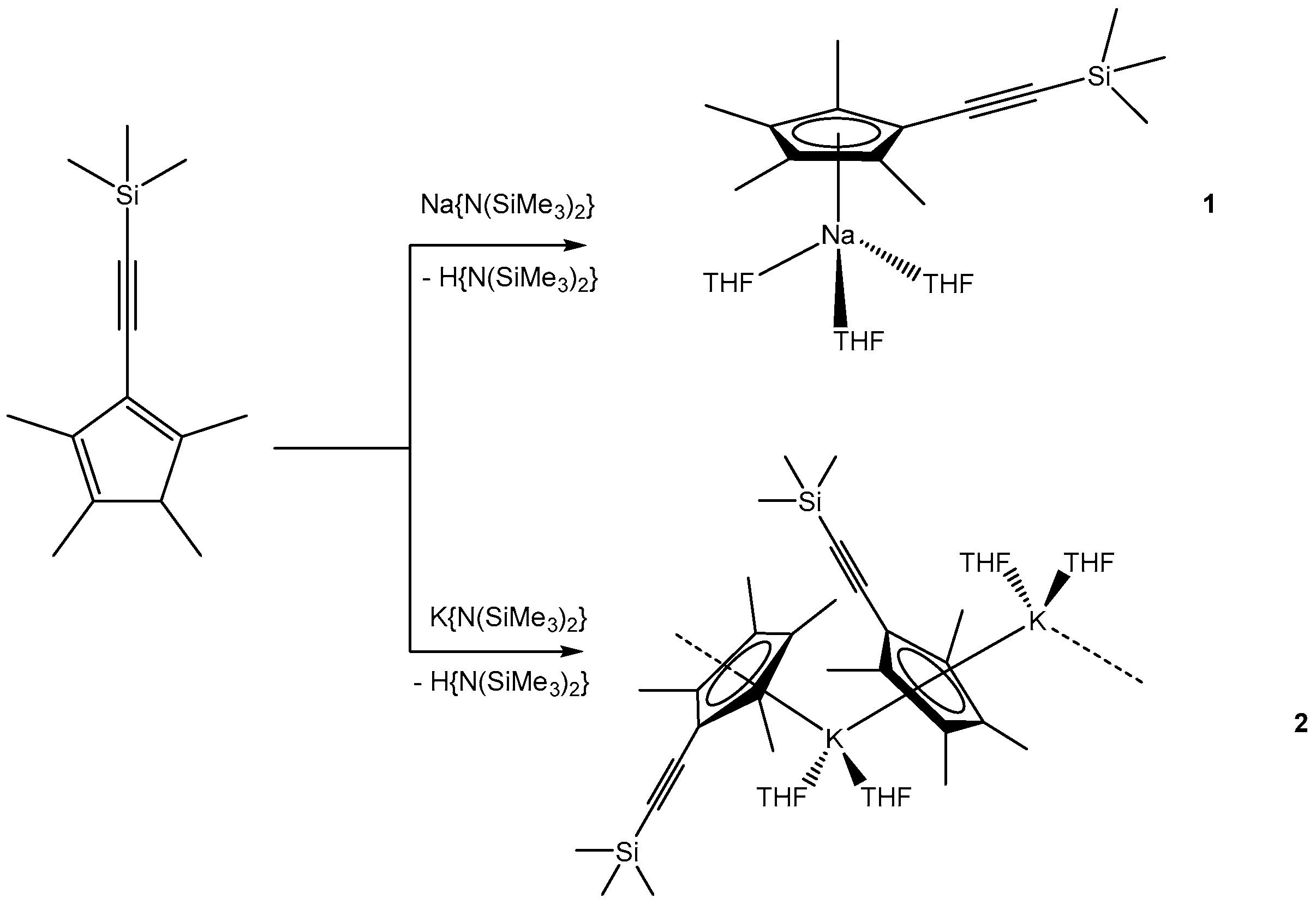
In the first metalation reaction, CpMe4(C≡CSiMe3)H was reacted with Na{N(SiMe3)2} in THF. Upon reaction, the solution turned dark red, indicating the formation of [NaCpMe4(C≡CSiMe3)(THF)3] (1) (Scheme 2). Single crystals suitable for X-ray diffraction formed in 50% yield upon cooling the concentrated solution to −30 °C.
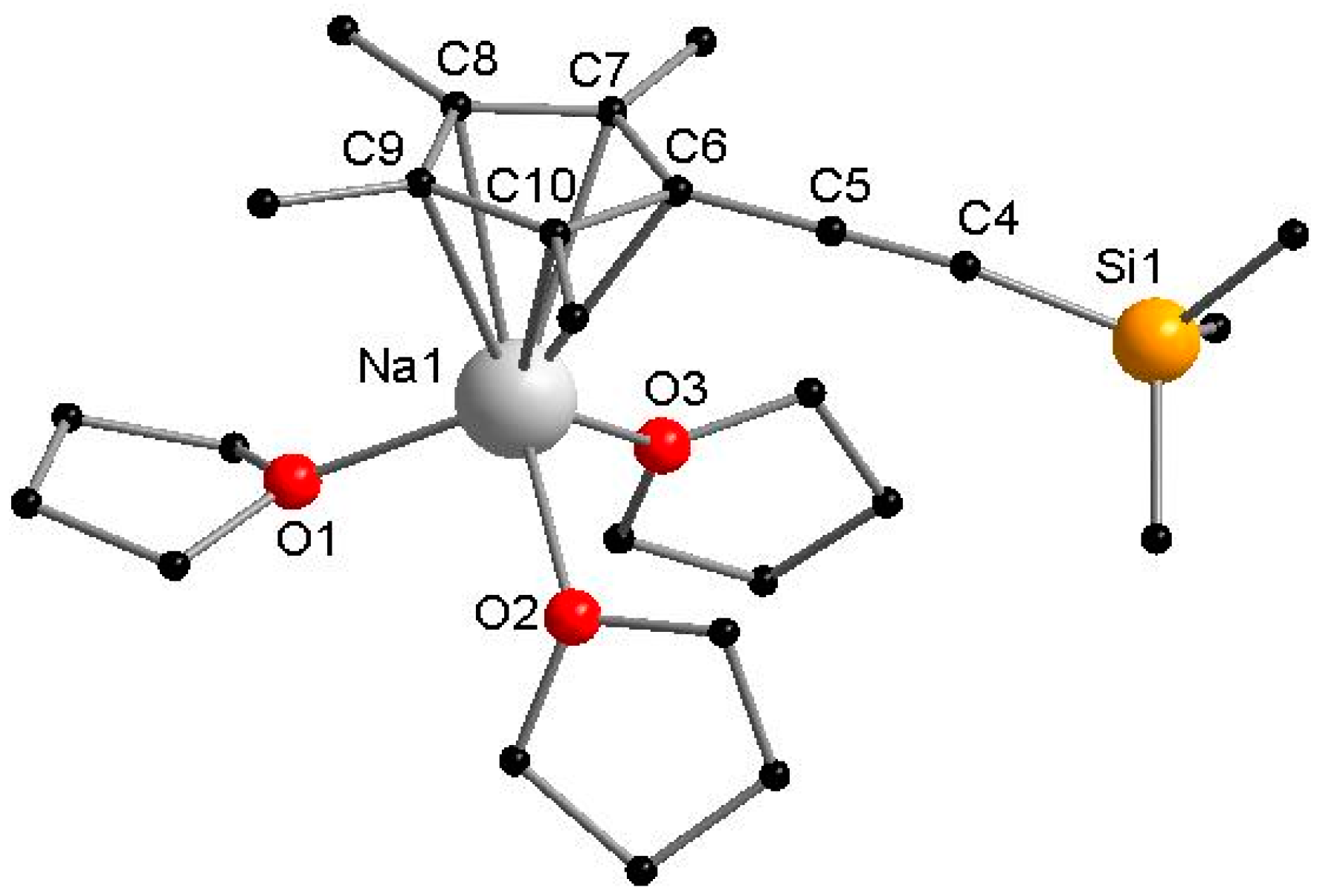
The sodium complex 1 crystallizes in the monoclinic space group P21/c with one molecule in the asymmetric unit (Figure 1). The molecular structure of 1 reveals a monomeric NaCpMe4(C≡CSiMe3) compound in the solid state, in which CpMe4(C≡CSiMe3)− coordinates in a η5 fashion to the metal center. Furthermore, three THF molecules are attached to the sodium atom. The coordination polyhedron thus forms a three-legged piano-stool configuration. The bond distances between the carbon atoms of the five-membered CpMe4(C≡CSiMe3) ring and the sodium atom (Na–C = 2.672–2.736 Å) are slightly elongated compared to NaCp* [25], which is probably caused by the steric demand of the rather larger TMS-ethynyl substituent. The O–Na–O angles average to 96.2°. Compound 1 was also characterized in solution by NMR methods. The resonances of the methyl groups are split into two signals (δ(1H) = 1.89 and 2.02 ppm; δ(13C) = 10.7 and 11.5 ppm). The resonance attributed to the Si(CH3)3 moiety is slightly upfield shifted from 0.22 ppm CpMe4(C≡CSiMe3)H to 0.12 ppm (1) in the 1H NMR spectrum. In the ATR-IR (ATR = Attenuated Total Reflection, IR = Infrared) spectrum, the C≡C triple bond of the ethynyl moiety in 1 shows a stretching band at 2118 cm−1, which is slightly shifted compared to CpMe4(C≡CSiMe3)H (2131 cm−1).
