
Identification of Protein Biomarkers for Differentiating Listeria monocytogenes Genetic Lineage III

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Bacterial Cultures
2.3. Protein Extraction with Phenol-Based Protocol
2.4. Protein Fractionation and In-Solution Digestion
2.5. Mass Spectrometry
2.6. Bioinformatics Analysis
2.7. Construction of Recombinant Plasmids and Protein Expression
2.8. Purification of Four Recombinant Proteins
2.9. Polyclonal Antibody Production
2.10. Analysis of Antibody Titer
2.11. Automated Western Blot (WES)
2.12. Statistical Analysis
3. Results
3.1. Mass Spectrometry
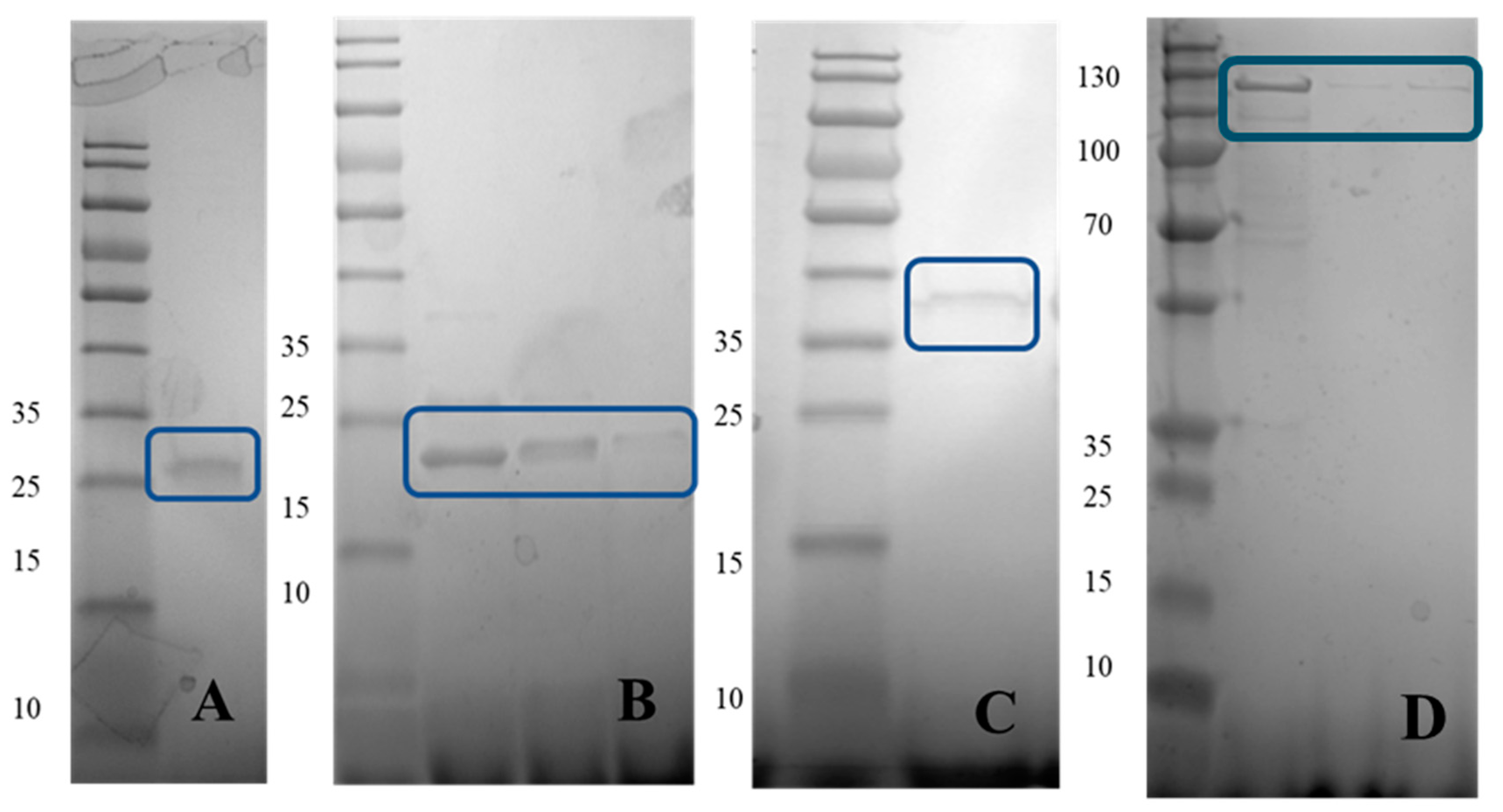
3.2. Expression and Purification of the Recombinant Proteins
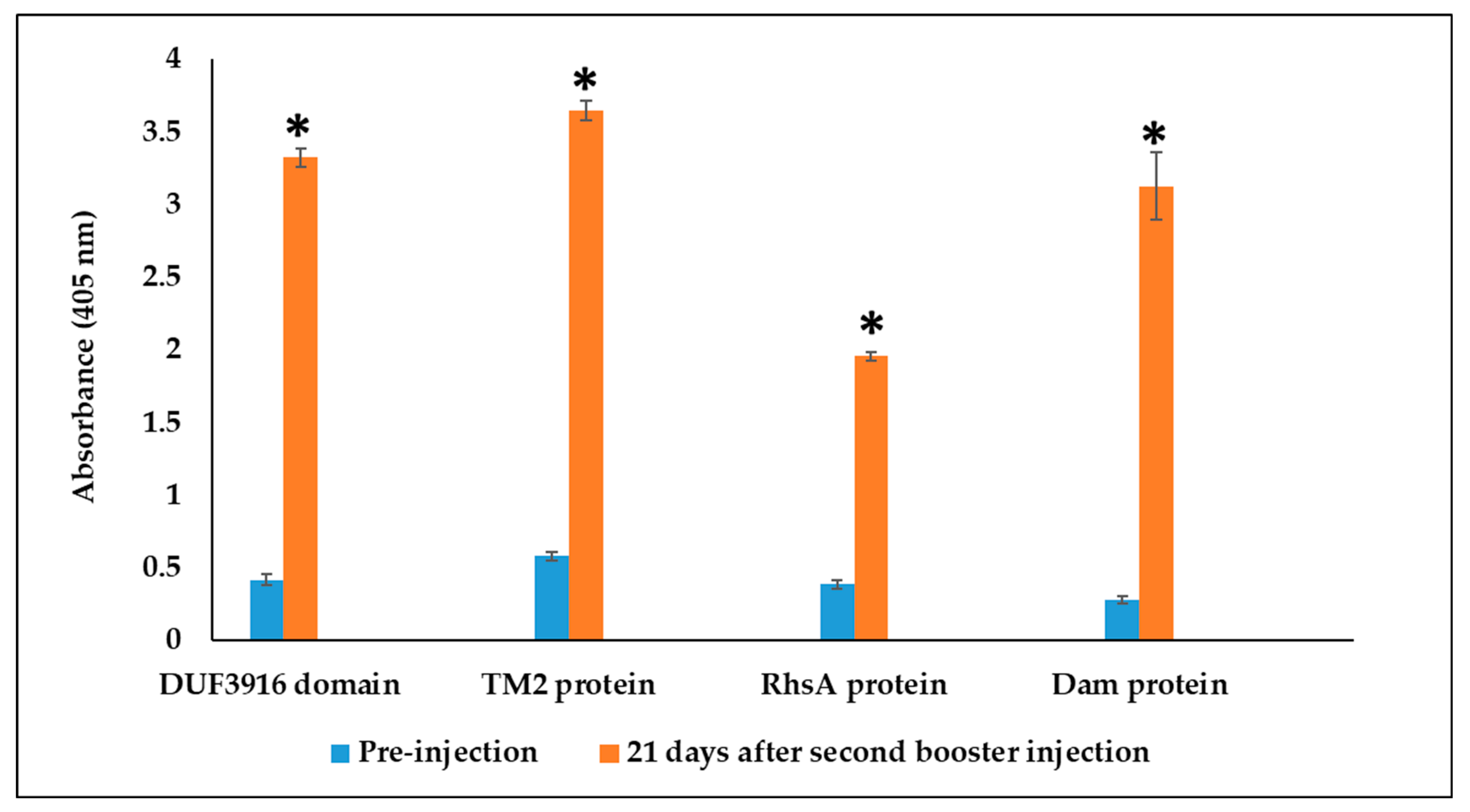
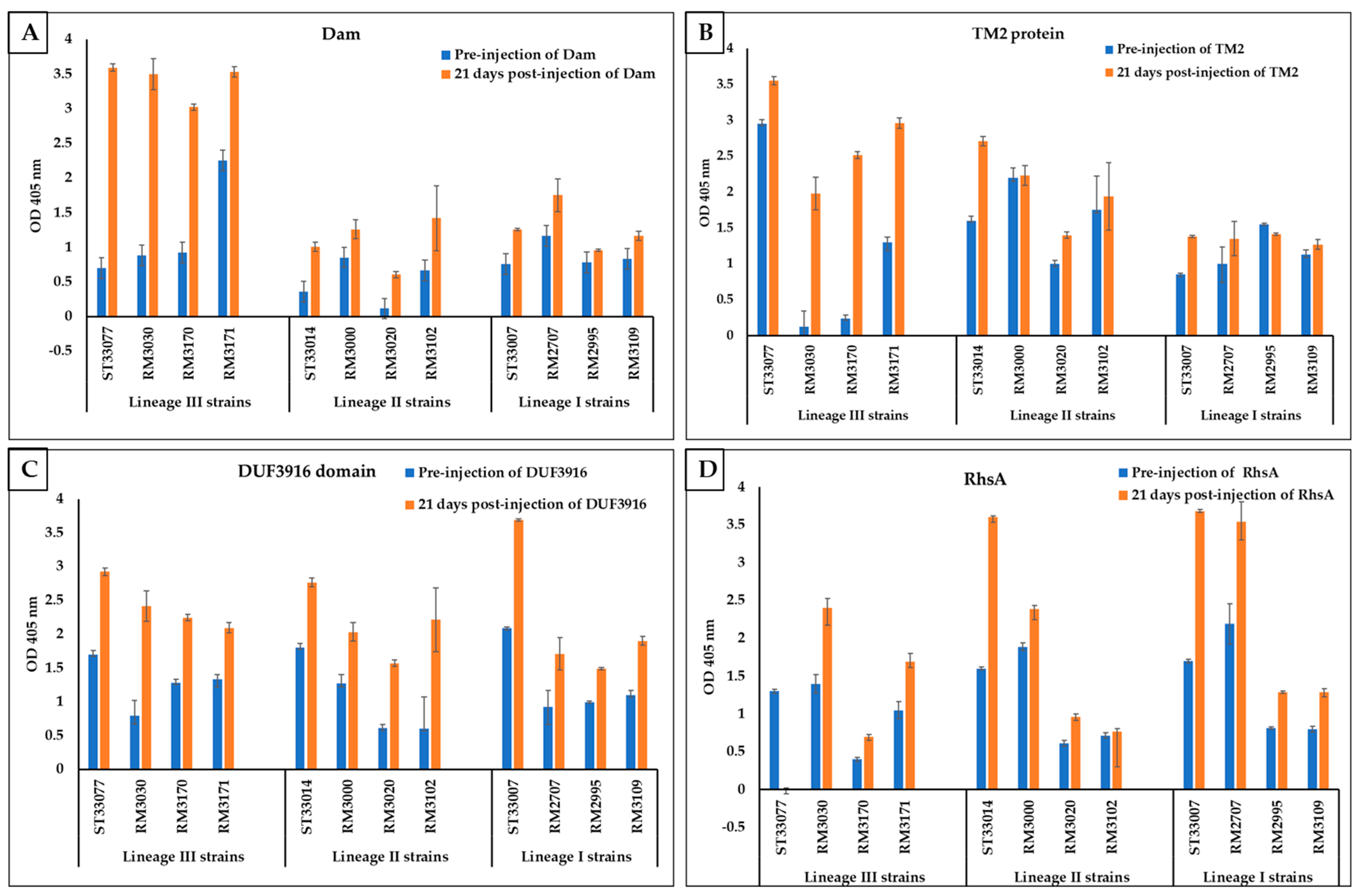
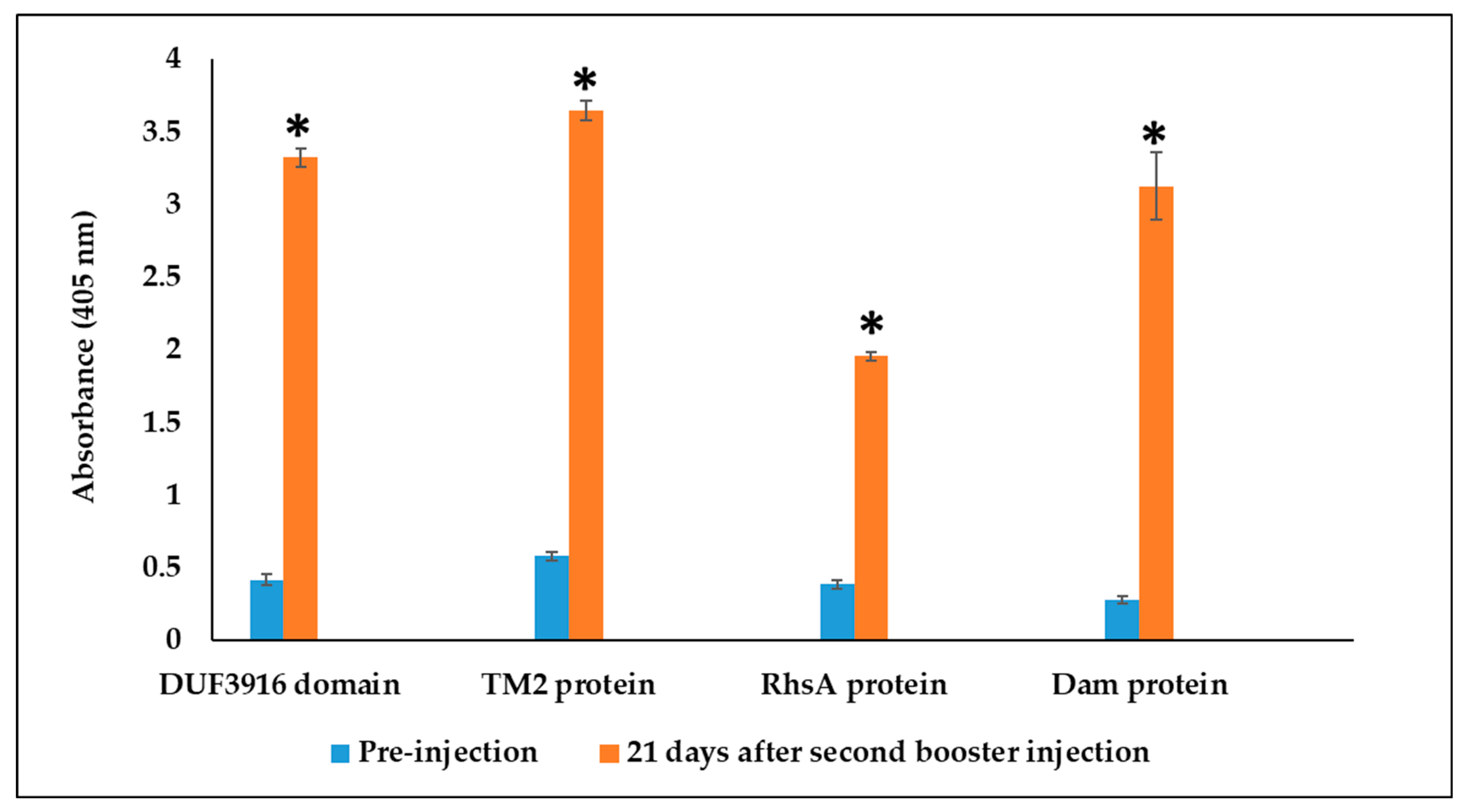
3.3. Rabbit Serum Antibody Response
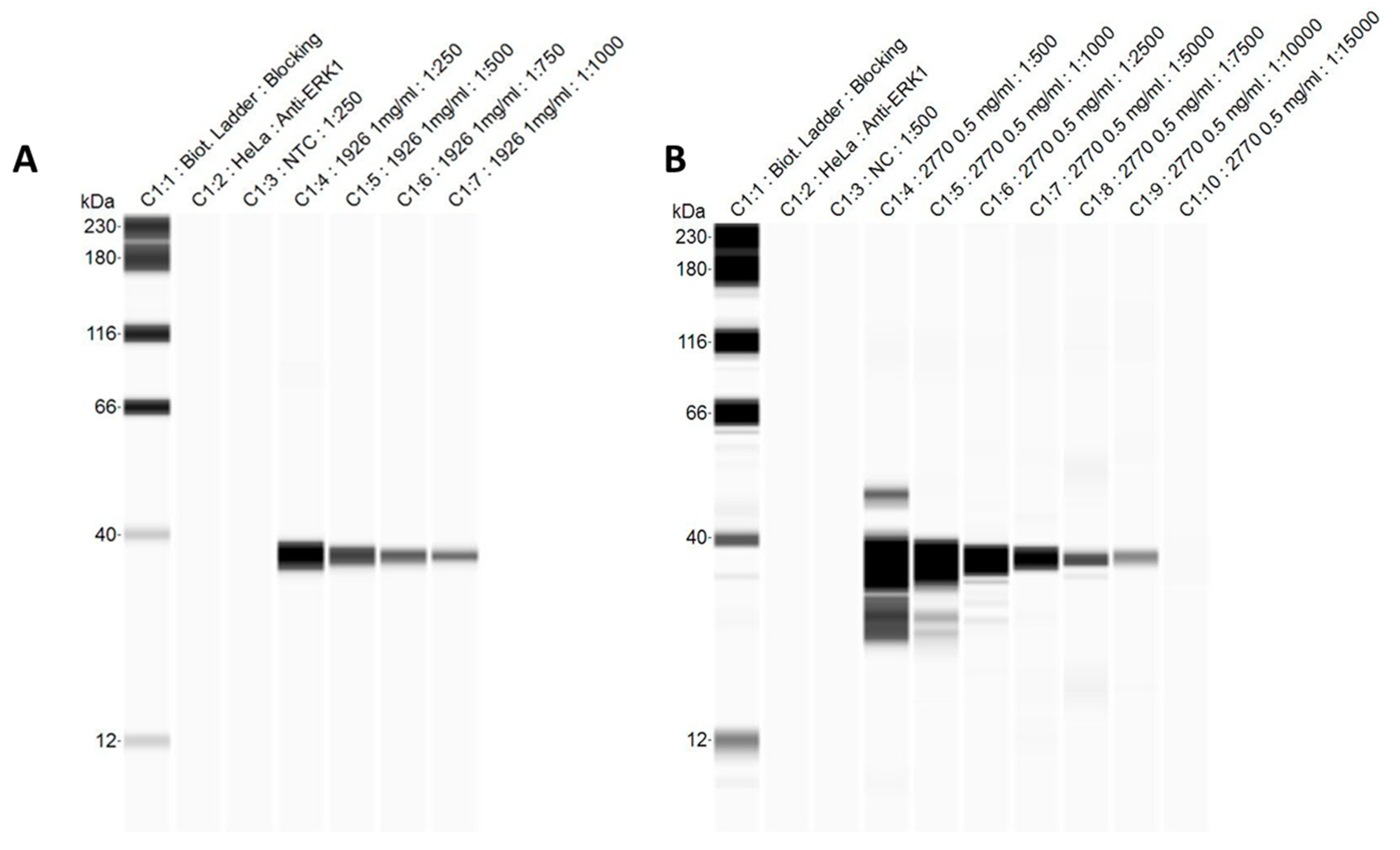
3.4. Automated Western Blots
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farber, J.M.; Peterkin, P.I. Listeria monocytogenes, a food-borne pathogen. Microbiol. Rev. 1991, 55, 476–511. [Google Scholar] [CrossRef] [PubMed]
- Murray, E.G.D.; Webb, R.A.; Swann, M.B.R. A disease of rabbits characterised by a large mononuclear leucocytosis, caused by a hitherto undescribed bacillus Bacterium monocytogenes (n.sp.). J. Pathol. Bacteriol. 1926, 29, 407–439. [Google Scholar] [CrossRef]
- Schlech, W.F.; Lavigne, P.M.; Bortolussi, R.A.; Allen, A.C.; Haldane, E.V.; Wort, A.J.; Hightower, A.W.; Johnson, S.E.; King, S.H.; Nicholls, E.S.; et al. Epidemic Listeriosis—Evidence for Transmission by Food. N. Engl. J. Med. 1983, 308, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Mateus, T.; Silva, J.; Maia, R.L.; Teixeira, P. Listeriosis during pregnancy: A public health concern. Int. Sch. Res. Not. 2013, 2013, 851712. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Dominguez-Bernal, G.; Goebel, W.; Gonzalez-Zorn, B.; Wehland, J.; Kreft, J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, W.F. Listeria, Listeriosis and Food Safety, 2nd edition. Int. J. Food Sci. Technol. 2001, 36, 114–115. [Google Scholar] [CrossRef]
