
Controlling Arteriogenesis and Mast Cells Are Central to Bioengineering Solutions for Critical Bone Defect Repair Using Allografts

 , ,
, ,
Abstract
:1. Introduction
1.1. Fracture Repair
1.2. Critical Size Bone Defects
1.3. Autograft and Allograft Interventions
2. Current Challenges and Approaches for Critical Bone Defect Repair
2.1. Growth Factor Delivery
2.2. Intermittent rPTH Administration
3. Mast Cells and Critical Defect Repair
3.1. Overview of Mast Cells
3.2. Mast Cells in Tissue Repair: Roles in Angiogenesis, Inflammation and Bone Repair
3.3. The Role of Mast Cells in Tissue Fibrosis
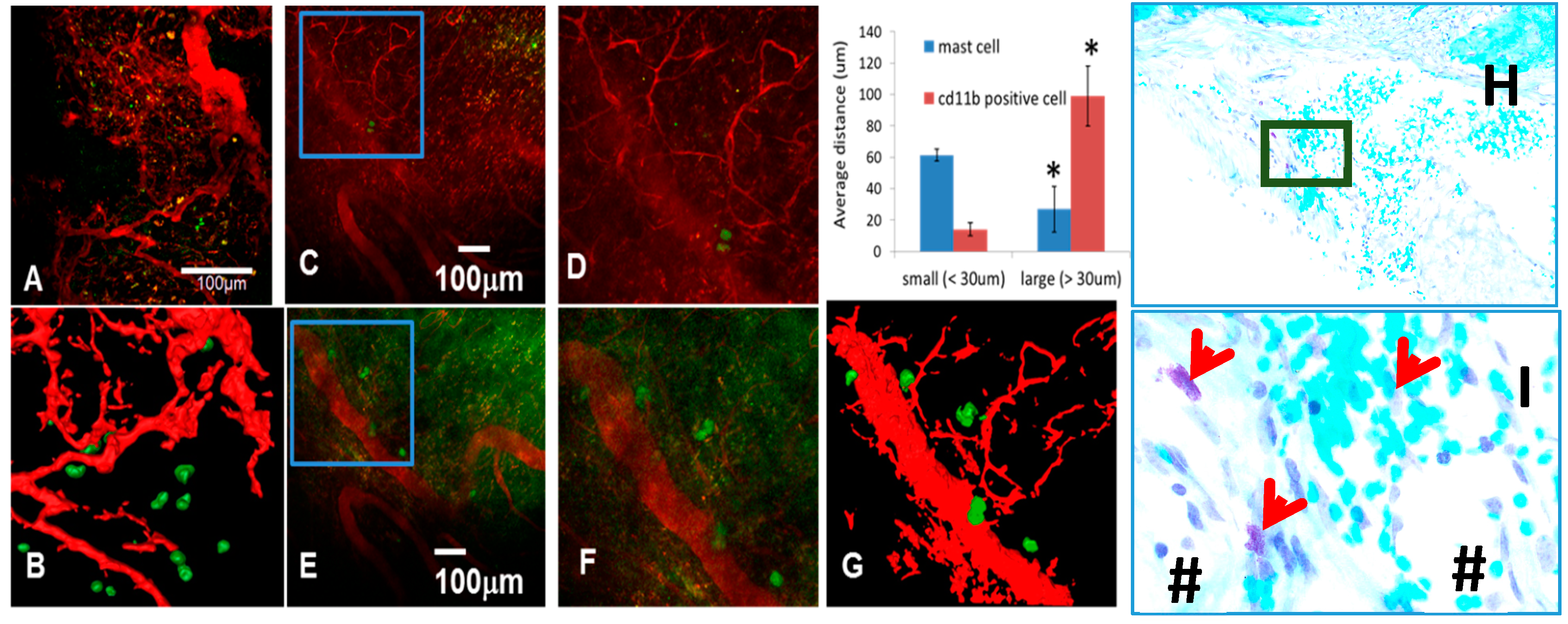
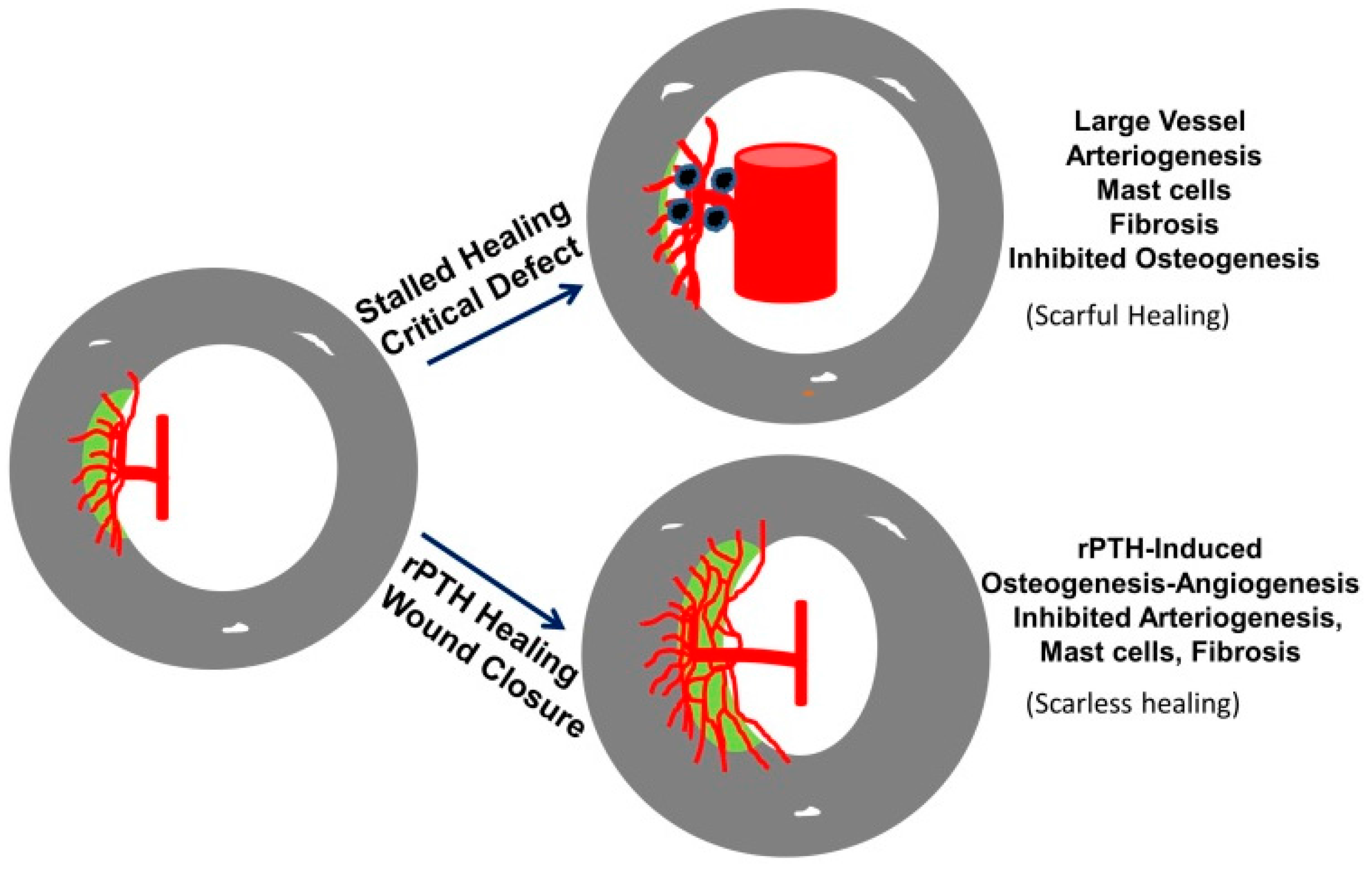
3.4. The Role of Large Vessel-Associated Mast Cells in Critical Defect Healing

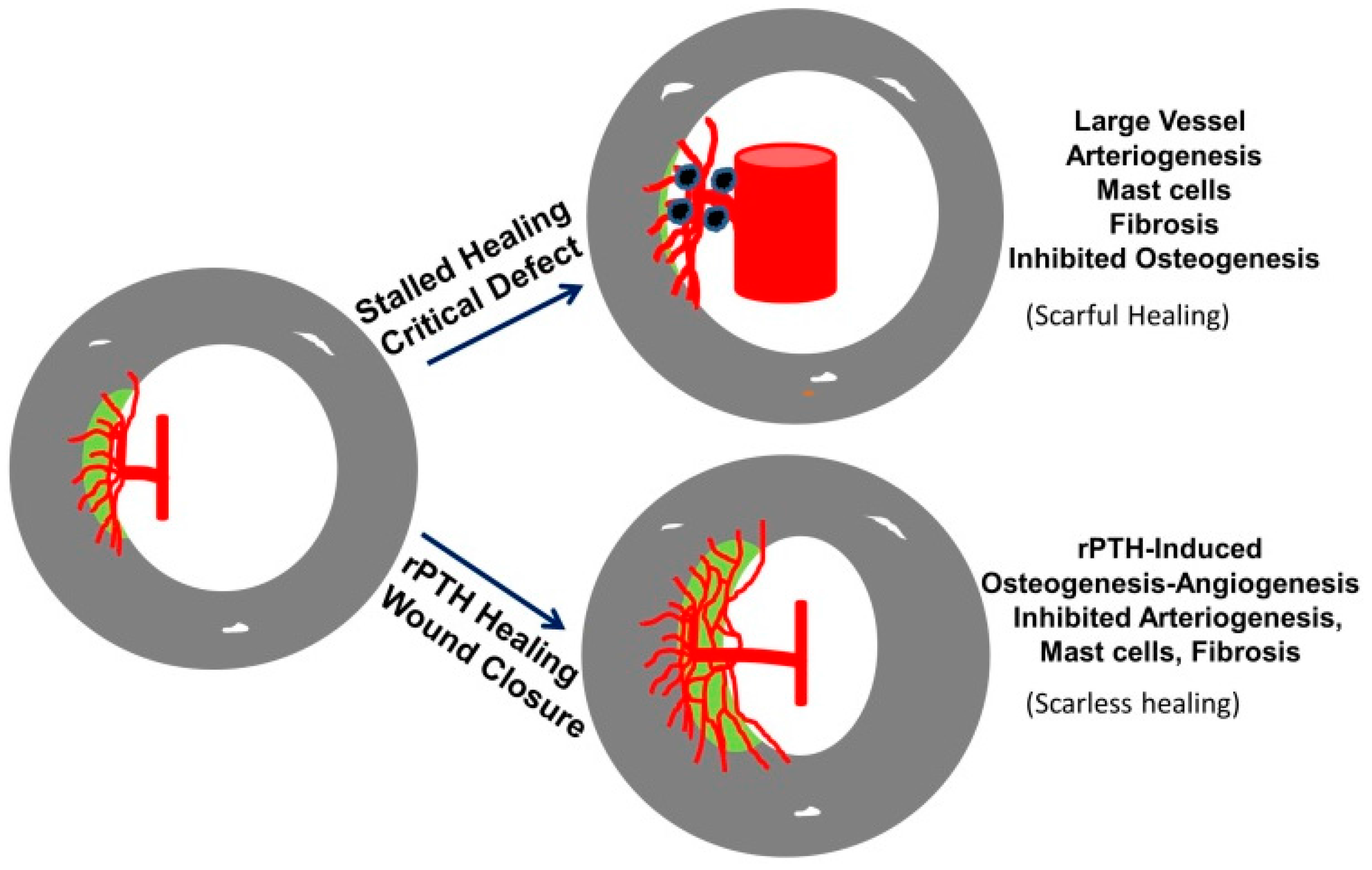
4. Conclusions

{kind=link}
{kind=link}
| Approach | Mechanism | Advantages | Disadvantages |
|---|---|---|---|
| Autografts | Osteoinduction; osteoconduction; and osteogenesis | Histoidentical; stimulates a robust regenerative response (high union ratios) | Finite amount; co-site morbidities; post-operative pain; and infection |
| Allografts | Osteoconduction | High availability and accessibility; circumvent donor site morbidity; reduced surgical time and site | Disease transmission; high failure rates; infection; immunologic reaction; and low union ratios |
| Coated Allografts | Release of inductive agents on an osteoconductive matrix | All advantages of allografts plus choice of coated agents; stimulates a robust regenerative response (high union ratios) | All disadvantages of allografts plus dependency on release of coated agents; laborious preparation; not FDA-approved |
| Intermittent rPTH | Anabolic effects on osteogenic cells; promotes small-vessel angiogenesis; inhibits arteriogenesis and mast cell accretion (early evidence) | FDA-approved adjuvant therapy; inhibits scar formation; stimulates a robust regenerative response (high union ratios) | Contra-indicated in a large target population (children and cancer patients) |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reizner, W.; Hunter, J.G.; O’Malley, N.T.; Southgate, R.D.; Schwarz, E.M.; Kates, S.L. A systematic review of animal models for staphylococcus aureus osteomyelitis. Eur. Cell Mater. 2014, 27, 196–212. [Google Scholar] [PubMed]
