A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen

 ,
,  ,
,  and
and
Abstract
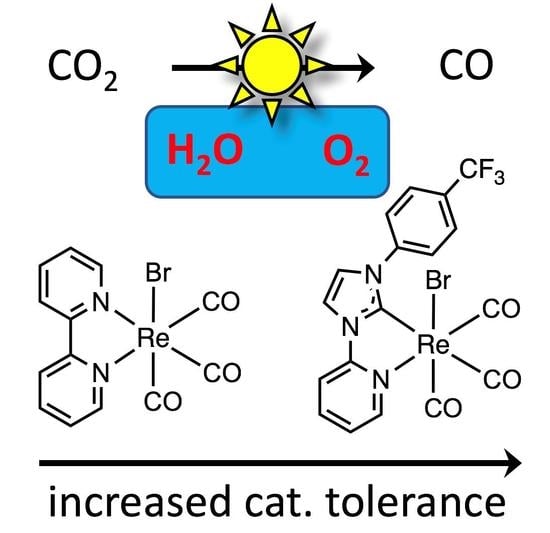
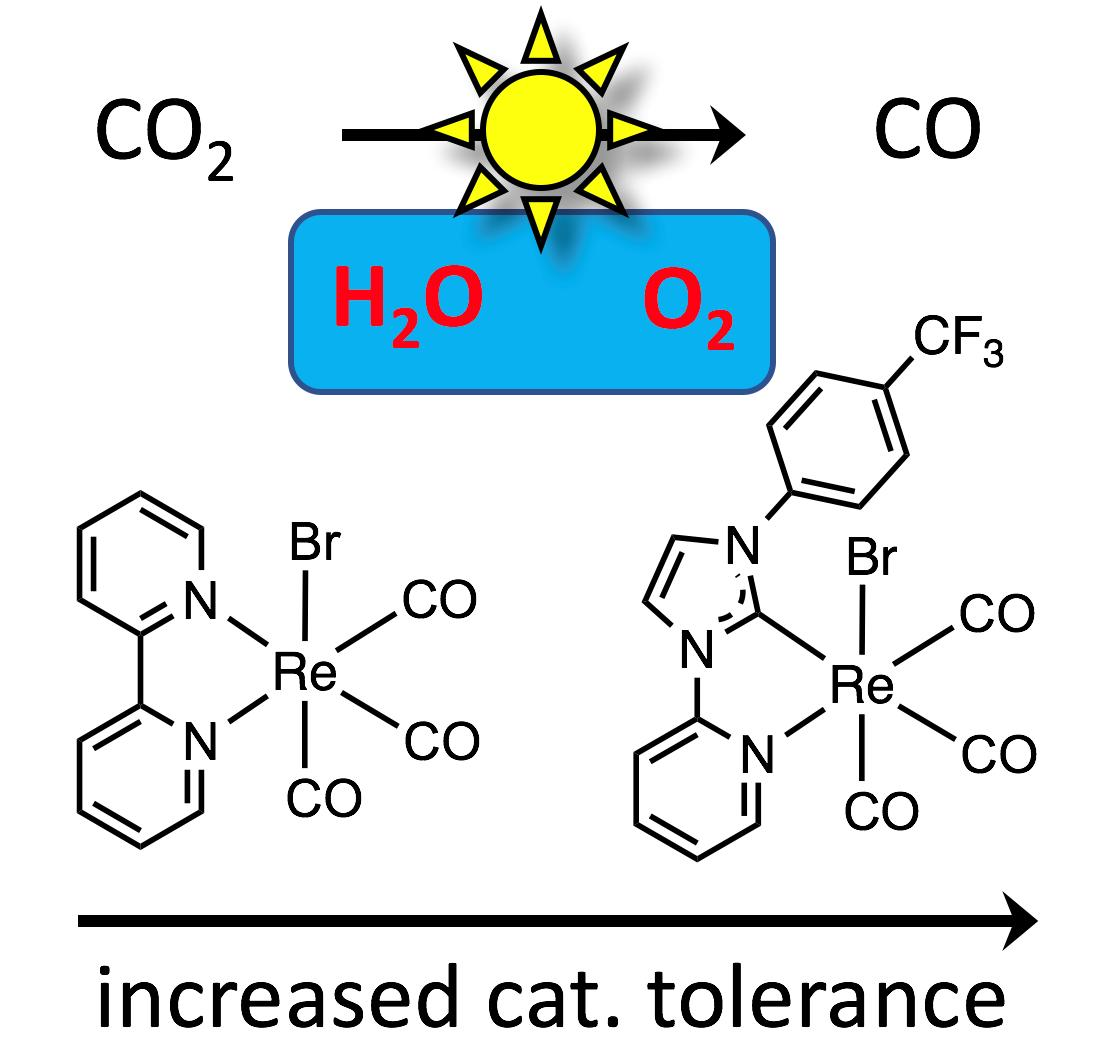
:
{kind=link}
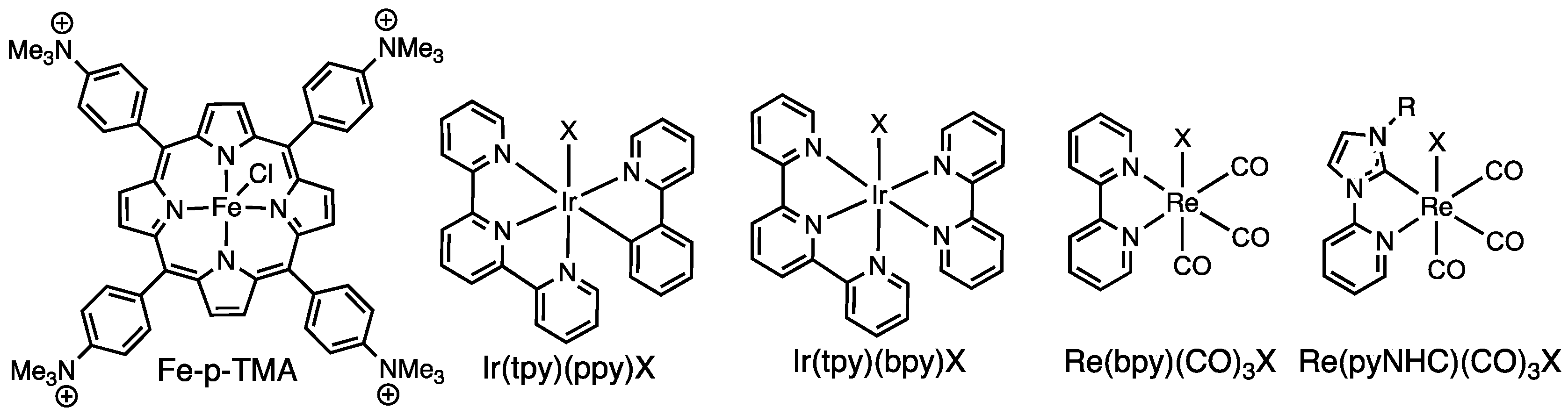{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
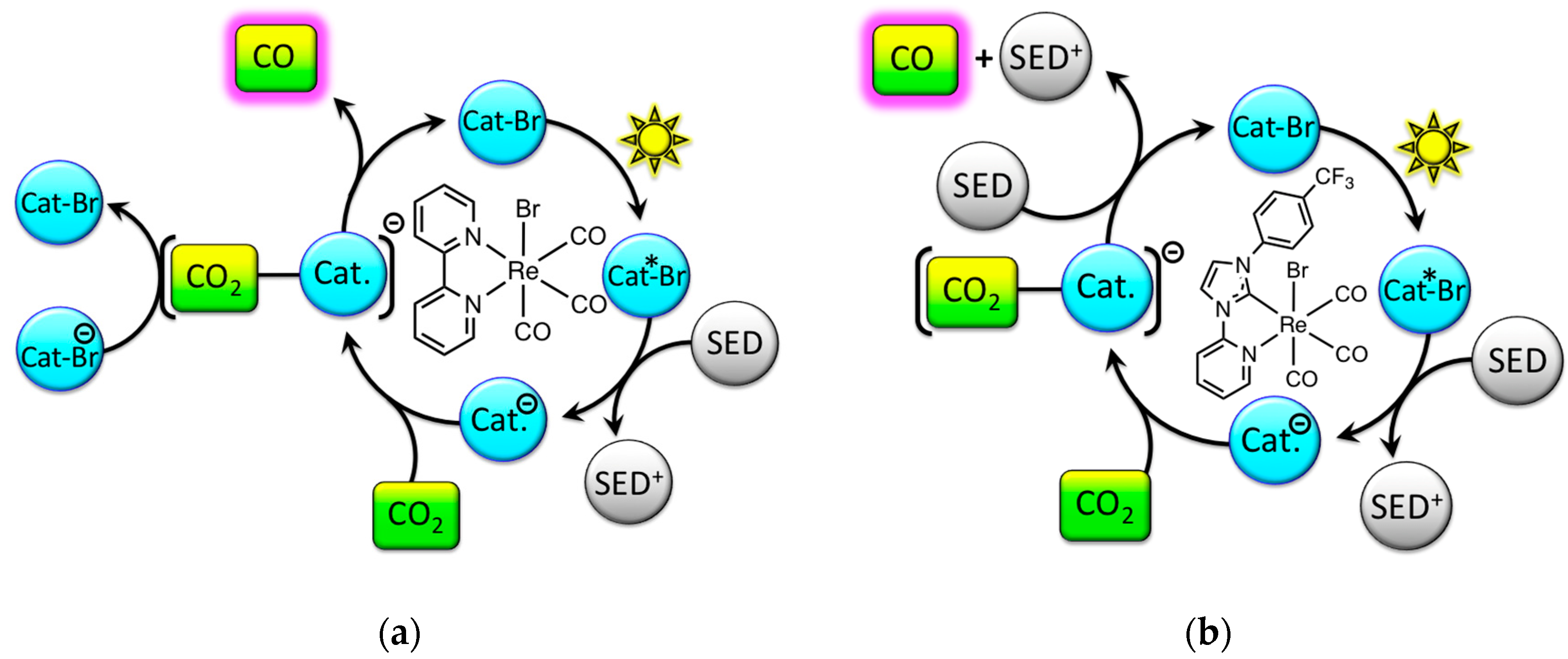
2. Results and Discussion
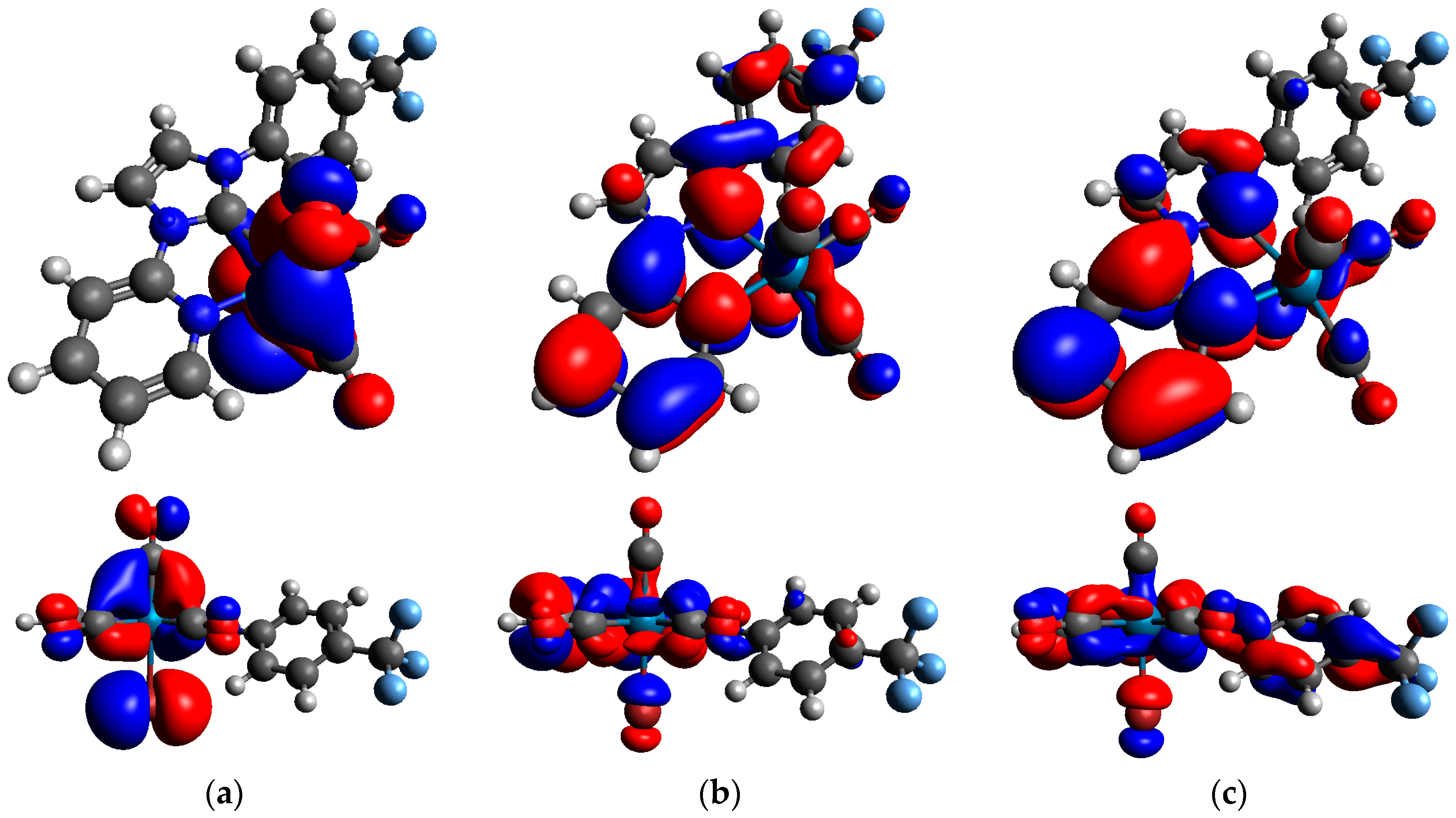
2.1. Computational Analysis: Role of the Ph-CF3 Substituent
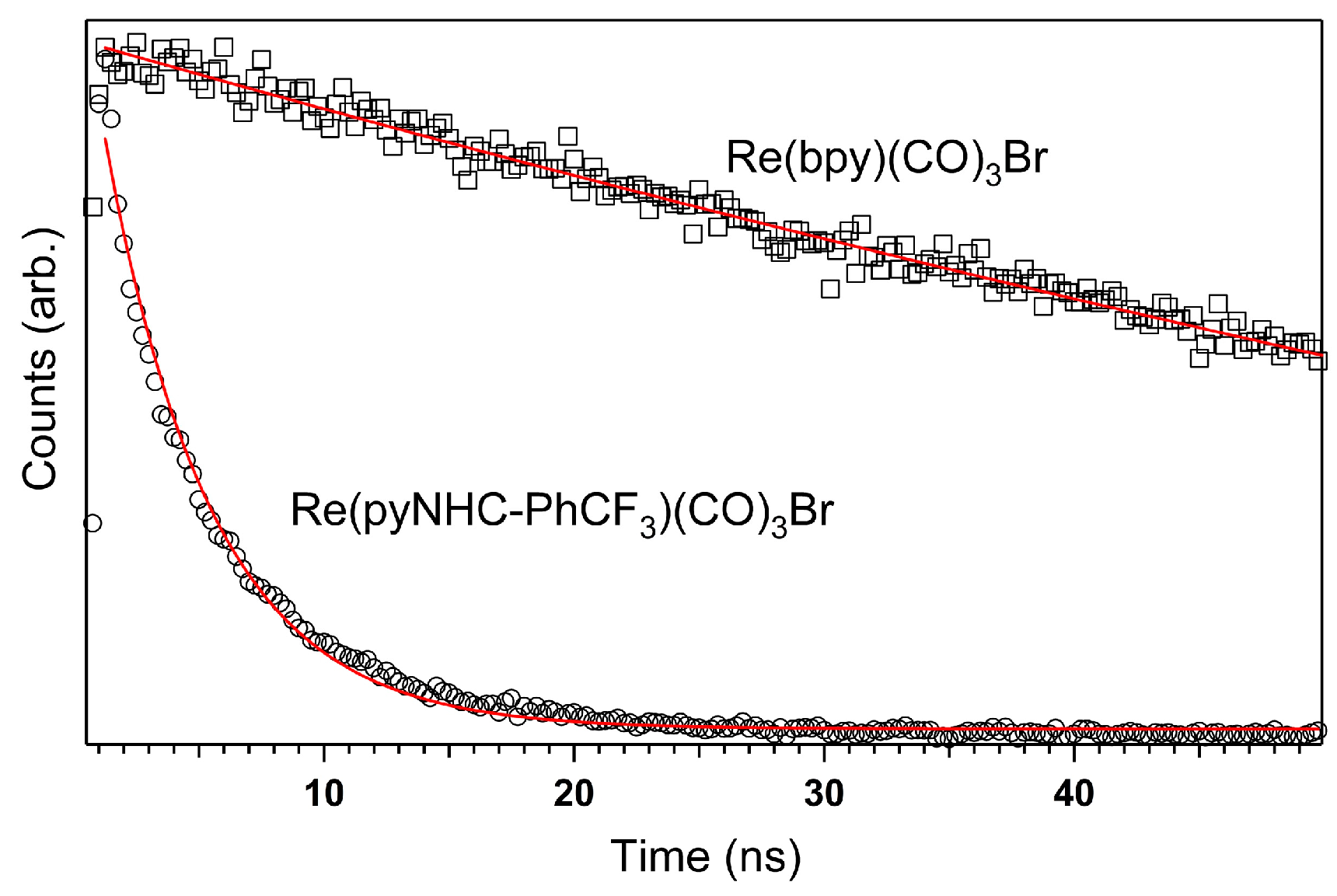
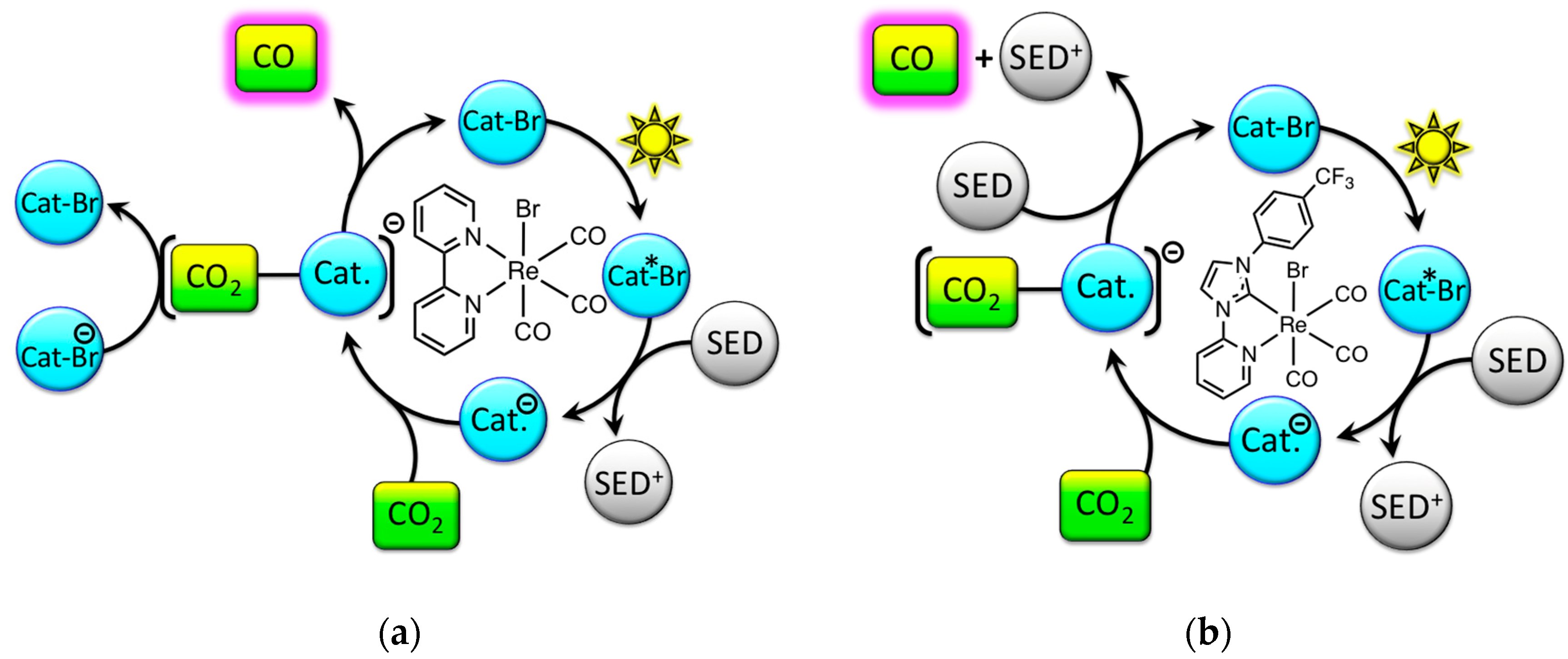
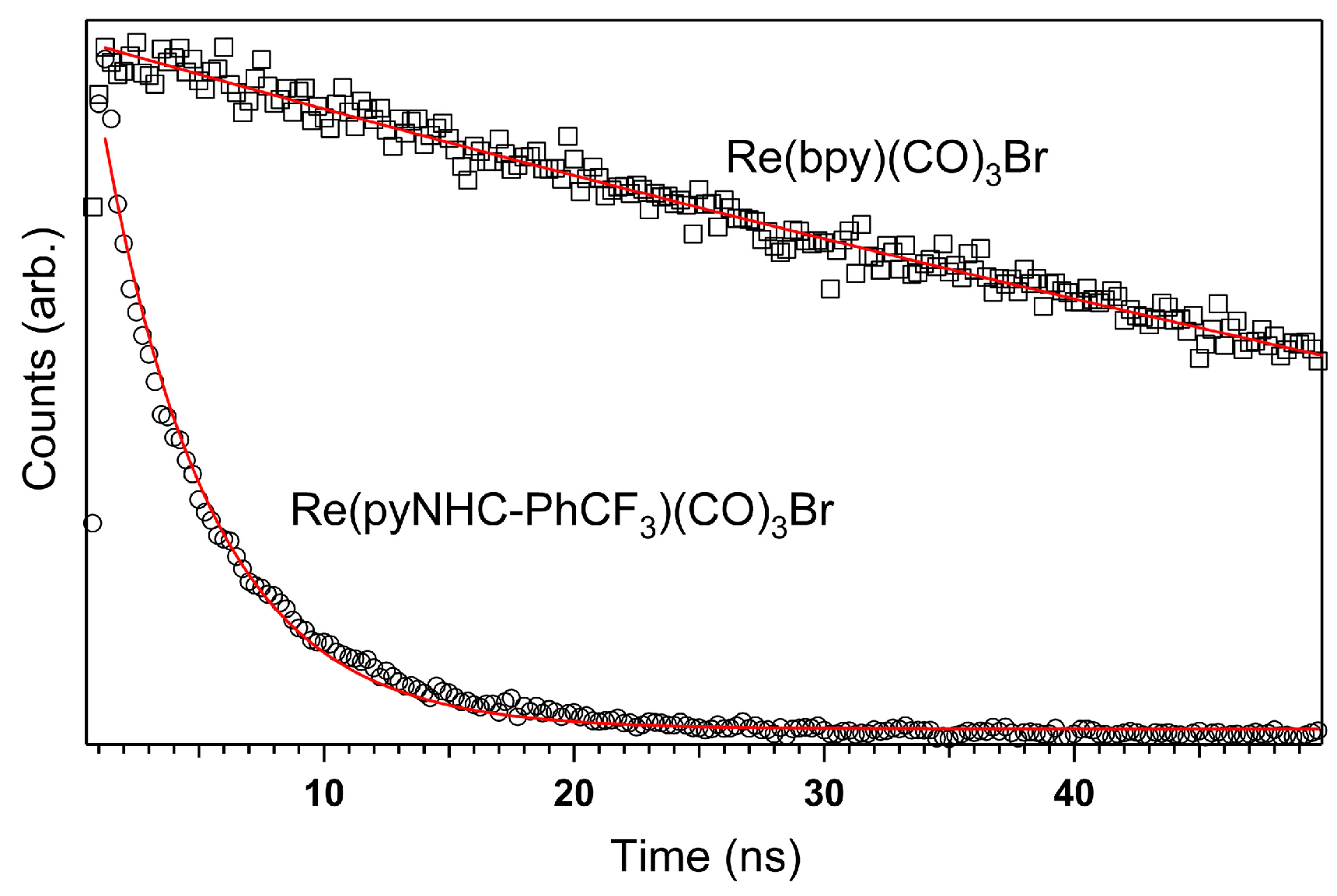
2.2. Excited-State Lifetime: Mechanistic Implications
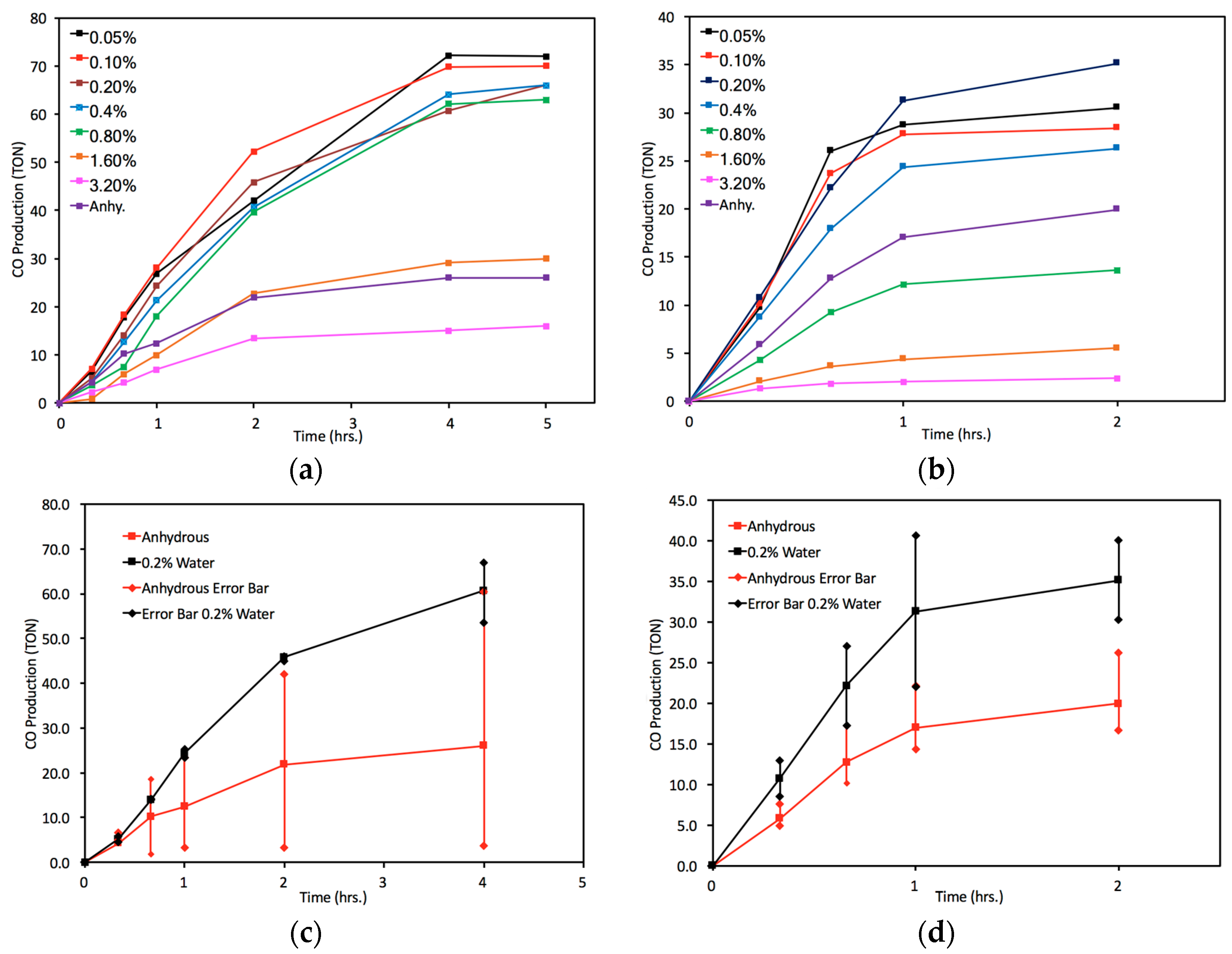
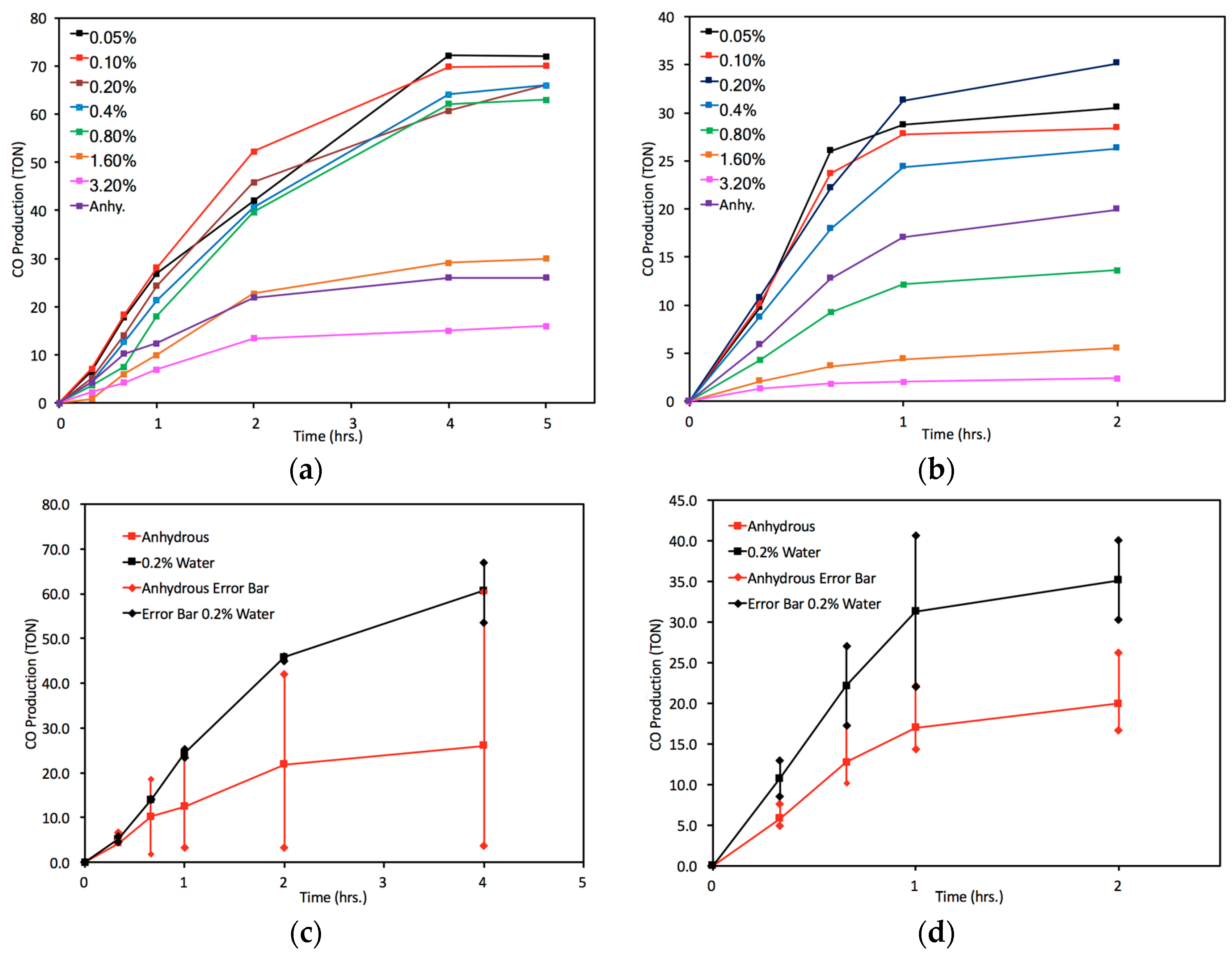
2.3. Catalyst Sensitivity: Water Concentration
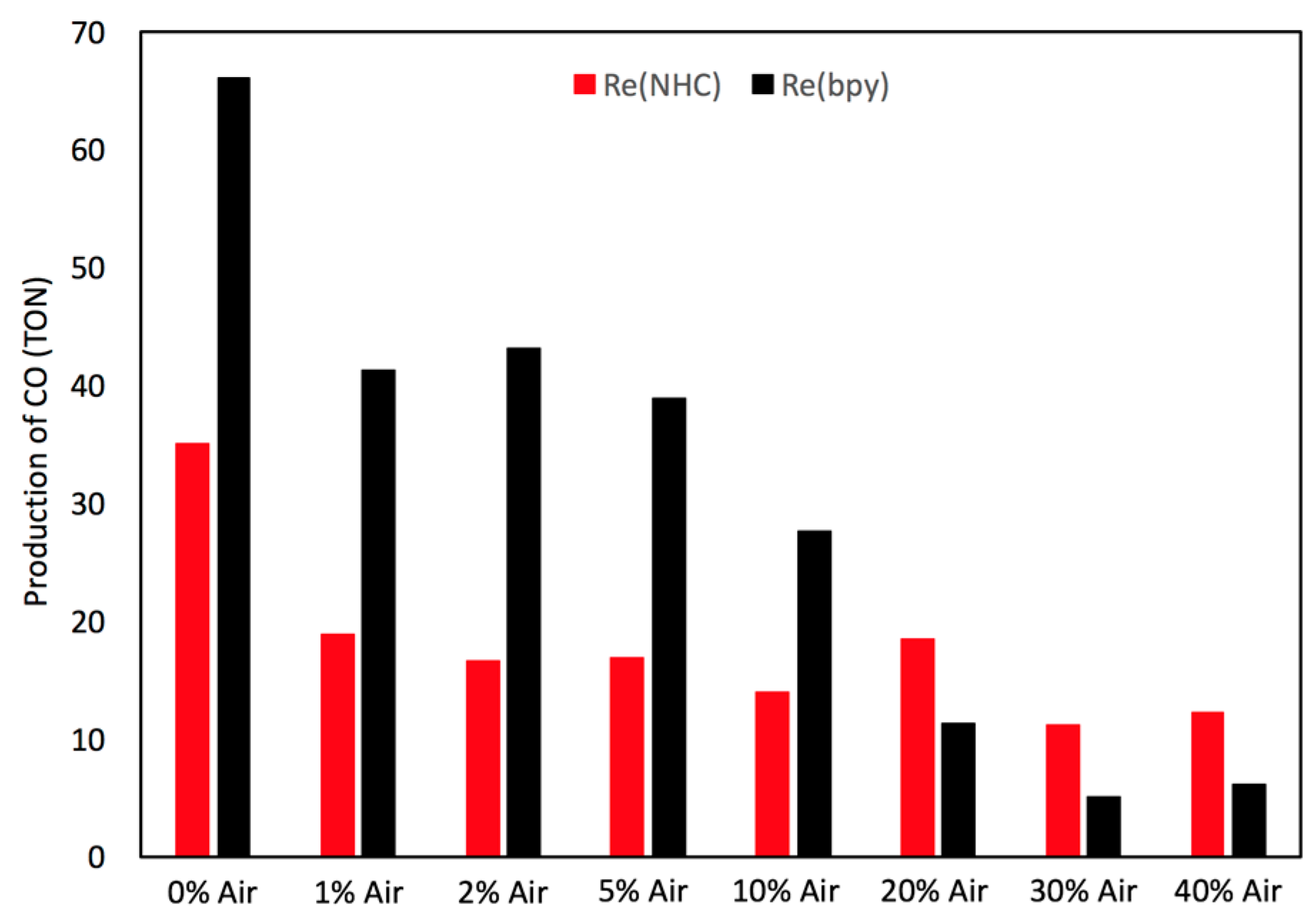
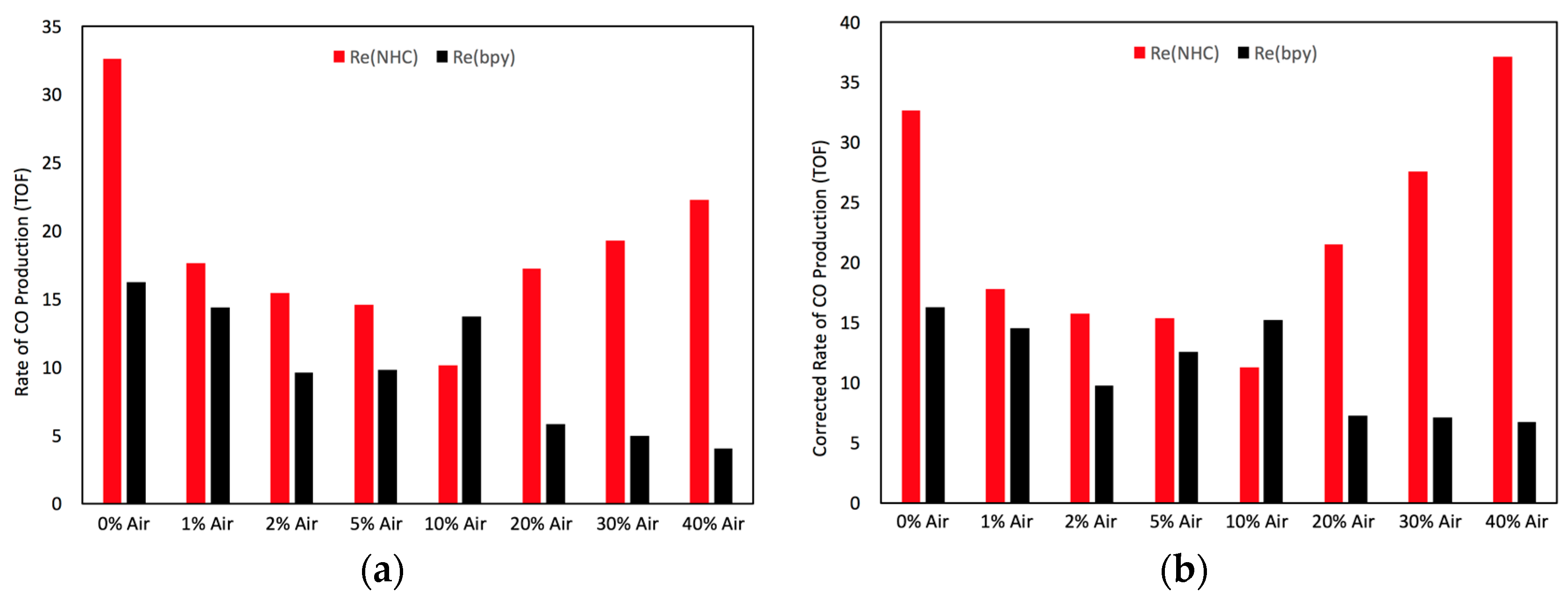
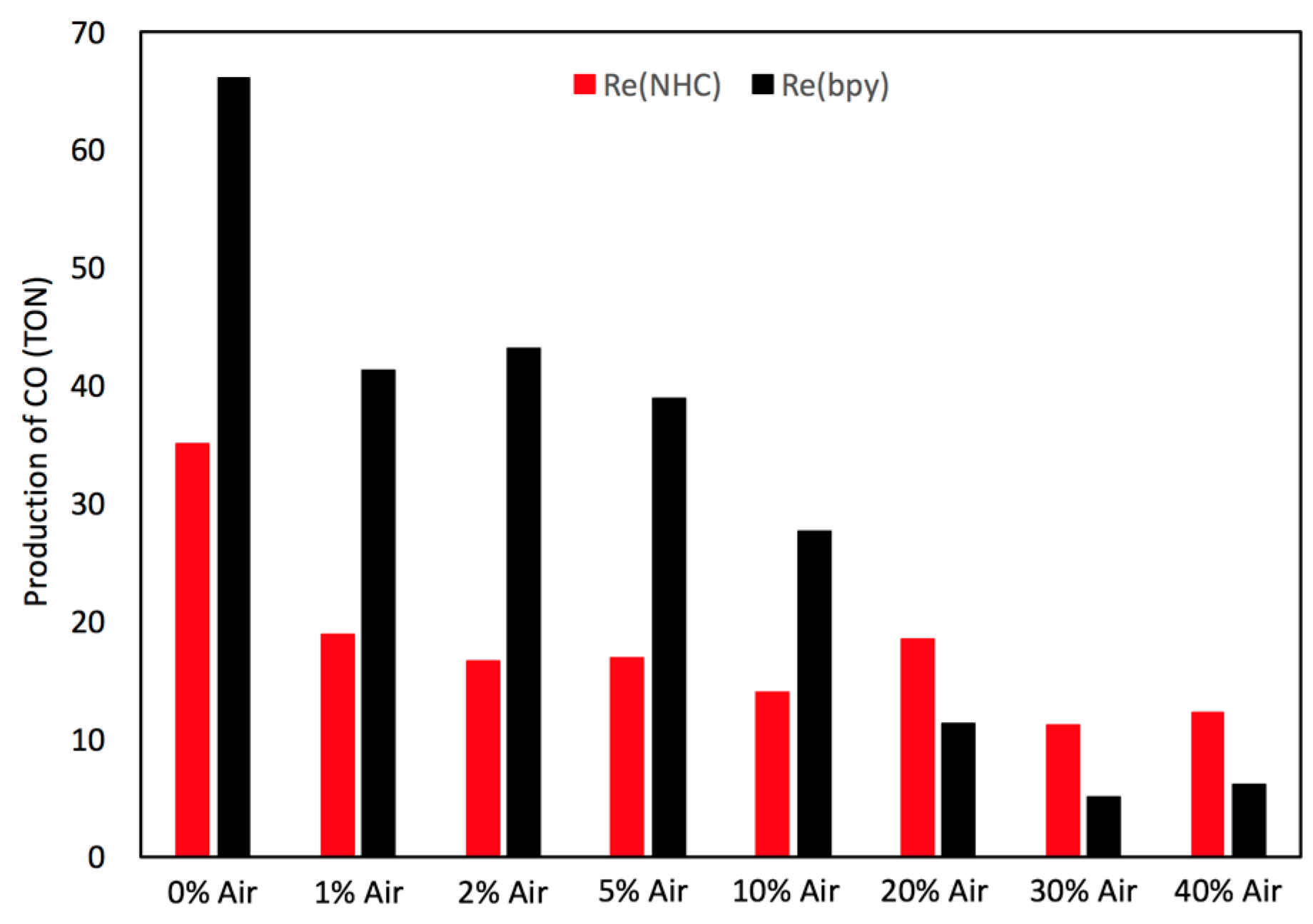
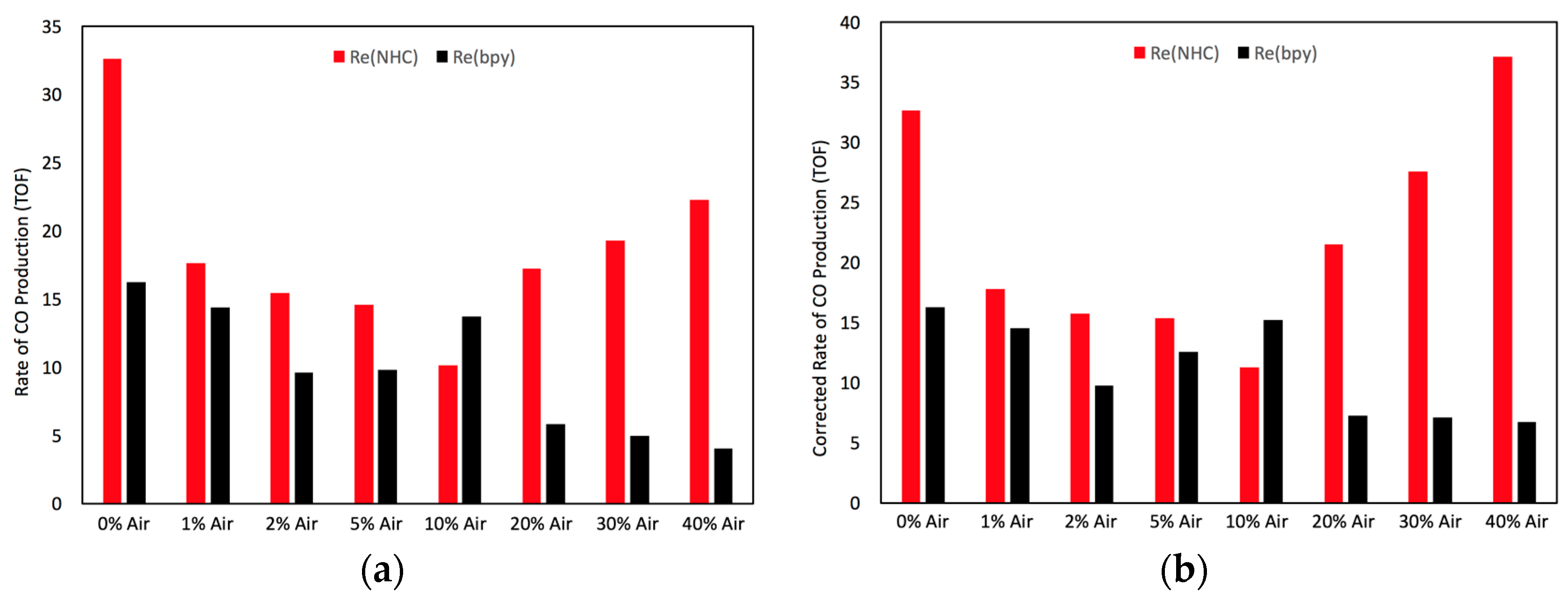
2.4. Catalyst Sensitivity: Air Concentration
3. Experimental Section
3.1. Computational Details
3.2. Electron Lifetime Measurement Information
3.3. Photocatalysis General Information
3.4. Water-Control Photocatalysis Procedure
3.5. Oxygen Control Photocatalysis Procedure