Next, we reacted CpMe4(C≡CSiMe3)H with K{N(SiMe3)2} in THF in order to compare the structural properties of different alkali metal complexes. Following the same synthetic and workup protocol as for 1, we isolated single crystals of [KCpMe4(C≡CSiMe3)(THF)2]n (2) in 19% yield (Scheme 2). The crystals were isolated by decantation from the mother liquor.
Compound 2 forms an infinite zig-zag chain in the solid state (Figure 2). It crystallizes in the chiral orthorhombic space group P212121 with one {KCpMe4(C≡CSiMe3)(THF)2} subunit in the asymmetric unit. No chirality is observed in the super sandwich structure [26]. Investigation of the molecular structure of 2 in the solid state reveals that every potassium ion features a bent metallocene structure similar to the motive found in [KCp*(THF)2]n [27]. Besides the two CpMe4(C≡CSiMe3)− ligands, two molecules of THF are bound to each metal atom. Bond lengths and angles are nearly identical for [KCp*(THF)2]n and 2. The Cp-centroid–K–Cp-centroid angle of 133.33(1)° is slightly smaller than in [KCp*(THF)2]n (137.9°). We suggest that the differences of the structures of compounds 1 and 2 are a result of the different ion radii. In solution, the NMR spectra reveal the expected signals. Thus, two singlets are observed for the protons of the methyl groups at 1.90 and 2.02 ppm in the 1H NMR spectrum. In the IR spectrum, the C≡C bond stretching frequency is detected at 2130 cm−1, which is in the range of CpMe4(C≡CSiMe3)H.
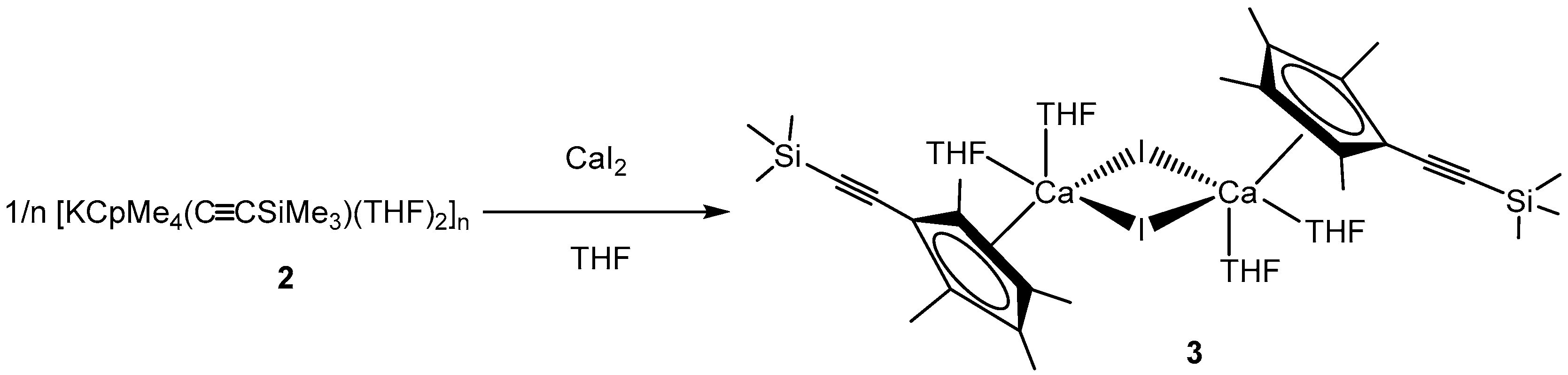
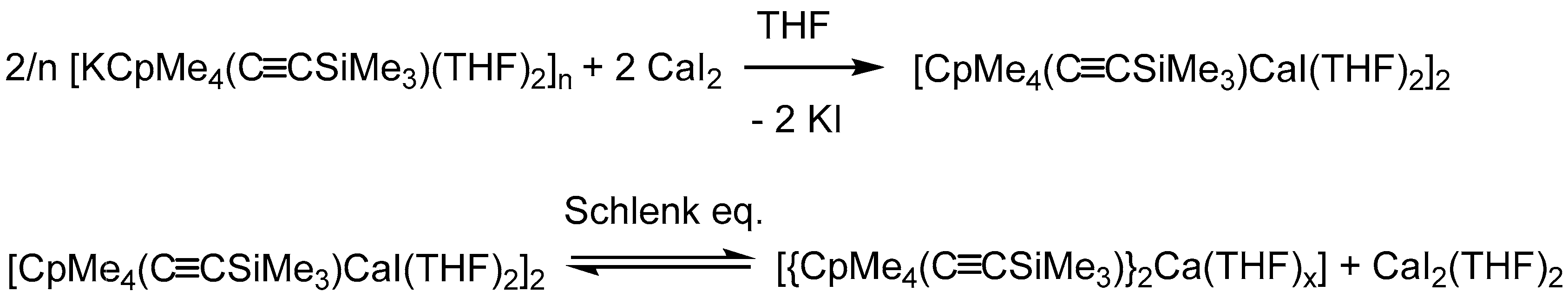
In order to determine their potential in group 2 chemistry, we aimed to investigate salt metathesis reactions with alkaline earth metal halides. Thus, 2 was reacted with CaI2 in a 2:1 ratio in THF to obtain the desired sandwich complex [{CpMe4(C≡CSiMe3)}2Ca(THF)2] as final product. Surprisingly, even after several attempts, only the iodide-bridged dimer [CaCpMe4(C≡CSiMe3)I(THF)2]2 (3) could be isolated as single product by crystallization. After adjusting the stochiometric ratio to 1:1, 3 was isolated as large yellow crystals in 29% yield (Scheme 3).