- Buchanan, R.L.; Stahl, H.G.; Whiting, R.C. Effects and Interactions of Temperature, pH, Atmosphere, Sodium Chloride, and Sodium Nitrite on the Growth of Listeria monocytogenes. J. Food Prot. 1989, 52, 844. [Google Scholar] [CrossRef] [PubMed]
- Matle, I.; Mbatha, K.R.; Madoroba, E. A review of Listeria monocytogenes from meat and meat products: Epidemiology, virulence factors, antimicrobial resistance and diagnosis. Onderstepoort J. Vet. Res. 2020, 87, e1–e20. [Google Scholar] [CrossRef]
- Nho, S.W.; Abdelhamed, H.; Reddy, S.; Karsi, A.; Lawrence, M.L. Identification of high-risk Listeria monocytogenes serotypes in lineage I (serotype 1/2a, 1/2c, 3a and 3c) using multiplex PCR. J. Appl. Microbiol. 2015, 119, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Nadon, C.A.; Woodward, D.L.; Young, C.; Rodgers, F.G.; Wiedmann, M. Correlations between Molecular Subtyping and Serotyping of Listeria monocytogenes. J. Clin. Microbiol. 2001, 39, 2704–2707. [Google Scholar] [CrossRef]
- Rasmussen, O.F.; Skouboe, P.; Dons, L.; Rossen, L.; Olsen, J.E. Listeria monocytogenes exists in at least three evolutionary lines: Evidence from flagellin, invasive associated protein and listeriolysin O genes. Microbiology 1995, 141, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.J.; Usgaard, T.; Evans, P. A targeted multilocus genotyping assay for lineage, serogroup, and epidemic clone typing of Listeria monocytogenes. Appl. Environ. Microbiol. 2010, 76, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Kuenne, C.; Billion, A.; Mraheil, M.A.; Strittmatter, A.; Daniel, R.; Goesmann, A.; Barbuddhe, S.; Hain, T.; Chakraborty, T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genom. 2013, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, C.; Gouin, E.; Jeannel, D.; Cossart, P.; Rocourt, J. Expression of ActA, Ami, InlB, and listeriolysin O in Listeria monocytogenes of human and food origin. Appl. Environ. Microbiol. 2002, 68, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Mead, P.S.; Dunne, E.F.; Graves, L.; Wiedmann, M.; Patrick, M.; Hunter, S.; Salehi, E.; Mostashari, F.; Craig, A.; Mshar, P.; et al. Nationwide outbreak of listeriosis due to contaminated meat. Epidemiol. Infect. 