- Harris, J.S.; Bemenderfer, T.B.; Wessel, A.R.; Kacena, M.A. A review of mouse critical size defect models in weight bearing bones. Bone 2013, 55, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Carano, R.A.; Filvaroff, E.H. Angiogenesis and bone repair. Drug Discov. Today 2003, 8, 980–989. [Google Scholar] [CrossRef]
- Kanczler, J.M.; Oreffo, R.O. Osteogenesis and angiogenesis: The potential for engineering bone. Eur. Cell Mater. 2008, 15, 100–114. [Google Scholar] [PubMed]
- Giannotti, S.; Bottai, V.; Dell’osso, G.; Pini, E.; de Paola, G.; Bugelli, G.; Guido, G. Current medical treatment strategies concerning fracture healing. Clin. Cases Miner. Bone Metab. 2013, 10, 116–120. [Google Scholar] [PubMed]
- Schipani, E.; Maes, C.; Carmeliet, G.; Semenza, G.L. Regulation of osteogenesis-angiogenesis coupling by HIFs and VEGF. J. Bone Miner. Res. 2009, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Bosse, M.J.; MacKenzie, E.J.; Kellam, J.F.; Burgess, A.R.; Webb, L.X.; Swiontkowski, M.F.; Sanders, R.W.; Jones, A.L.; McAndrew, M.P.; Patterson, B.M.; et al. An analysis of outcomes of reconstruction or amputation after leg-threatening injuries. N. Engl. J. Med. 2002, 347, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Marino, J.T.; Ziran, B.H. Use of solid and cancellous autologous bone graft for fractures and nonunions. Orthop. Clin. N. Am. 2010, 41, 15–26, table of contents. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Enneking, W.F. Allograft bone decreases in strength in vivo over time. Clin. Orthop. Relat. Res. 2005, 36–42. [Google Scholar] [CrossRef]
- Kleinhans, C.; Mohan, R.R.; Vacun, G.; Schwarz, T.; Haller, B.; Sun, Y.; Kahlig, A.; Kluger, P.; Finne-Wistrand, A.; Walles, H.; et al. A perfusion bioreactor system efficiently generates cell-loaded bone substitute materials for addressing critical size bone defects. Biotechnol. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Verrier, S.; Alini, M. Strategies to stimulate mobilization and homing of endogenous stem and progenitor cells for bone tissue repair. Front. Bioeng. Biotechnol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Gothard, D.; Smith, E.L.; Kanczler, J.M.; Rashidi, H.; Qutachi, O.; Henstock, J.; Rotherham, M.; el Haj, A.; Shakesheff, K.M.; Oreffo, R.O. Tissue engineered bone using select growth factors: A comprehensive review of animal studies and clinical translation studies in man. Eur. Cell Mater. 2014, 28, 166–207; discussion 207–168. [Google Scholar] [PubMed]