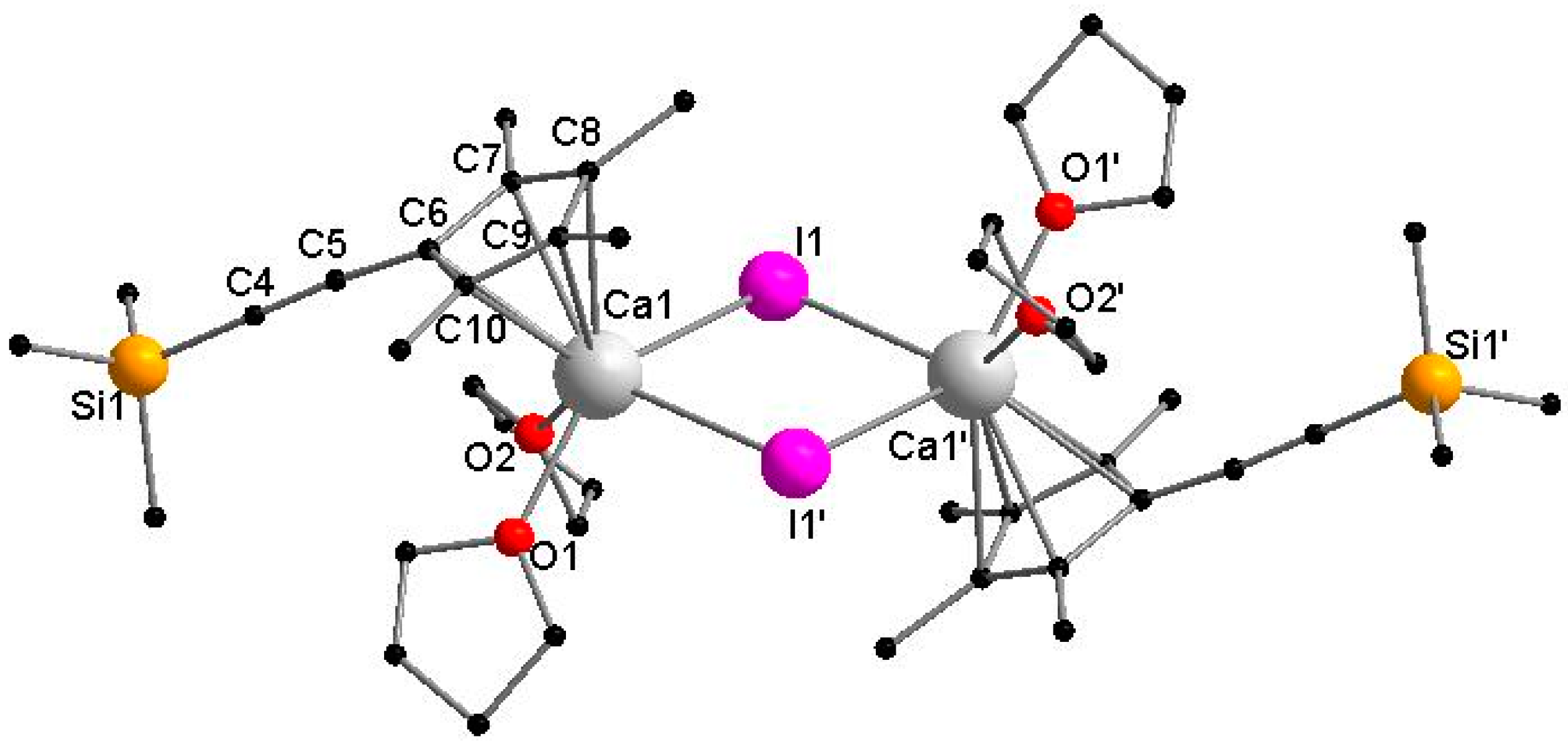
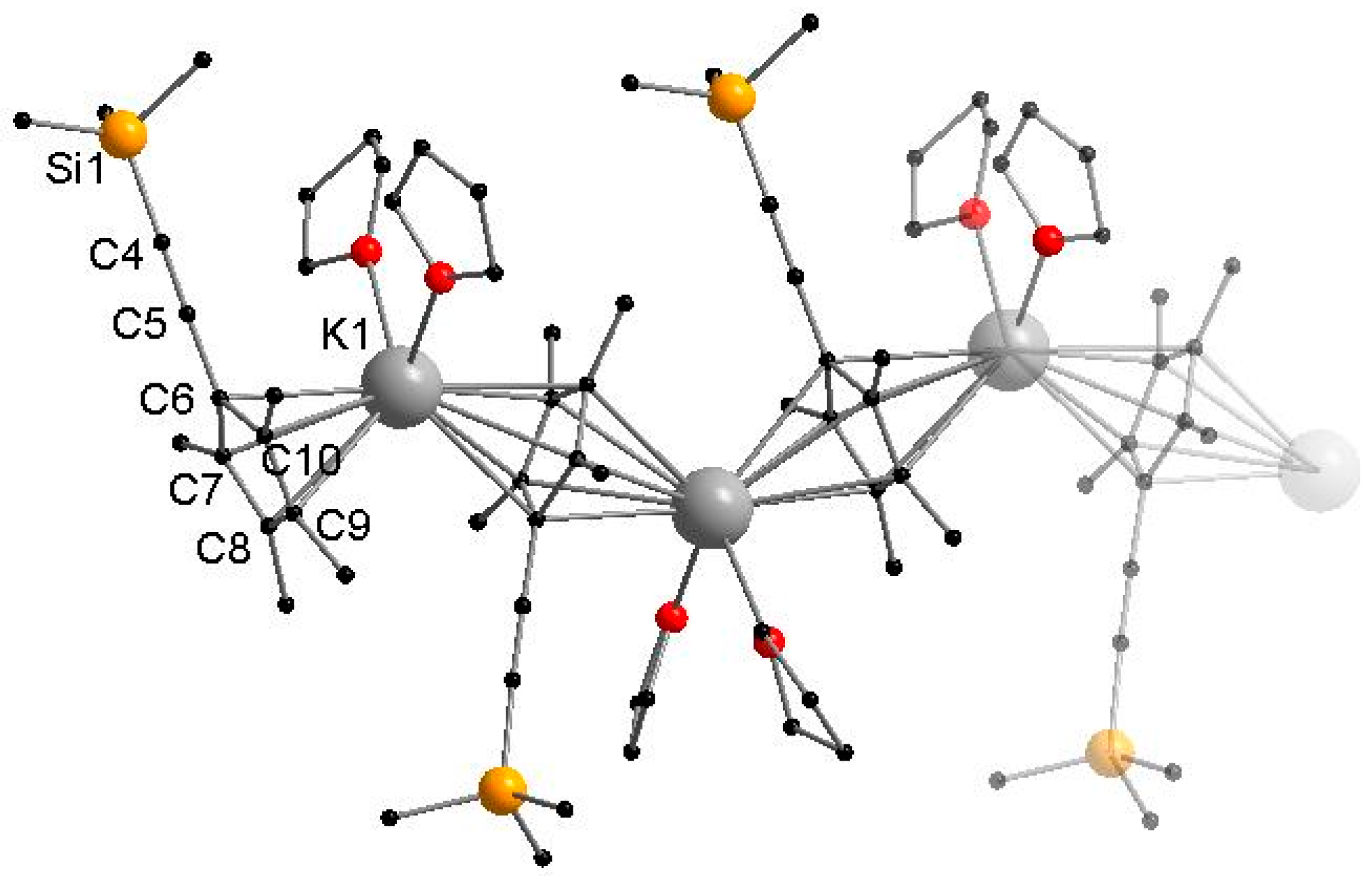
The calcium complex 3 crystallizes from toluene in the monoclinic space group P21/n with half of a molecule in the asymmetric unit. Compound 3 forms a halide-bridged dimer, in which both calcium atoms are coordinated by a single CpMe4(C≡CSiMe3) ring, two THF molecules and two bridging iodine atoms (Figure 3). A crystallographic C2 axis is observed along I and I′. The two bridging iodide anions show a short and a slightly elongated Ca–I bond (Ca1–I1 3.0920(7) Å and Ca1–I1′ 3.2039(7) Å). The average cyclopentadienyl carbon calcium distances in 3 (2.6948 Å) are comparable to those in [Cp*CaI(THF)2]2 (2.67 Å) [28]. As a result of the steric demand, the two ethynyl substituents point to opposite directions. Compared to the sodium compound 1 and potassium compound 2, the C≡C bond stretching frequency in the IR spectrum is shifted to a slightly lower wavenumber (2108 cm−1). In the 1H and 13C NMR spectra of 3 in THF-d8, two sets of signals were identified, although single crystals of 3 were used for these studies. In the 1H NMR spectrum, the methyl protons display four singlet resonances. These signals form two pairs of signals. The integral ratio of the first pair (1.89 ppm, 1.95 ppm) to the second pair (1.98 ppm, 2.04 ppm) can be ascertained to 63:37. In contrast, there is only one resonance for the Si(CH3)3 moieties, which may be a result of overlaid signals. The obvious anisochrony of the resonances of the methyl group suggests a Schlenk equilibrium as observed for [Cp*CaI(THF)2]2. According to Scheme 4, each set can be assigned to either 3 or [{CpMe4(C≡CSiMe3)}2Ca(THF)x], respectively.
According to McCormick et al., separation of the analogous [Cp*CaI(THF)2]2 by solvent extraction is very difficult, since all components of the Schlenk equilibrium have a similar solvation behavior [16]. Thus, isolation can only be accomplished by crystallization. Unfortunately, we were not able to isolate [{CpMe4(C≡CSiMe3)}2Ca(THF)x] as a crystalline material. However, besides 3, the metallocene can be identified by mass spectrometry of a solution of crystalline compound 3 dissolved in THF. By changing the solvent for the 1H NMR spectra from THF-d8 to C6D6 and the temperature, we expected a shift of the Schlenk equilibrium. By using C6D6 instead of THF-d8 as solvent, a downfield shift of the methyl resonance is observed (set 1: 2.03, 2.18 ppm; set 2: 2.35, 2.45 ppm). Furthermore, in 1H NMR in C6D6 at room temperature, the intensity ratio of the two sets of resonances changed to 15:85, whereas at 333 K only two singlets at 2.25 and 2.40 ppm are displayed. These observations indicate a dynamic process in solution. Although loss of THF in organo-alkaline-earth metal complexes is fairly common [29,30] and is also known for organolanthanide systems [31], we could not detect any free THF in NMR experiments.