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Robert, M. Running the clock: CO2 catalysis in the age of anthropocene. ACS Energy Lett. 2016, 281–282. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.I.; Sutton, S.; Delcamp, J.H.; Graham, J.D. Photocatalytic water splitting and carbon dioxide reduction. In Handbook of Climate Change Mitigation and Adaptation; Chen, W.-Y., Suzuki, T., Lackner, M., Eds.; Springer: New York, NY, USA, 2016; pp. 2709–2756. [Google Scholar]
- Berardi, S.; Drouet, S.; Francas, L.; Gimbert-Surinach, C.; Guttentag, M.; Richmond, C.; Stoll, T.; Llobet, A. Molecular artificial photosynthesis. Chem. Soc. Rev. 2014, 43, 7501–7519. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 2009, 42, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Morikawa, T.; Kajino, T.; Ishitani, O. A highly efficient mononuclear iridium complex photocatalyst for CO2 reduction under visible light. Angew. Chem. Int. Ed. 2013, 52, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Morikawa, T. [Ir(tpy)(bpy)Cl] as a photocatalyst for CO2 reduction under visible-light irradiation. ChemPhotoChem 2017. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+–Co2+ combinations as homogeneous catalysts. J. Chem. Soc. Chem. Commun. 1983, 536–538. [Google Scholar] [CrossRef]