- Amini, A.R.; Laurencin, C.T.; Nukavarapu, S.P. Bone tissue engineering: Recent advances and challenges. Crit. Rev. Biomed. Eng. 2012, 40, 363–408. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Segar, C.E.; Chu, Y.; Wang, T.W.; Lin, Y.; Yang, C.; Du, X.; Ogle, R.C.; Cui, Q.; Botchwey, E.A. Bioactive lipid coating of bone allografts directs engraftment and fate determination of bone marrow-derived cells in rat gfp chimeras. Biomaterials 2015, 64, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Krieger, J.; Huang, C.; Das, A.; Francis, M.P.; Ogle, R.; Botchwey, E. Enhanced osseous integration of human trabecular allografts following surface modification with bioactive lipids. Drug Deliv. Transl. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Koefoed, M.; Tiyapatanaputi, P.; Gromov, K.; Goater, J.J.; Carmouche, J.; Zhang, X.; Rubery, P.T.; Rabinowitz, J.; Samulski, R.J.; et al. Remodeling of cortical bone allografts mediated by adherent rAAV-RANKL and VEGF gene therapy. Nat. Med. 2005, 11, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Yazici, C.; Takahata, M.; Reynolds, D.G.; Xie, C.; Samulski, R.J.; Samulski, J.; Beecham, E.J.; Gertzman, A.A.; Spilker, M.; Zhang, X.; et al. Self-complementary AAV2.5-BMP2-coated femoral allografts mediated superior bone healing versus live autografts in mice with equivalent biomechanics to unfractured femur. Mol. Ther. 2011, 19, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Pelled, G.; Ben-Arav, A.; Hock, C.; Reynolds, D.G.; Yazici, C.; Zilberman, Y.; Gazit, Z.; Awad, H.; Gazit, D.; Schwarz, E.M. Direct gene therapy for bone regeneration: Gene delivery, animal models, and outcome measures. Tissue Eng. Part. B Rev. 2010, 16, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ben-Arav, A.; Pelled, G.; Zilberman, Y.; Kimelman-Bleich, N.; Gazit, Z.; Schwarz, E.M.; Gazit, D. Adeno-associated virus-coated allografts: A novel approach for cranioplasty. J. Tissue Eng. Regen. Med. 2012, 6, e43–e50. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Yano, K.; Wakitani, S.; Matsumoto, T.; Nakamura, H.; Takaoka, K. Repair of critical long bone defects using frozen bone allografts coated with an rhBMP-2-retaining paste. J. Orthop. Sci. 2012, 17, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.B.; Sabatino, C.T.; Badura, J.M.; Sietsema, D.L.; Marotta, J.S. Improved healing efficacy in canine ulnar segmental defects with increasing recombinant human bone morphogenetic protein-2/allograft ratios. J. Orthop. Trauma 2008, 22, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Baas, J.; Elmengaard, B.; Jensen, T.B.; Jakobsen, T.; Andersen, N.T.; Soballe, K. The effect of pretreating morselized allograft bone with rhBMP-2 and/or pamidronate on the fixation of porous Ti and HA-coated implants. Biomaterials 2008, 29, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Sharmin, F.; Adams, D.; Pensak, M.; Dukas, A.; Lieberman, J.; Khan, Y. Biofunctionalizing devitalized bone allografts through polymer-mediated short and long term growth factor delivery. J. Biomed. Mater. Res. A 2015, 103, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xie, C.; Lin, A.S.; Ito, H.; Awad, H.; Lieberman, J.R.; Rubery, P.T.; Schwarz, E.M.; O’Keefe, R.J.; Guldberg, R.E. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: Implications for functional tissue engineering. J. Bone Miner. Res. 2005, 20, 2124–2137. [Google Scholar] [CrossRef] [PubMed]
- Street, J.; Bao, M.; deGuzman, L.; Bunting, S.; Peale, F.V., Jr.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.; Neumeyer, K.; Kirkpatrick, P. Teriparatide. Nat. Rev. Drug Discov. 2003, 2, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, J.J.; Mitlak, B.H.; Wang, O.; Genant, H.K.; Eriksen, E.F. Recombinant human parathyroid hormone (1–34) [Teriparatide] improves both cortical and cancellous bone structure. J. Bone Miner. Res. 2003, 18, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.; Zhou, H.; Cosman, F.; Nieves, J.; Dempster, D.W.; Hodsman, A.B. Effects of a one-month treatment with PTH(1–34) on bone formation on cancellous, endocortical, and periosteal surfaces of the human ilium. J. Bone Miner. Res. 2007, 22, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Q.; Lang, T.F.; Arnaud, C.D.; Modin, G.W.; Lane, N.E. Daily treatment with parathyroid hormone is associated with an increase in vertebral cross-sectional area in postmenopausal women with glucocorticoid-induced osteoporosis. Osteoporos. Int. 2003, 14, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Hodsman, A.B.; Bauer, D.C.; Dempster, D.W.; Dian, L.; Hanley, D.A.; Harris, S.T.; Kendler, D.L.; McClung, M.R.; Miller, P.D.; Olszynski, W.P.; et al. Parathyroid hormone and teriparatide for the treatment of osteoporosis: A review of the evidence and suggested guidelines for its use. Endocr. Rev. 2005, 26, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Girotra, M.; Rubin, M.R.; Bilezikian, J.P. The use of parathyroid hormone in the treatment of osteoporosis. Rev. Endocr. Metab. Disord. 