2005, 134, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Breidt, F.; Jr Kathariou, S. Competition of Listeria monocytogenes serotype 1/2a and 4b strains in mixed-culture biofilms. Appl. Environ. Microbiol. 2009, 75, 5846–5852. [Google Scholar] [CrossRef] [PubMed]
- Borucki, M.K.; Call, D.R. Listeria monocytogenes serotype identification by PCR. J. Clin. Microbiol. 2003, 41, 5537–5540. [Google Scholar] [CrossRef]
- Tresse, O.; Shannon, K.; Pinon, A.; Malle, P.; Vialette, M.; Midelet-Bourdin, G. Variable adhesion of Listeria monocytogenes isolates from food-processing facilities and clinical cases to inert surfaces. J. Food Prot. 2007, 70, 1569–1578. [Google Scholar] [CrossRef]
- Gorski, L.; Palumbo, J.D.; Nguyen, K.D. Strain-specific differences in the attachment of Listeria monocytogenes to alfalfa sprouts. J. Food Prot. 2004, 67, 2488–2495. [Google Scholar] [CrossRef] [PubMed]
- Gorski, L.; Palumbo, J.D.; Mandrell, R.E. Attachment of Listeria monocytogenes to radish tissue is dependent upon temperature and flagellar motility. Appl. Environ. Microbiol. 2003, 69, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hurkman, W.J.; Tanaka, C.K. Solubilization of plant membrane proteins for analysis by two-dimensional gel electrophoresis. Plant Physiol. 1986, 81, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, H.; Nho, S.W.; Turaga, G.; Banes, M.M.; Karsi, A.; Lawrence, M.L. Protective efficacy of four recombinant fimbrial proteins of virulent Aeromonas hydrophila strain ML09-119 in channel catfish. Vet. Microbiol. 2016, 197, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Aarts, H.J.; Hakemulder, L.E.; Van Hoef, A.M. Genomic typing of Listeria monocytogenes strains by automated laser fluorescence analysis of amplified fragment length polymorphism fingerprint patterns. Int. J. Food Microbiol. 1999, 49, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bultet, L.; Nicholson, P.; Rychener, L.; Dreyer, M.; Gozel, B.; Origgi, F.C.; Oevermann, A.; Frey, J.; Falquet, L. Genetic Separation of Listeria monocytogenes Causing Central Nervous System Infections in Animals. Front. Cell. Infect. Microbiol. 