- Huckaba, A.J.; Sharpe, E.A.; Delcamp, J.H. Photocatalytic reduction of CO2 with Re–pyridyl–NHCs. Inorg. Chem. 2016, 55, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Sahara, G.; Ishitani, O. Efficient photocatalysts for CO2 reduction. Inorg. Chem. 2015, 54, 5096–5104. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Bonin, J.; Robert, M. Non-sensitized selective photochemical reduction of CO2 to CO under visible light with an iron molecular catalyst. Chem. Commun. 2017, 53, 2830–2833. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, N.P.; Dulaney, H.A.; Huckaba, A.J.; Jurss, J.W.; Delcamp, J.H. Electrocatalytic reduction of CO2 to CO with Re-pyridyl-NHCs: Proton source influence on rates and product selectivities. Inorg. Chem. 2016, 55, 6085–6094. [Google Scholar] [CrossRef] [PubMed]
- Cope, J.D.; Liyanage, N.P.; Kelley, P.J.; Denny, J.A.; Valente, E.J.; Webster, C.E.; Delcamp, J.H.; Hollis, T.K. Electrocatalytic reduction of CO2 with CCC-NHC pincer nickel complexes. Chem. Commun. 2017, 53, 9442–9445. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Shaw, T.W.; Stanton, C.J., 3rd; Majetich, G.F.; Bocarsly, A.B.; Schaefer, H.F., 3rd. NHC-containing manganese(I) electrocatalysts for the two-electron reduction of CO2. Angew. Chem. Int. Ed. 2014, 53, 5152–5155. [Google Scholar] [CrossRef]
- Jin, T.; He, D.; Li, W.; Stanton, C.J.; Pantovich, S.A.; Majetich, G.F.; Schaefer, H.F.; Agarwal, J.; Wang, D.; Li, G. CO2 reduction with Re(I)–NHC compounds: Driving selective catalysis with a silicon nanowire photoelectrode. Chem. Commun. 2016, 52, 14258–14261. [Google Scholar] [CrossRef] [PubMed]
- Stanton, C.J., 3rd; Machan, C.W.; Vandezande, J.E.; Jin, T.; Majetich, G.F.; Schaefer, H.F., 3rd; Kubiak, C.P.; Li, G.; Agarwal, J. Re(I)NHC complexes for electrocatalytic conversion of CO2. Inorg. Chem. 2016, 55, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Stanton, C.J., 3rd; Vandezande, J.E.; Majetich, G.F.; Schaefer, H.F., 3rd; Agarwal, J. Mn–NHC electrocatalysts: Increasing π-acidity lowers the reduction potential and increases the turnover frequency for CO2 reduction. Inorg. Chem. 2016, 55, 9509–9512. [Google Scholar] [CrossRef] [PubMed]