2006, 7, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Boonen, S.; Marin, F.; Mellstrom, D.; Xie, L.; Desaiah, D.; Krege, J.H.; Rosen, C.J. Safety and efficacy of teriparatide in elderly women with established osteoporosis: Bone anabolic therapy from a geriatric perspective. J. Am. Geriatr. Soc. 2006, 54, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Kaback, L.A.; Soung-Do, Y.; Naik, A.; Geneau, G.; Schwarz, E.M.; Rosier, R.N.; O’Keefe, R.J.; Drissi, H. Teriparatide (1–34 human PTH) regulation of osterix during fracture repair. J. Cell Biochem. 2008, 105, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhao, X.; Yang, C.; Crane, J.; Xian, L.; Lu, W.; Wan, M.; Cao, X. Parathyroid hormone induces differentiation of mesenchymal stromal/stem cells by enhancing bone morphogenetic protein signaling. J. Bone Miner. Res. 2012, 27, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Carpio, L.; Gladu, J.; Goltzman, D.; Rabbani, S.A. Induction of osteoblast differentiation indexes by PTHrP in MG-63 cells involves multiple signaling pathways. Am. J. Physiol. Endocrinol. Metab. 2001, 281, 489–499. [Google Scholar]
- Daugaard, H.; Elmengaard, B.; Andreassen, T.T.; Baas, J.; Bechtold, J.E.; Soballe, K. The combined effect of parathyroid hormone and bone graft on implant fixation. J. Bone Jt. Surg. Br. 2011, 93, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.G.; Takahata, M.; Lerner, A.L.; O’Keefe, R.J.; Schwarz, E.M.; Awad, H.A. Teriparatide therapy enhances devitalized femoral allograft osseointegration and biomechanics in a murine model. Bone 2011, 48, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Washimi, Y.; Kanaji, A.; Tajima, K.; Ishimura, D.; Yamada, H. The effect of bisphosphonate and intermittent human parathyroid hormone 1–34 treatments on cortical bone allografts in rabbits. J. Endocrinol. Investig. 2012, 35, 139–145. [Google Scholar] [PubMed]