2018, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Bibb, W.F.; Gellin, B.G.; Weaver, R.; Schwartz, B.; Plikaytis, B.D.; Reeves, M.W.; Pinner, R.W.; Broome, C.V. Analysis of clinical and food-borne isolates of Listeria monocytogenes in the United States by multilocus enzyme electrophoresis and application of the method to epidemiologic investigations. Appl. Environ. Microbiol. 1990, 56, 2133–2141. [Google Scholar] [CrossRef] [PubMed]
- Piffaretti, J.C.; Kressebuch, H.; Aeschbacher, M.; Bille, J.; Bannerman, E.; Musser, J.M.; Selander, R.K.; Rocourt, J. Genetic characterization of clones of the bacterium Listeria monocytogenes causing epidemic disease. Proc. Natl. Acad. Sci. USA 1989, 86, 3818–3822. [Google Scholar] [CrossRef]
- Levenson, V.V. DNA methylation as a universal biomarker. Expert Rev. Mol. Diagn. 2010, 10, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.J.; Bereman, M.S.; Durand, S.; Valaskovic, G.A.; MacCoss, M.J. Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC-MS/MS of complex proteomic samples. J. Am. Soc. Mass. Spectrom. 2013, 24, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Y.; Li, Y.; Bai, B.; Wang, X.; Tan, H.; Liu, T.; Beach, T.G.; Peng, J.; Wu, Z. Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J. Proteome Res. 2015, 14, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Jakobsdottir, J.; van der Lee, S.J.; Bis, J.C.; Chouraki, V.; Li-Kroeger, D.; Yamamoto, S.; Grove, M.L.; Naj, A.; Vronskaya, M.; Salazar, J.L.; et al. Rare Functional Variant in TM2D3 is Associated with Late-Onset Alzheimer’s Disease. PLOS Genet. 2016, 12, e1006327. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.L.; Yang, S.-A.; Lin, Y.Q.; Li-Kroeger, D.; Marcogliese, P.C.; Deal, S.L.; Neely, G.G.; Yamamoto, S. Alzheimer’s disease-associated TM2D genes regulate Notch signaling and neuronal function in Drosophila. bioRxiv 2021, 17, e1009962. [Google Scholar] [CrossRef] [PubMed]