3. Experimental
3.1. General Procedures
All manipulations were performed under rigorous exclusion of moisture and oxygen in flame-dried Schlenk-type glassware or in an argon-filled MBraun glovebox (Garching, Germany). THF was distilled from potassium and benzophenone prior to use. Hydrocarbon solvents (diethyl ether, n-pentane) were dried using an MBraun solvent purification system (SPS-800). Deuterated solvents were obtained from Carl Roth GmbH (99.5 atom % Deuterium) (Karlsruhe, Germany). NMR spectra were recorded on a BrukerAvance II 300 MHz or Avance 400 MHz (Bruker Biospin, Rheinstetten, Germany). 1H and 13C{1H} chemical shifts were referenced to the residual 1H and 13C{1H} resonances of the deuterated solvents and are reported relative to tetramethylsilane. IR spectra were obtained on a Bruker Tensor 37 (Bruker Optik, Ettlingen, Germany). Elemental analyses were carried out with an Elementar Micro Cube (Elementar Analysensysteme GmbH, Langenselbold, Germany). Mass spectra were recorded on a LTQ Orbitrap XL Q Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) equipped with an HESI II probe. The instrument was calibrated in the m/z range 74–1822 using premixed calibration solutions (Thermo Fisher Scientific, San Jose, CA, USA). A constant spray voltage of 4.6 kV, a dimensionless sheath gas of 8, and a dimensionless auxiliary gas flow rate of 2 were applied. The capillary temperature and the S-lens RF level were set to 320 °C and 62.0, respectively. Trimethyl((2,3,4,5-tetramethylcyclopentadien-1-yl)ethynyl)silane (CpMe4(C≡CSiMe3)H) was prepared according to literature procedures [23]. Na{N(SiMe3)2}, K{N(SiMe3)2} and CaI2 were purchased from Sigma-Aldrich (Schnelldorf, Germany) and used as received.
3.1.1. [NaCpMe4(C≡CSiMe3)(THF)3] (1)
Na{N(SiMe3)2} (462 mg, 2.52 mmol) was dissolved in THF (10 mL) and CpMe4(C≡CSiMe3)H (550 mg, 2.52 mmol) was slowly added by using a syringe. The solution instantly turned dark red upon addition. After complete addition, the solvent was reduced to approximately 5 mL and the flask was stored at −30°C. Colorless needles of [NaCpMe4(C≡CSiMe3)(THF)3] formed after 6 h. The needles were filtered off, washed with precooled n-hexane and dried under vacuum. Yield: 305 mg (50%, single crystals).
1H NMR (THF-d8, 300 MHz): δ [ppm] = 0.07 (s, 9H, Si(CH3)3), 1.67–1.71 (m, THF), 1.89 (s, 6H, CpMe4(C≡CSiMe3)–CH3), 2.02 (s, 6H, CpMe4(C≡CSiMe3)–CH3), 3.53–3.56 (m, THF). 13C{1H} NMR (THF-d8, 75 MHz): δ [ppm] = 0.79 (Si(CH3)3), 10.7 (CpMe4(C≡CSiMe3)–CH3), 11.5 (CpMe4(C≡CSiMe3)–CH3), 88.2 (C≡C), 92.6 (C≡C), 109.2 (C–CH3), 113.3 (C–CH3), 113.4 (C–C≡C). IR: ῦ (cm−1) = 2959 (w), 2118 (w), 1586 (vw), 1438 (w), 1376 (vw), 1247 (s), 1078 (w), 996 (w), 859 (w), 837 (vs), 757 (s), 695 (w), 663 (w), 527 (vw). Elemental Analysis: calcd. (%) for C26H45NaO3Si: C 68.38, H 9.93; found: C 67.60, H 8.94.
3.1.2. [KCpMe4(C≡CSiMe3)(THF)2]n (2)
[KCpMe4(C≡CSiMe3)(THF)2]n was synthesized in a similar way as [NaCpMe4(C≡CSiMe3)(THF)3] from K{N(SiMe3)2} (435 mg, 2.18 mmol) and CpMe4(C≡CSiMe3)H (476 mg, 2.18 mmol). Yield: 105 mg (19%, single crystals).
1H NMR (THF-d8, 300 MHz): δ [ppm] = 0.10 (s, 9H, Si(CH3)3), 1.71–1.75 (m, THF), 1.90 (s, 6H, CpMe4(C≡CSiMe3)–CH3), 2.02 (s, 6H, CpMe4(C≡CSiMe3)–CH3), 3.57–3.59 (m, THF). 13C{1H} NMR (THF-d8, 75 MHz): δ [ppm] = 0.87 (Si(CH3)3), 10.6 (CpMe4(C≡CSiMe3)–CH3), 11.4 (CpMe4(C≡CSiMe3)–CH3), 88.3 (C≡C), 93.5 (C≡C), 109.9 (C–CH3), 114.2 (C–CH3), 128.0 (C–C≡C). IR: ῦ (cm−1) = 2961 (w), 2129 (w), 1581 (w), 1422 (w), 1376 (vw), 1247 (s), 1073 (w), 861 (w), 838 (vs), 757 (s), 696 (w), 663 (s), 528 (w). Elemental Analysis: calcd. (%) for C22H37KO2Si: C 65.94, H 9.31; found: C 65.86, H 8.217.
3.1.3. [CpMe4(C≡CSiMe3)CaI(THF)2]2 (3)
[KCpMe4(C≡CSiMe3)(THF)2]n (312 mg, 0.78 mmol) and CaI2 (229 mg, 0.78 mmol) were placed in a Schlenk flask and THF (10 mL) was added. The mixture was stirred overnight at room temperature and all volatiles were removed under reduced pressure. The residue was extracted with toluene (10 mL) and filtered. The orange solution was concentrated to approximately 5 mL and stored at −30 °C. Blocks of [CpMe4(C≡CSiMe3)CaI(THF)2]2 formed within a period of one week. The crystals were filtered off, washed with n-pentane (5 mL) and dried under vacuum. Yield: 121 mg (29%, single crystals).
1H NMR (THF-d8, 300 MHz): δ [ppm] = 0.11 (s, Si(CH3)3), 1.67–1.71 (m, THF), 1.89 (s, CpMe4(C≡CSiMe3)a–CH3), 1.95 (s, CpMe4(C≡CSiMe3)a–CH3), 1.98 (s, CpMe4(C≡CSiMe3)b–CH3), 2.04 (s, CpMe4(C≡CSiMe3)b–CH3), 3.53–3.55 (m, THF). 1H NMR (C6D6, 300 MHz): δ [ppm] = 0.29 (s, Si(CH3)3), 1.40–1.49 (m, THF), 2.03 (s, CpMe4(C≡CSiMe3)a–CH3), 2.18 (s, CpMe4(C≡CSiMe3)a–CH3), 2.35 (s, CpMe4(C≡CSiMe3)b–CH3), 2.45 (s, CpMe4(C≡CSiMe3)b–CH3), 3.77–3.91 (m, THF). 13C{1H} NMR (THF-d8, 75 MHz)*: δ [ppm] = 0.07 (Si(CH3)3), 10.8 (CpMe4(C≡CSiMe3)a–CH3), 11.5 (CpMe4(C≡CSiMe3)b–CH3), 11.6 (CpMe4(C≡CSiMe3)b–CH3), 11.7 (CpMe4(C≡CSiMe3)a–CH3), 91.8, 97.8, 98.1, 107.6, 114.6, 116.1, 117.8, 118.0. 13C{1H} NMR (C6D6, 75 MHz) (some signals could not be assigned to the corresponding nuclei): δ [ppm] = 0.75 (Si(CH3)3), 10.9 (CpMe4(C≡CSiMe3)a–CH3), 11.8 (CpMe4(C≡CSiMe3)a–CH3), 12.4 (CpMe4(C≡CSiMe3)b–CH3), 12.5 (CpMe4(C≡CSiMe3)b–CH3), 86.9, 92.6, 99.5, 107.0, 115.6, 117.3, 119.4. Elemental Analysis: calcd. (%) for C44H74Ca2I2O4Si2: C 49.99, H 7.06; found: C 49.58, H 7.105. ESI-MS: m/z = [[CaCpMe4(C≡CSiMe3)I(THF)2]2–SiMe4 − H+] = calcd. 967.168; found 966.906, m/z = [CaCpMe4(C≡CSiMe3)2–SiMe4 + H+] = calcd. 387.182; found 387.142.
3.2. X-ray Crystallographic Studies of 1–3
Suitable crystals 1–3 were covered in mineral oil (Aldrich) and mounted onto a glass fiber. The crystals were transferred directly into the cold stream of a Stoe IPDS 2 or StadiVari diffractometer (STOE & Cie GmbH, Darmstadt, Germany).