- Takahata, M.; Schwarz, E.M.; Chen, T.; O’Keefe, R.J.; Awad, H.A. Delayed short course treatment with teriparatide (PTH(1–34) ) improves femoral allograft healing by enhancing intramembranous bone formation at the graft-host junction. J. Bone Miner. Res. 2012, 27, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, R.S.; Xie, C.; Tyler, W.; Calvi, L.M.; Awad, H.A.; Zuscik, M.J.; O’Keefe, R.J.; Schwarz, E.M. PTH enhanced structural allograft healing is associated with decreased angiopoietin-2 mediated arteriogenesis, mast cell accumulation and fibrosis. J. Bone Miner. Res. 2013, 28, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Abzhanov, A.; Rodda, S.J.; McMahon, A.P.; Tabin, C.J. Regulation of skeletogenic differentiation in cranial dermal bone. Development 2007, 134, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Sheyn, D.; Cohn-Yakubovich, D.; Kallai, I.; Su, S.; Da, X.; Pelled, G.; Tawackoli, W.; Cook-Weins, G.; Schwarz, E.M.; Gazit, D.; et al. Pth promotes allograft integration in a calvarial bone defect. Mol. Pharm. 2013, 10, 4462–4471. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.L.; Kakar, S.; Vora, S.; Morgan, E.F.; Gerstenfeld, L.C.; Einhorn, T.A. Stimulation of fracture-healing with systemic intermittent parathyroid hormone treatment. J. Bone Jt. Surg. Am. 2008, 90, S120–S127. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.G.; Shaikh, S.; Papuga, M.O.; Lerner, A.L.; O’Keefe, R.J.; Schwarz, E.M.; Awad, H.A. MuCT-based measurement of cortical bone graft-to-host union. J. Bone Miner. Res. 2009, 24, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Rubery, P.T.; Bukata, S.V. Teriparatide may accelerate healing in delayed unions of type III odontoid fractures: A report of 3 cases. J. Spinal. Disord. Tech. 2010, 23, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Deshpande, S.S.; Donneys, A.; Rodriguez, J.J.; Nelson, N.S.; Felice, P.A.; Chepeha, D.B.; Buchman, S.R. Parathyroid hormone reverses radiation induced hypovascularity in a murine model of distraction osteogenesis. Bone 2013, 56, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. The controversial role of mast cells in tumor growth. Int. Rev. Cell Mol. Biol. 2009, 275, 89–131. [Google Scholar] [PubMed]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [PubMed]