- Kajkowski, E.M.; Lo, C.F.; Ning, X.; Walker, S.; Sofia, H.J.; Wang, W.; Edris, W.; Chanda, P.; Wagner, E.; Vile, S.; et al. μ-Amyloid peptide-induced apoptosis regulated by a novel protein containing a G protein activation module. J. Biol. Chem. 2001, 276, 18748–18756. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, F.C. Escherichia coli and Salmonella: Cellular and Molecular Biology; ASM Press: Washington, DC, USA, 1996. [Google Scholar]
- Heithoff, D.M.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. An Essential Role for DNA Adenine Methylation in Bacterial Virulence. Science 1999, 284, 967–970. [Google Scholar] [CrossRef] [PubMed]
- García-Del Portillo, F.; Pucciarelli, M.G.; Casadesús, J. DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1999, 96, 11578–11583. [Google Scholar] [CrossRef] [PubMed]
- Julio, S.M.; Heithoff, D.M.; Provenzano, D.; Klose, K.E.; Sinsheimer, R.L.; Low, D.A.; Mahan, M.J. DNA adenine methylase is essential for viability and plays a role in the pathogenesis of Yersinia pseudotuberculosis and Vibrio cholerae. Infect. Immun. 2001, 69, 7610–7615. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.-F.; Dougherty, B.A.; Merrick, J.M. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Tost, J. DNA methylation: An introduction to the biology and the disease-associated changes of a promising biomarker. Methods Mol. Biol. 2009, 507, 3–20. [Google Scholar] [PubMed]
- Lin, R.-J.; Capage, M.; Hill, C. A repetitive DNA sequence, rhs, responsible for duplications within the Escherichia coli K-12 chromosome. J. Mol. Biol. 1984, 177, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Sandt, C.; Feulner, G.; Vlazny, D.; Gray, J.A.; Hill, C. Rhs elements of Escherichia coli K-12: Complex composites of shared and unique components that have different evolutionary histories. J. Bacteriol. 1993, 175, 2799–2808. [Google Scholar] [CrossRef] [PubMed]
- Poole, S.J.; Diner, E.J.; Aoki, S.K.; Braaten, B.A.; t’Kint de Roodenbeke, C.; Low, D.A.; Hayes, C.S. Identification of Functional Toxin/Immunity Genes Linked to Contact-Dependent Growth Inhibition (CDI) and Rearrangement Hotspot (Rhs) Systems. PLOS Genet. 2011, 7, e1002217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Iyer, L.M.; Aravind, L. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011, 39, 4532–4552. [Google Scholar] [CrossRef] [PubMed]