- Boudreaux, C.M.; Liyanage, N.P.; Shirley, H.; Siek, S.; Gerlach, D.L.; Qu, F.; Delcamp, J.H.; Papish, E.T. Ruthenium(II) complexes of pyridinol and N-heterocyclic carbene derived pincers as robust catalysts for selective carbon dioxide reduction. Chem. Commun. 2017, 53, 11217–11220. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.A., III; Clavier, H.; Giudice, S.; Scott, N.M.; Stevens, E.D.; Bordner, J.; Samardjiev, I.; Hoff, C.D.; Cavallo, L.; Nolan, S.P. Determination of N-heterocyclic carbene (NHC) steric and electronic parameters using the [(NHC)Ir(CO)2Cl] system. Organometallics 2008, 27, 202–210. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; Inesi, A. Advances in the knowledge of N-heterocyclic carbenes properties. The backing of the electrochemical investigation. Catalysts 2016, 6, 178. [Google Scholar] [CrossRef]
- Cavallo, L.; Correa, A.; Costabile, C.; Jacobsen, H. Steric and electronic effects in the bonding of N-heterocyclic ligands to transition metals. J. Organomet. Chem. 2005, 690, 5407–5413. [Google Scholar] [CrossRef]
- Takeda, H.; Koike, K.; Inoue, H.; Ishitani, O. Development of an Efficient Photocatalytic System for CO2 Reduction Using Rhenium(I) Complexes Based on Mechanistic Studies. J. Am. Chem. Soc. 2008, 130, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Mukuta, T.; Simpson, P.V.; Vaughan, J.G.; Skelton, B.W.; Stagni, S.; Massi, M.; Koike, K.; Ishitani, O.; Onda, K. Photochemical processes in a rhenium(I) tricarbonyl N-heterocyclic carbene complex studied by time-resolved measurements. Inorg. Chem. 2017, 56, 3404–3413. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.G.; Reid, B.L.; Wright, P.J.; Ramchandani, S.; Skelton, B.W.; Raiteri, P.; Muzzioli, S.; Brown, D.H.; Stagni, S.; Massi, M. Photophysical and photochemical trends in tricarbonyl rhenium(I) N-heterocyclic carbene complexes. Inorg. Chem. 2014, 53, 3629–3641. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.G.; Reid, B.L.; Ramchandani, S.; Wright, P.J.; Muzzioli, S.; Skelton, B.W.; Raiteri, P.; Brown, D.H.; Stagni, S.; Massi, M. The photochemistry of rhenium(I) tricarbonyl N-heterocyclic carbene complexes. Dalton Trans. 2013, 42, 14100–14114. [Google Scholar] [CrossRef] [PubMed]