- Rodewald, H.R.; Feyerabend, T.B. Widespread immunological functions of mast cells: Fact or fiction? Immunity 2012, 37, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wulff, B.C.; Wilgus, T.A. Mast cell activity in the healing wound: More than meets the eye? Exp. Dermatol 2013, 22, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Oschatz, C.; Maas, C.; Lecher, B.; Jansen, T.; Bjorkqvist, J.; Tradler, T.; Sedlmeier, R.; Burfeind, P.; Cichon, S.; Hammerschmidt, S.; et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 2011, 34, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kunder, C.A.; St John, A.L.; Abraham, S.N. Mast cell modulation of the vascular and lymphatic endothelium. Blood 2011, 118, 5383–5393. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, A.M.; Beaven, M.A. Regulation of mast cell responses in health and disease. Crit. Rev. Immunol. 2011, 31, 475–529. [Google Scholar] [CrossRef] [PubMed]
- Weller, K.; Foitzik, K.; Paus, R.; Syska, W.; Maurer, M. Mast cells are required for normal healing of skin wounds in mice. FASEB J. 2006, 20, 2366–2368. [Google Scholar] [CrossRef] [PubMed]
- Egozi, E.I.; Ferreira, A.M.; Burns, A.L.; Gamelli, R.L.; Dipietro, L.A. Mast cells modulate the inflammatory but not the proliferative response in healing wounds. Wound Repair Regen. 2003, 11, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Bankaitis, E.; Heimbach, L.; Li, N.; Abrink, M.; Pejler, G.; An, L.; Diaz, L.A.; Werb, Z.; Liu, Z. Dual targets for mouse mast cell protease-4 in mediating tissue damage in experimental bullous pemphigoid. J. Biol. Chem. 2011, 286, 37358–37367. [Google Scholar] [CrossRef] [PubMed]
- Takato, H.; Yasui, M.; Ichikawa, Y.; Waseda, Y.; Inuzuka, K.; Nishizawa, Y.; Tagami, A.; Fujimura, M.; Nakao, S. The specific chymase inhibitor TY-51469 suppresses the accumulation of neutrophils in the lung and reduces silica-induced pulmonary fibrosis in mice. Exp. Lung Res. 2011, 37, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Younan, G.; Suber, F.; Xing, W.; Shi, T.; Kunori, Y.; Abrink, M.; Pejler, G.; Schlenner, S.M.; Rodewald, H.R.; Moore, F.D., Jr.; et al. The inflammatory response after an epidermal burn depends on the activities of mouse mast cell proteases 4 and 5. J. Immunol. 2010, 185, 7681–7690. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Nishikori, Y.; Kakizoe, E.; Shimoura, K.; Niibayashi, T.; Shimbori, C.; Tanaka, T.; Okunishi, H. Pathophysiological role of skin mast cells in wound healing after scald injury: Study with mast cell-deficient W/W(V) mice. Int. Arch. Allergy Immunol. 2010, 151, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Liebler, J.M.; Powers, M.R.; Galey, T.; Ahmadi, P.; Huang, X.N.; Ansel, J.C.; Butterfield, J.H.; Planck, S.R.; Rosenbaum, J.T. Mast cells are a major source of basic fibroblast growth factor in chronic inflammation and cutaneous hemangioma. Am. J. Pathol 1995, 147, 564–573. [Google Scholar] [PubMed]