- Koskiniemi, S.; Lamoureux, J.G.; Nikolakakis, K.C.; t’Kint de Roodenbeke, C.; Kaplan, M.D.; Low, D.A.; Hayes, C.S. Rhs proteins from diverse bacteria mediate intercellular competition. Proc. Natl. Acad. Sci. USA 2013, 110, 7032–7037. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, K.; Lee, K.H. Overexpression of cloned RhsA sequences perturbs the cellular translational machinery in Escherichia coli. J. Bacteriol. 2011, 193, 4869–4880. [Google Scholar] [CrossRef] [PubMed]
- Beekman, C.; Janson, A.A.; Baghat, A.; van Deutekom, J.C.; Datson, N.A. Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS ONE 2018, 13, e0195850. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain No. | Serotype | Genetic Lineage | Source | Reference |
|---|---|---|---|---|
| NRRL B-33077 | 4a | III | Unknown | USDA-ARS |
| RM3030 | 4c | III | Bull | [19] |
| RM3170 | 4c | III | Unknown | [19] |
| RM3171 | 4a | III | Unknown | [19] |
| NRRL B-33014 | II | Unknown | USDA-ARS | |
| RM3000 | 1/2c | II | Soil | [19] |
| RM3020 | 1/2c | II | Unknown | [19] |
| RM3102 | 1/2a | II | Monkey | [19] |
| RM3109 | 1/2b | I | Unknown | [20] |
| NRRL B-33007 | I | Unknown | USDA-ARS | |
| RM2707 | 1/2b | I | Cheese | [20] |
| RM2995 | 1/2b | I | Cow brain | [19] |
| E. coli Strain or Plasmid | Description | Reference or Source |
|---|---|---|
| NovaBlue BL21(DE3) C41 (DE3) | endA1 hsdR17 (rK12–mK12+) supE44 thi-1 recA1 gyrA96 relA1 lac F′[proA+B+lacIqZΔM15:Tn10] (TetR) F−ompT hsdS gal; expression host F–ompT hsdSB (rB-mB-) gal dcm Derived from Bl21 (DE3) to help plasmid stability | Novagen EMD Millipore Lucigen |
| pET-28a pET28a_1897 pET28a_2770 pET28a_1129 pET28a_1926 | Expression vector; KmR pET28a with cloned ST33077_1897 pET28a with cloned ST33077_2770 pET28a with cloned ST33077_1129 pET28a with cloned ST33077_1926 | Novagen This study This study This study This study |
| ORF | Primers | Primer Sequence (5′ to 3′) | Restriction Enzymes | Amplicon Size (bp) |