All structures were solved by using the program SHELXS/T [32]. The remaining non-hydrogen atoms were located from successive difference Fourier map calculations. The refinements were carried out by using full-matrix least-squares techniques on F2 by using the program SHELXL [32]. The hydrogen atom contributions of all of the compounds were calculated, but not refined. In each case, the locations of the largest peaks in the final difference Fourier map calculations, as well as the magnitude of the residual electron densities, were of no chemical significance.
3.2.1. [NaCpMe4(C≡CSiMe3)(THF)3]
C26H45NaO3Si, Mr = 456.70, monoclinic, P21/c (No. 14), a = 8.1123(16) Å, b = 14.126(3) Å, c = 24.766(5) Å, β = 99.36(3)°, V = 2800.4(10) Å3, T = 100 K, Z = 4, Z' = 1, μ(Mo Kα) = 0.121, 12,871 reflections measured, 5468 unique (Rint = 0.1124) which were used in all calculations. The final wR2 was 0.2846 (all data) and R1 was 0.0931 (I > 4σ(I)).
3.2.2. [KCpMe4(C≡CSiMe3)(THF)2]n (2)
C22H37KO2Si, Mr = 400.70, orthorhombic, P212121 (No. 19), a = 10.287(2) Å, b = 11.429(2) Å, c = 20.495(4) Å, V = 2409.6(8) Å3, T = 100 K, Z = 4, Z' = 1, μ(Mo Kα) = 0.282, 19,379 reflections measured, 4731 unique (Rint = 0.0584) which were used in all calculations. The final wR2 was 0.1043 (all data) and R1 was 0.0414 (I > 4σ(I)).
3.2.3. [CaCpMe4(C≡CSiMe3)I(THF)2]2 (3)
C44H74Ca2I2O4Si2, Mr = 1057.17, monoclinic, P21/n (No. 14), a = 9.4746(4) Å, b = 14.9924(8) Å, c = 21.7467(8) Å, β = 101.490(3)°, V = 3027.1(2) Å3, T = 220 K, Z = 2, Z' = 0.5, μ(Mo Kα) = 1.278, 14,525 reflections measured, 5910 unique (Rint = 0.0286) which were used in all calculations. The final wR2 was 0.1181 (all data) and R1 was 0.0392 (I > 4σ(I)).
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre and the relevant codes are: 1541197–1541199. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: (+(44)1223-336-033; email: [email protected]).
4. Conclusions
The trimethylsilylethynyl-substituted cyclopentadienyl ligand CpMe4(C≡CSiMe3)− was introduced into the chemistry of the s-block metals. The sodium and potassium derivatives were obtained by deprotonation of the corresponding cyclopentadiene with Na{N(SiMe3)3} and K{N(SiMe3)3}. Whereas the sodium compound is monomeric, the potassium species forms a zig-zag chain in the solid state. Determination of the versatility in alkaline earth chemistry was carried out by subsequent reaction of compound 2 with CaI2, which resulted in the iodide bridged dimer [CpMe4(C≡CSiMe3)CaI(THF)2]2. The solid-state structure shows similarities to organolanthanide compounds, whereas in solution a Schlenk equilibrium typical for heavier organometallic group 2 compounds was observed. The corresponding metallocene [{CpMe4(C≡CSiMe3)}2Ca(THF)x] was detected by NMR spectroscopy and mass spectrometry but could not be isolated as a crystalline solid.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/2/28/s1, Crystallographic data, NMR Spectra, IR Spectra, Mass spectra, cif and cif-checked files.
Author Contributions
Tim Seifert and Peter W. Roesky conceived and designed the experiments, analyzed the data and wrote the paper; Tim Seifert performed the experiments; Peter W. Roesky contributed reagents/materials/analysis tools.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Thiele, J. Ueber Abkömmlinge des Cyclopentadiëns. Eur. J. Inorg. Chem. 1901, 34, 68–71. [Google Scholar] [CrossRef]
- Fischer, E.; Jira, R.Z. Di-cyclopentadienyl-kobalt (II). Naturforschung B 1953, 8, 327–328. [Google Scholar]
- Fischer, E.; Hafner, W.; Stahl, H.Z. Über Cyclopentadienyl-metall-carbonyl-wasserstoffe des Chroms, Molybdäns und Wolframs. Anorg. Allg. Chem. 1955, 282, 47–62. [Google Scholar] [CrossRef]
- Ziegler, K.; Froitzheim-Kühlhorn, H.; Hafner, K. Metallorganische Verbindungen XXI: Metallverbindungen des Cyclopentadiens. Chem. Ber. 1956, 89, 434–443. [Google Scholar] [CrossRef]
- Panda, T.K.; Gamer, M.T.; Roesky, P.W. An improved synthesis of sodium and potassium cyclopentadienide. Organometallics 2003, 22, 877–878. [Google Scholar] [CrossRef]
- Borisov, A.; Makhaev, V. The synthesis of potassium and sodium cyclopentadienides by the interaction of cyclopentadiene with alkalis. Organomet. Chem. USSR 1989, 2, 680. [Google Scholar]
- Panda, T.K.; Gamer, M.T.; Roesky, P.W.; Yoo, H.; Berry, D.H. Sodium and Potassium Cyclopentadienide. In Inorganic Syntheses; John Wiley & Sons, Inc.: New Jersey, NJ, USA, 2014; Volume 36. [Google Scholar]
- Kohl, F.X.; Jutzi, P. An improved synthesis of 1,2,3,4,5-pentamethylcyclopentadiene. J. Organomet. Chem. 1983, 243, 119–121. [Google Scholar] [CrossRef]
- DeVries, L. Communications-Preparation of 1,2,3,4,5-Pentamethyl-cyclopentadiene, 1,2,3,4,5,5-Hexamethyl-cyclopentadiene, and 1,2,3,4,5-Pentamethyl-cyclopentadienylcarbinol. J. Org. Chem. 1960, 25, 1838. [Google Scholar] [CrossRef]
- Sawtelle, S.M.; Johnston, R.F.; Cook, C.C. An investigation of electron transfer properties of (Cp)Mn(CO)3 and substituted Cp derivatives. Inorg. Chim. Acta 1994, 221, 85–92. [Google Scholar] [CrossRef]