- Feldt, S.M.; Lohse, P.W.; Kessler, F.; Nazeeruddin, M.K.; Grätzel, M.; Boschloo, G.; Hagfeldt, A. Regeneration and recombination kinetics in cobalt polypyridine based dye-sensitized solar cells, explained using Marcus theory. Phys. Chem. Chem. Phys. 2013, 15, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Last, G.V.; Schmick, M.T. A review of major non-power-related carbon dioxide stream compositions. Environ. Earth Sci. 2015, 74, 1189–1198. [Google Scholar] [CrossRef]
- Nakajima, T.; Tamaki, Y.; Ueno, K.; Kato, E.; Nishikawa, T.; Ohkubo, K.; Yamazaki, Y.; Morimoto, T.; Ishitani, O. Photocatalytic reduction of low concentration of CO2. J. Am. Chem. Soc. 2016, 138, 13818–13821. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scurseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, A.; et al. Gaussian09; revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Dill, J.D.; Pople, J.A. Self-consistent molecular orbital methods. XV. Extended gaussian-type basis sets for lithium, beryllium, and boron. J. Chem. Phys. 1975, 62, 2921–2923. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Agarwal, J.; Johnson, R.P.; Li, G. Reduction of CO2 on a tricarbonyl rhenium(I) complex: Modeling a catalytic cycle. J. Phys. Chem. A 2011, 115, 2877–2881. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Fujita, E.; Schaefer, H.F., 3rd; Muckerman, J.T. Mechanisms for CO production from CO2 using reduced rhenium tricarbonyl catalysts. J. Am. Chem. Soc. 2012, 134, 5180–5186. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpenter, C.A.; Brogdon, P.; McNamara, L.E.; Tschumper, G.S.; Hammer, N.I.; Delcamp, J.H. A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen. Inorganics 2018, 6, 22. https://doi.org/10.3390/inorganics6010022
Carpenter CA, Brogdon P, McNamara LE, Tschumper GS, Hammer NI, Delcamp JH. A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen. Inorganics. 2018; 6(1):22. https://doi.org/10.3390/inorganics6010022
Chicago/Turabian StyleCarpenter, Casey A., Phillip Brogdon, Louis E. McNamara, Gregory S. Tschumper, Nathan I. Hammer, and Jared H. Delcamp. 2018. "A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen" Inorganics 6, no. 1: 22. https://doi.org/10.3390/inorganics6010022
APA StyleCarpenter, C. A., Brogdon, P., McNamara, L. E., Tschumper, G. S., Hammer, N. I., & Delcamp, J. H. (2018). A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen. Inorganics, 6(1), 22. https://doi.org/10.3390/inorganics6010022