|---|---|---|---|---|
| NRRL B 33077_1897 | Forward Reverse | AAAGTCGACTTTTCCTATAAACCAAA * AAAGGATCCGGTATGGGGAATAAGGA | SalI BamHI | 534 |
| NRRL B 33077_2770 | Forward Reverse | AAAGGATCCATTTTCATGTTTAATAA AAAGTCGACTTGGGCATCATTCGCTT | BamHI SalI | 567 |
| NRRL B 33077_1926 | Forward Reverse | AAAGTCGACCACCACCCCTGCCTGAT AAAGGATCCGGATCCTTTATTCGTGCAATTAA | SalI BamHI | 920 |
| NRRL B 33077_1129 | Forward Reverse | AAAGTCGACGGCACCGTCTTCGTGGT AAAGCTAGCAAGGACCTGAAGACACA | SalI NheI | 2900 |
| ORF | Lineage I (%Identity × %Coverage) * | Lineage II (%Identity × %Coverage) |
|---|---|---|
| ST33077_0135 | NS † | NS |
| ST33077_0258 | WP_003740445.1 (47.77) | WP_012951247.1 (47.32) |
| ST33077_0314 | WP_003725659.1 (10.08) | NS |
| ST33077_0492 | NS | NS |
| ST33077_0493 | WP_023550422.1 (19.28) | NS |
| ST33077_0937 | NS | NS |
| ST33077_1129 | WP_061385917.1 (2.68) | NS |
| ST33077_1328 | WP_003725868.1 (28.53) | WP_014602121.1 (28.15) |
| ST33077_1569 | WP_003728189.1 (16.05) | WP_014601042.1 (10.39) |
| ST33077_1816 | NS | NS |
| ST33077_1897 | NS | NS |
| ST33077_1926 | NS | WP_049962080.1 (26.68) |
| ST33077_2218 | NS | NS |
| ST33077_2271 | WP_003724612.1 (8.91) | WP_003731898.1 (8.91) |
| ST33077_2323 | NS | NS |
| ST33077_2398 | NS | NS |
| ST33077_2419 | NS | NS |
| ST33077_2617 | WP_021496264.1 (8.47) | NS |
| ST33077_2739 | NS | NS |
| ST33077_2770 | WP_003728337.1 (9.27) | NS |
| ORF | NCBI ID * | Similarity † (%) | Function |
|---|---|---|---|
| ST33077_0135 | NS ‡ | 0 | |
| ST33077_0258 | WP_012581893.1 | 97 | hypothetical protein |
| ST33077_0314 | WP_039381474.1 | 10.8 | guanylate kinase |
| ST33077_0492 | WP_070220410.1 | 100 | hypothetical protein |
| ST33077_0493 | WP_070220412.1 | 100 | hypothetical protein |
| ST33077_0937 | NS | 0 | |
| ST33077_1129 | WP_014589151.1 | 94.2029 | type IV secretion protein RhsA |
| ST33077_1328 | WP_003739362.1 | 28.534 | oxidoreductase |
| ST33077_1569 | WP_070221254.1 | 100 | hypothetical protein |
| ST33077_1816 | NS | 0 | |
| ST33077_1897 | WP_012582068.1 | 95 | hypothetical protein |