- Ye, B.; Cramer, N. Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C–H functionalization. Science 2012, 338, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.P.; Norton, J.R.; Buccella, D.; Sites, L.A.; Kleinbach, S.S.; Jarem, D.A.; Bocage, K.M.; Nataro, C. Synthesis, Electrochemistry, and Reactivity of Half-Sandwich Ruthenium Complexes Bearing Metallocene-Based Bisphosphines. Organometallics 2009, 28, 3804–3814. [Google Scholar] [CrossRef]
- Powell, C.E.; Humphrey, M.G. Nonlinear optical properties of transition metal acetylides and their derivatives. Coord. Chem. Rev. 2004, 248, 725–756. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Lo, K.K.; Wong, K.M.C. Luminescent polynuclear metal acetylides. J. Organomet. Chem. 1999, 578, 3–30. [Google Scholar] [CrossRef]
- Dias, H.R.; Flores, J.A.; Wu, J.; Kroll, P. Monomeric copper(I), silver(I), and gold(I) alkyne complexes and the coinage metal family group trends. J. Am. Chem. Soc. 2009, 131, 11249–11255. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Dash, C.; Celik, M.A.; Yousufuddin, M.; Frenking, G.; Dias, H.R. Tris(alkyne) and Bis(alkyne) Complexes of Coinage Metals: Synthesis and Characterization of (cyclooctyne)3M+ (M = Cu, Ag) and (cyclooctyne)2Au+ and Coinage Metal (M = Cu, Ag, Au) Family Group Trends. Organometallics 2013, 32, 3135–3144. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Wang, L.; Pagni, R.M. Sonogashira coupling and cyclization reactions on alumina: A route to aryl alkynes, 2-substituted-benzo [b] furans and 2-substituted-indoles. Tetrahedron 2001, 57, 8017–8028. [Google Scholar] [CrossRef]
- Yi, C.; Hua, R. Efficient copper-free PdCl2(PCy3)2-catalyzed sonogashira coupling of aryl chlorides with terminal alkynes. J. Org. Chem. 2006, 71, 2535–2537. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Dai, M.; Chen, J.; Yang, Z. Copper-free Sonogashira coupling reaction with PdCl2 in water under aerobic conditions. J. Org. Chem. 2005, 70, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC)“click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Crowley, J.D.; Bandeen, P.H. A multicomponent CuAAC “click” approach to a library of hybrid polydentate 2-pyridyl-1,2,3-triazole ligands: New building blocks for the generation of metallosupramolecular architectures. Dalton Trans. 2010, 39, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, H.; Lin, C.; Liu, Z.; Wang, C.; Zhang, Y. Cobalt-Catalyzed Cyclization of Aliphatic Amides and Terminal Alkynes with Silver-Cocatalyst. J. Am. Chem. Soc. 2015, 137, 12990–12996. [Google Scholar] [CrossRef] [PubMed]
- Pudelski, J.K.; Callstrom, M.R. Structure, reactivity, and electronic properties of [4]ferrocenophanes and [4]ruthenocenophanes prepared via a novel heteroannular cyclization reaction. Organometallics 1994, 13, 3095–3109. [Google Scholar] [CrossRef]
- Yao, X.; Li, C.J. Water-Triggered and Gold(I)-Catalyzed Cascade Addition/Cyclization of Terminal Alkynes with ortho-Alkynylaryl Aldehyde. Org. Lett. 2006, 8, 1953–1955. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, C.; Dinnebier, R.E.; Olbrich, F.; Smaalen, S.V. Disordered crystal structure of pentamethylcyclopentadienylsodium as seen by high-resolution X-ray powder diffraction. Acta Crystallogr. Sect. B Struct. Sci. 2001, 57, 673–679. [Google Scholar] [CrossRef]
- Pidcock, E. Achiral molecules in non-centrosymmetric space groups. Chem. Commun. 2005, 3457–3459. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Brady, J.C.; Fujimoto, C.H.; Giarikos, D.G.; Ziller, J.W. The bent metallocene geometries of potassium polyalkyl cyclopentadienyl THF solvates: Monosolvated [(THF)K(μ-C5Me5)]n, disolvated [(THF)2K(μ-C5Me5)]n and the tethered olefin complex [(THF)K(μ-C5Me4SiMe2CH2CH=CH2)]n. J. Organomet. Chem. 2002, 649, 252–257. [Google Scholar] [CrossRef]
- McCormick, M.J.; Sockwell, S.C.; Davies, C.E.; Hanusa, T.P.; Huffman, J.C. Synthesis of a monopentamethylcyclopentadienyl halide complex of calcium. The X-ray crystal structure of [(Me5C5)Ca(μ-I)(THF)2]2. Organometallics 1989, 8, 2044–2049. [Google Scholar] [CrossRef]
- McCormick, M.; Williams, R.; Levine, L.; Hanusa, T. Solution synthesis of calcium, strontium and barium metallocenes. Polyhedron 1988, 7, 725–730. [Google Scholar] [CrossRef]
- Westerhausen, M.Z. Recent Developments in the Organic Chemistry of Calcium–An Element with Unlimited Possibilities in Organometallic Chemistry? Anorg. Allg. Chem. 2009, 635, 13–32. [Google Scholar] [CrossRef]
- Evans, W.J.; Meadows, J.H.; Hanusa, T.P. Organolanthanide and organoyttrium hydride chemistry. 6. Direct synthesis and proton NMR spectral analysis of the trimetallic yttrium and yttrium–zirconium tetrahydride complexes, {[(C5H5)2YH]3H}{Li(THF)4} and {[(CH3C5H4)2YH]2[(CH3C5H4)2ZrH]H}. J. Am. Chem. Soc. 1984, 106, 4454–4460. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Preparation of the ligand CpMe4(C≡CSiMe3)H [23].
Scheme 1.
Preparation of the ligand CpMe4(C≡CSiMe3)H [23].
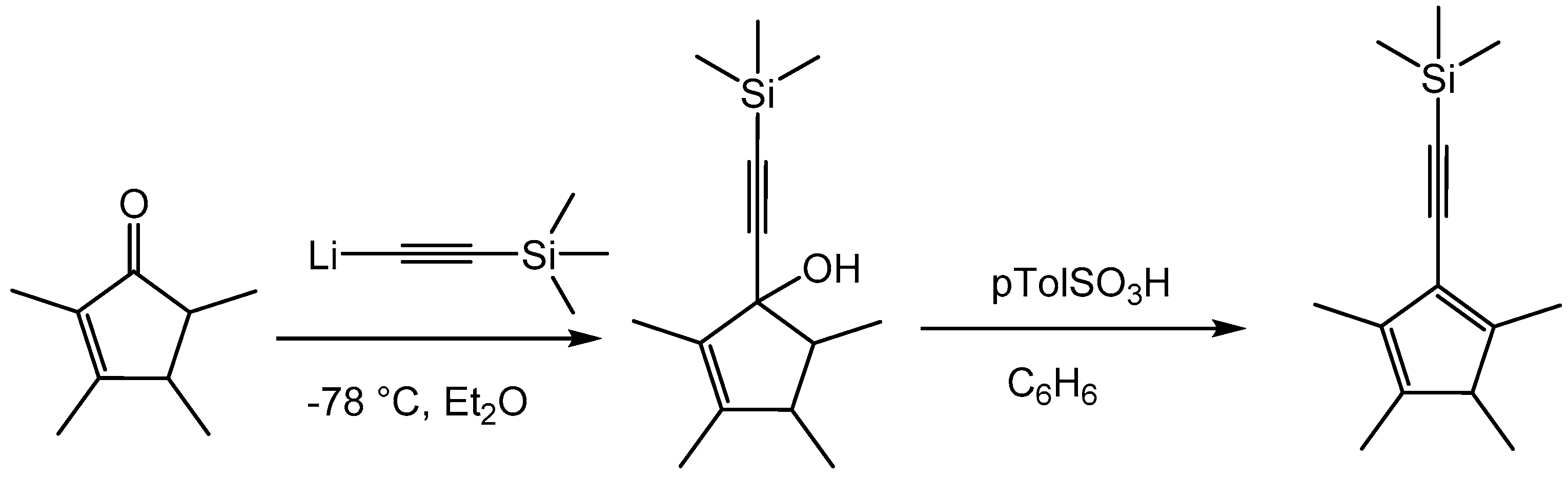
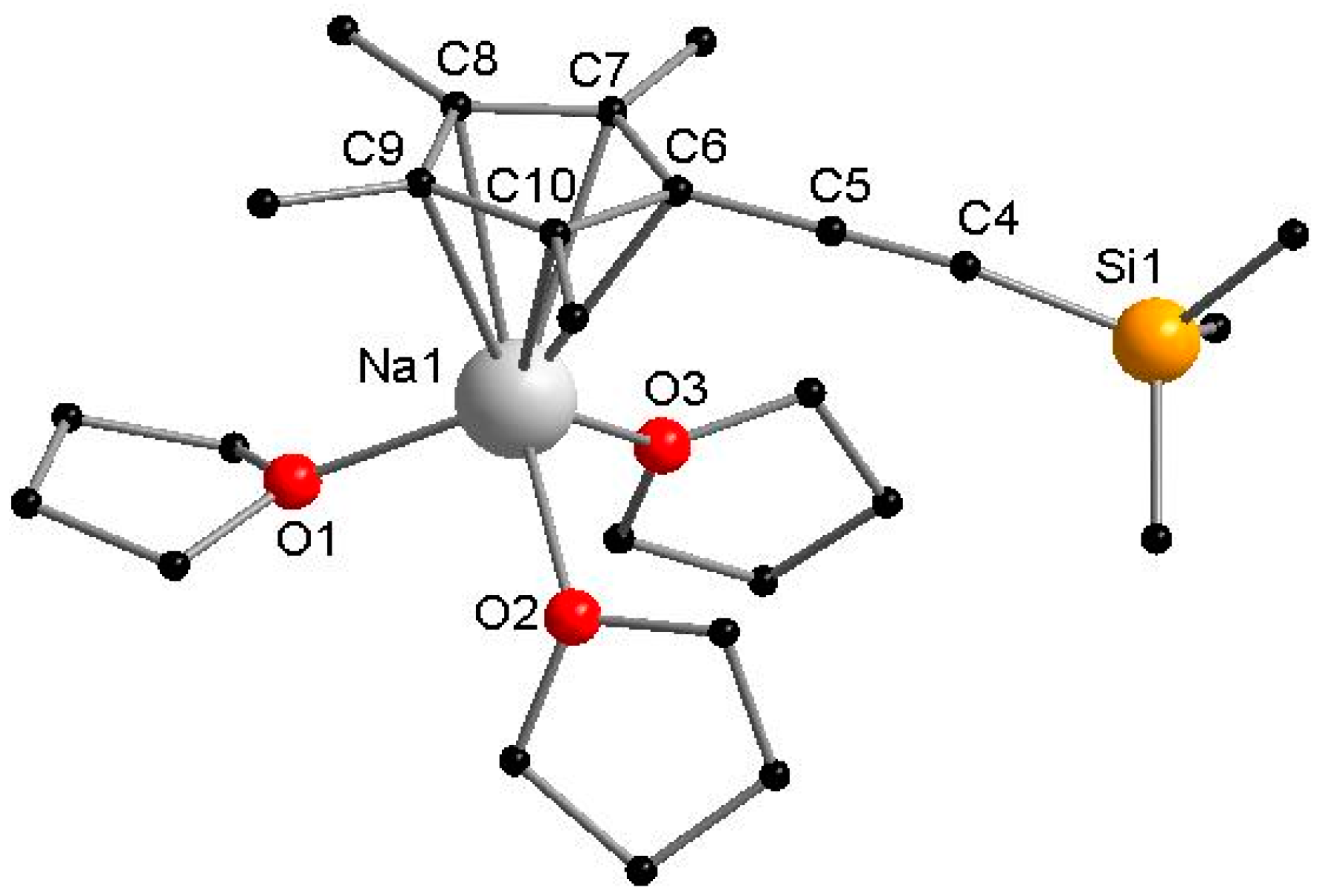
Figure 1.