| ST33077_1926 | WP_077954308.1 | 100 | DNA methyltransferase |
| ST33077_2218 | NS | 0 | |
| ST33077_2271 | WP_070779485.1 | 9.54548 | peptide ABC transporter substrate-binding protein |
| ST33077_2323 | WP_070219794.1 | 100 | hypothetical protein |
| ST33077_2398 | WP_070219932.1 | 100 | hypothetical protein |
| ST33077_2419 | WP_070219871.1 | 100 | hypothetical protein |
| ST33077_2617 | WP_070284542.1 | 13.1763 | hypothetical protein |
| ST33077_2739 | NS | 0 | |
| ST33077_2770 | WP_003728958.1 | 90 | membrane protein |
| Protein | Number of Peptides | % Coverage |
|---|---|---|
| ST33077_0135 | 1 | 19.697 |
| ST33077_0258 | 1 | 8.21918 |
| ST33077_0314 | 1 | 9.42029 |
| ST33077_0492 | 7 | 6.7623 |
| ST33077_0493 | 1 | 4.8913 |
| ST33077_0937 | 3 | 15.6522 |
| ST33077_1129 | 1 | 0.87069 |
| ST33077_1328 | 12 | 8.73016 |
| ST33077_1569 | 7 | 19.4915 |
| ST33077_1816 | 1 | 9.56522 |
| ST33077_1897 | 3 | 21.4689 |
| ST33077_1926 | 31 | 24.6528 |
| ST33077_2218 | 1 | 3.76712 |
| ST33077_2271 | 1 | 6.96203 |
| ST33077_2323 | 16 | 7.22892 |
| ST33077_2398 | 1 | 7.69231 |
| ST33077_2419 | 4 | 20.7547 |
| ST33077_2617 | 1 | 12.5 |
| ST33077_2739 | 1 | 19.1781 |
| ST33077_2770 | 100 | 29.8913 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomaa, B.; Lu, J.; Abdelhamed, H.; Banes, M.; Pechanova, O.; Pechan, T.; Arick, M.A., II; Karsi, A.; Lawrence, M.L. Identification of Protein Biomarkers for Differentiating Listeria monocytogenes Genetic Lineage III. Foods 2024, 13, 1302. https://doi.org/10.3390/foods13091302
Gomaa B, Lu J, Abdelhamed H, Banes M, Pechanova O, Pechan T, Arick MA II, Karsi A, Lawrence ML. Identification of Protein Biomarkers for Differentiating Listeria monocytogenes Genetic Lineage III. Foods. 2024; 13(9):1302. https://doi.org/10.3390/foods13091302
Chicago/Turabian StyleGomaa, Basant, Jingjun Lu, Hossam Abdelhamed, Michelle Banes, Olga Pechanova, Tibor Pechan, Mark A. Arick, II, Attila Karsi, and Mark L. Lawrence. 2024. "Identification of Protein Biomarkers for Differentiating Listeria monocytogenes Genetic Lineage III" Foods 13, no. 9: 1302. https://doi.org/10.3390/foods13091302