Molecular structure of 1 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Na1–C6 2.691(4), Na1–C7 2.673(4), Na1–C8 2.672(4), Na1–C9 2.717(4), Na1–C10 2.736(4), C4–C5 1.223(6), Na1–O1 2.289(3), Na1–O2 2.321(3), Na1–O3 2.293(3), O1–Na1–O2 96.70(2), O2–Na1–O3 95.75(12), O3–Na1–O1 96.14(12).
Figure 1.
Molecular structure of 1 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Na1–C6 2.691(4), Na1–C7 2.673(4), Na1–C8 2.672(4), Na1–C9 2.717(4), Na1–C10 2.736(4), C4–C5 1.223(6), Na1–O1 2.289(3), Na1–O2 2.321(3), Na1–O3 2.293(3), O1–Na1–O2 96.70(2), O2–Na1–O3 95.75(12), O3–Na1–O1 96.14(12).
Scheme 2.
Synthesis of [NaCpMe4(C≡CSiMe3)(THF)3] and [KCpMe4(C≡CSiMe3)(THF)2]n.
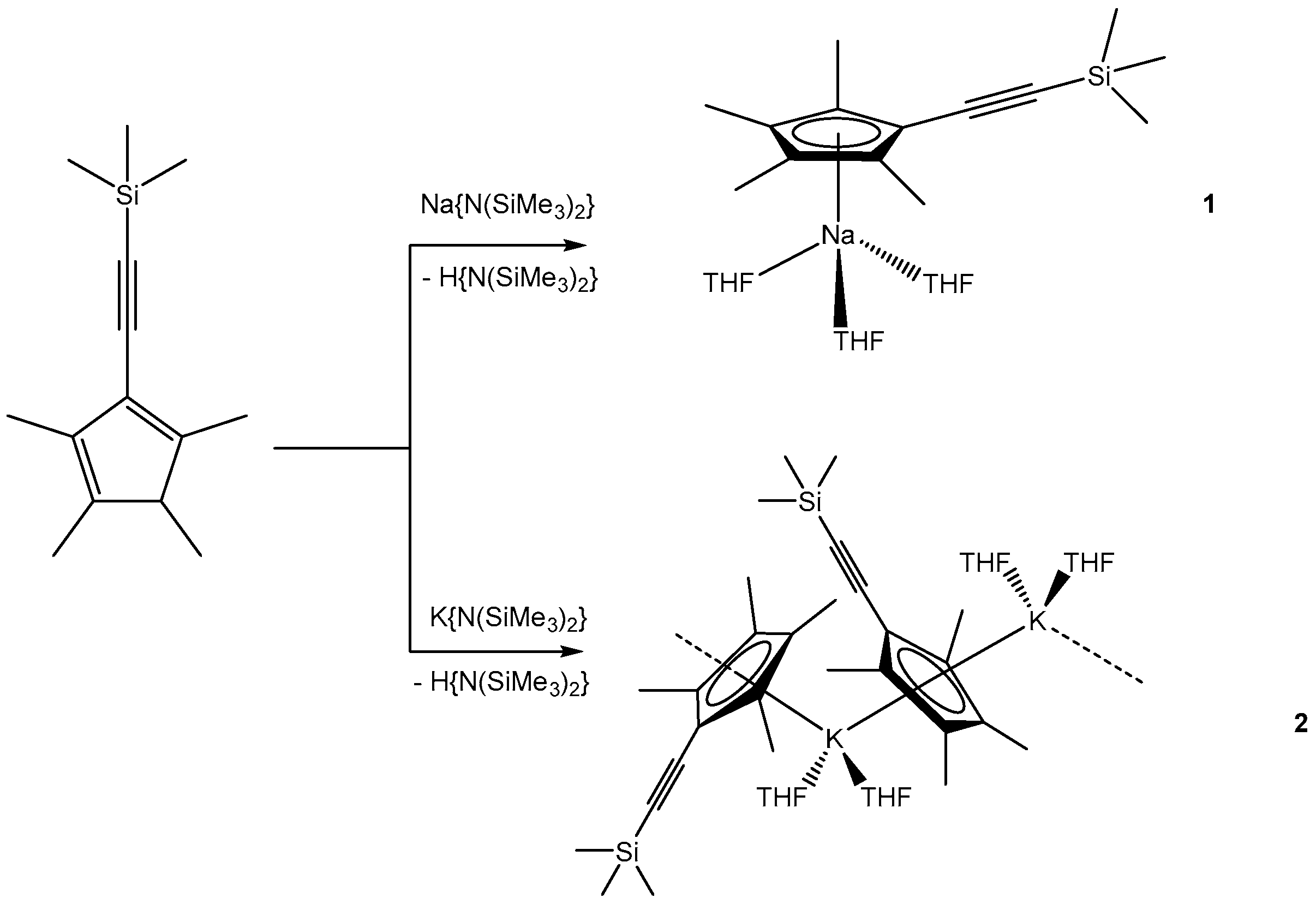
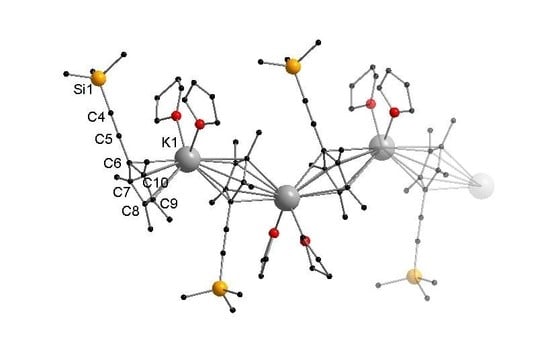
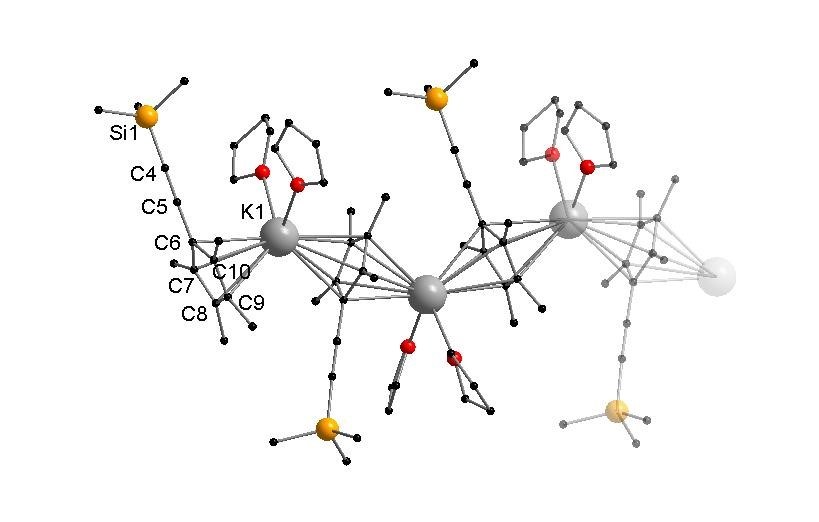
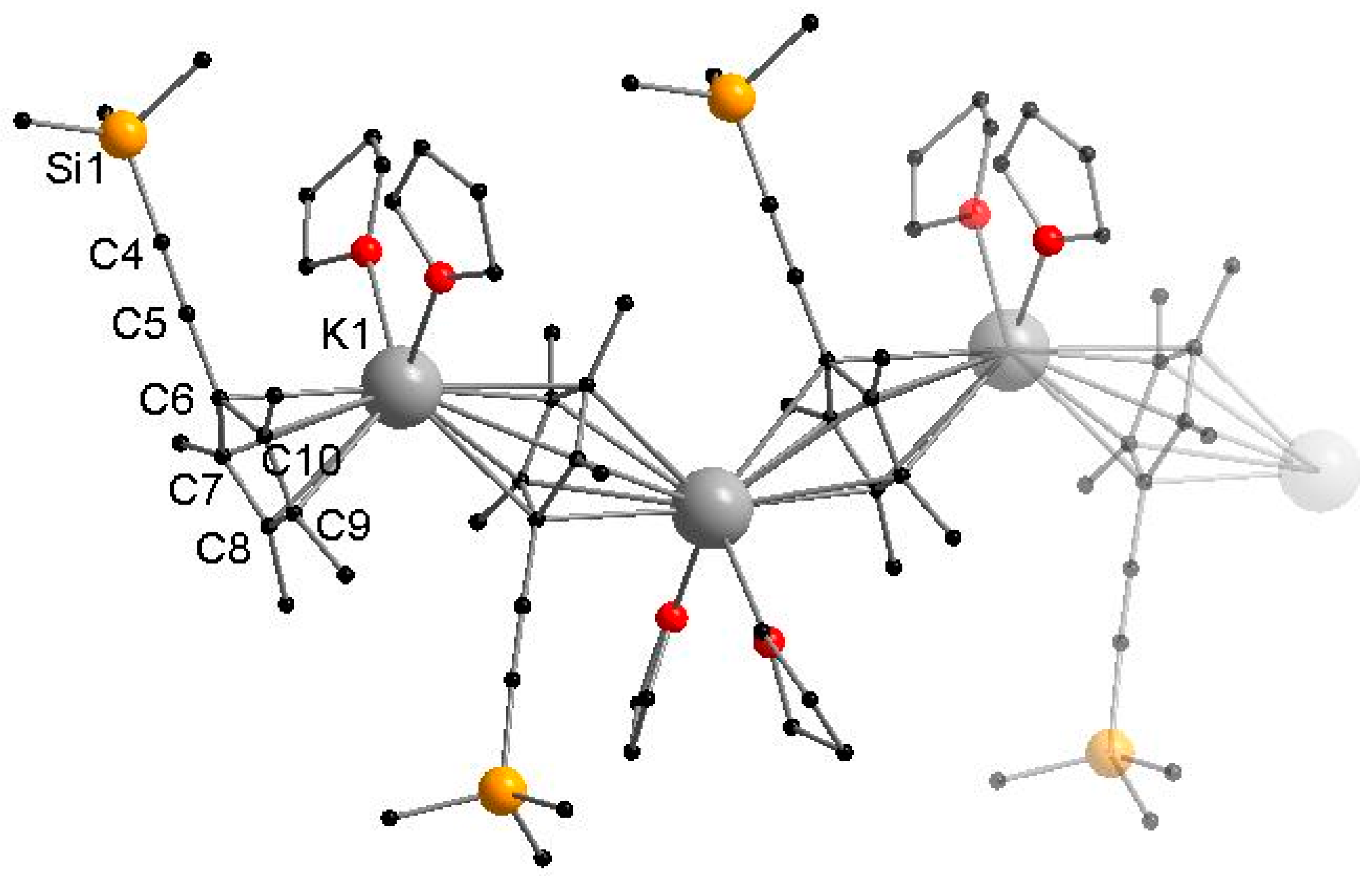
Figure 2.
Cutout of the molecular structure of 2 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: K1–C6 3.075(3), K1–C6′ 3.020(3), K1–C7 3.020(3), K1–C7′ 3.103(3), K1–C8 3.054(3), K1–C8′ 3.070(3), K1–C9 3.002(3), K1–C9′ 3.054(3), K1–C10 3.036(3), K1–C10′ 3.011(3), C4–C5 1.212(5), K1–O1 2.796(3), K1–O2 2.720(2), CpMe4(C≡CSiMe3)-Centroid–K1–CpMe4(C≡CSiMe3)-Centroid 133.33(1), O1–K1–O2 90.20(8).
Figure 2.
Cutout of the molecular structure of 2 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: K1–C6 3.075(3), K1–C6′ 3.020(3), K1–C7 3.020(3), K1–C7′ 3.103(3), K1–C8 3.054(3), K1–C8′ 3.070(3), K1–C9 3.002(3), K1–C9′ 3.054(3), K1–C10 3.036(3), K1–C10′ 3.011(3), C4–C5 1.212(5), K1–O1 2.796(3), K1–O2 2.720(2), CpMe4(C≡CSiMe3)-Centroid–K1–CpMe4(C≡CSiMe3)-Centroid 133.33(1), O1–K1–O2 90.20(8).
Scheme 3.
Conversion of 2 with CaI2 yields in 3.
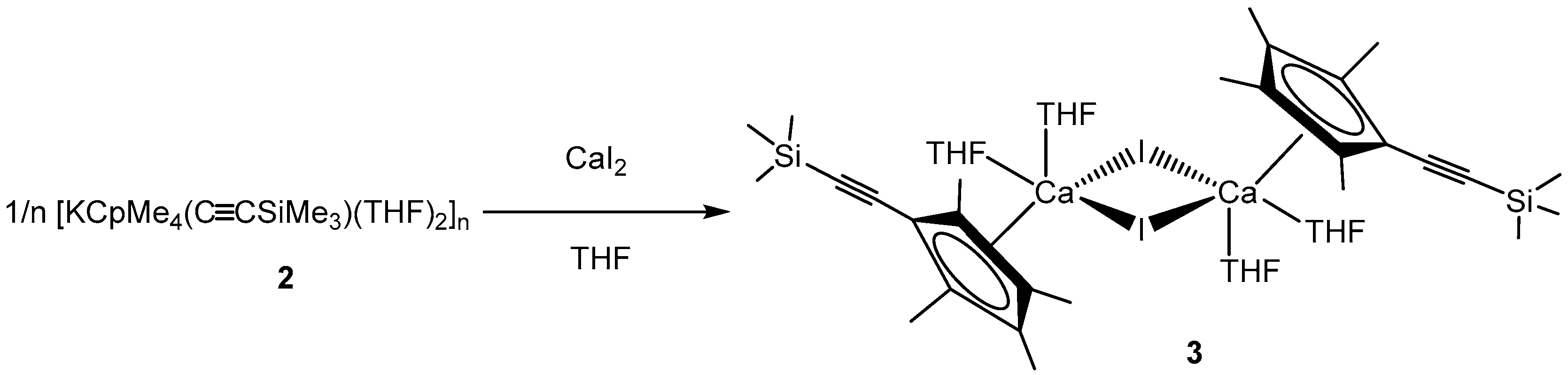
Figure 3.
Molecular structure of 3 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ca1–I1 3.0920(7), Ca1–I1′ 3.2039(7), Ca1–O1 2.369(3), Ca1–O2 2.410(3), Ca1–C6 2.634(3), Ca1–C7 2.686(4), Ca1–C8 2.734(5), Ca1–C9 2.733(5), Ca1–C10 2.687(4), C4–C5 1.199(5), I1–Ca1–I1′ 83.109(2), Ca1–I1–Ca1′ 96.891(2), O1–Ca1–O2 75.01(10), CpMe4(C≡CSiMe3)-Centroid–Ca1–I1 109.320(1), CpMe4(C≡CSiMe3)-Centroid–Ca1–I1′ 115.625(1).
Figure 3.
Molecular structure of 3 in the solid state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ca1–I1 3.0920(7), Ca1–I1′ 3.2039(7), Ca1–O1 2.369(3), Ca1–O2 2.410(3), Ca1–C6 2.634(3), Ca1–C7 2.686(4), Ca1–C8 2.734(5), Ca1–C9 2.733(5), Ca1–C10 2.687(4), C4–C5 1.199(5), I1–Ca1–I1′ 83.109(2), Ca1–I1–Ca1′ 96.891(2), O1–Ca1–O2 75.01(10), CpMe4(C≡CSiMe3)-Centroid–Ca1–I1 109.320(1), CpMe4(C≡CSiMe3)-Centroid–Ca1–I1′ 115.625(1).

Scheme 4.
Supposed Schlenk equilibrium between 3 and [{CpMe4(C≡CSiMe3)}2Ca(THF)x] + CaI2(THF)2.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seifert, T.; Roesky, P.W. Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand. Inorganics 2017, 5, 28. https://doi.org/10.3390/inorganics5020028
AMA Style
Seifert T, Roesky PW. Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand. Inorganics. 2017; 5(2):28. https://doi.org/10.3390/inorganics5020028
Chicago/Turabian StyleSeifert, Tim, and Peter W. Roesky. 2017. "Alkali and Alkaline Earth Metal Complexes Ligated by an Ethynyl Substituted Cyclopentadienyl Ligand" Inorganics 5, no. 2: 28. https://doi.org/10.3390/inorganics5020028
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.