- Qu, Z.; Huang, X.; Ahmadi, P.; Stenberg, P.; Liebler, J.M.; Le, A.C.; Planck, S.R.; Rosenbaum, J.T. Synthesis of basic fibroblast growth factor by murine mast cells. Regulation by transforming growth factor beta, tumor necrosis factor alpha, and stem cell factor. Int. Arch. Allergy Immunol. 1998, 115, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Kayton, R.J.; Ahmadi, P.; Liebler, J.M.; Powers, M.R.; Planck, S.R.; Rosenbaum, J.T. Ultrastructural immunolocalization of basic fibroblast growth factor in mast cell secretory granules. Morphological evidence for bFGF release through degranulation. J. Histochem. Cytochem. 1998, 46, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Grutzkau, A.; Kruger-Krasagakes, S.; Baumeister, H.; Schwarz, C.; Kogel, H.; Welker, P.; Lippert, U.; Henz, B.M.; Moller, A. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: Implications for the biological significance of VEGF206. Mol. Biol. Cell. 1998, 9, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Blair, R.J.; Meng, H.; Marchese, M.J.; Ren, S.; Schwartz, L.B.; Tonnesen, M.G.; Gruber, B.L. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. J. Clin. Invest. 1997, 99, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, R.; Lindholm, S.; Liukko, P. Fracture healing and mast cells. I. The periosteal callus in rats. Acta Orthop. Scand. 1967, 38, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Banovac, K.; Renfree, K.; Makowski, A.L.; Latta, L.L.; Altman, R.D. Fracture healing and mast cells. J. Orthop. Trauma 1995, 9, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L. Mast cells in the pathogenesis of fibrosis. Curr. Rheumatol. Rep. 2003, 5, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Yokoyama, Y.; Amano, H.; Matsushima, Y.; Kan, C.; Ishikawa, O. Effect of activated human mast cells and mast cell-derived mediators on proliferation, type I collagen production and glycosaminoglycans synthesis by human dermal fibroblasts. Eur. J. Dermatol. 2002, 12, 340–346. [Google Scholar] [PubMed]
- Hirai, S.; Ohyane, C.; Kim, Y.I.; Lin, S.; Goto, T.; Takahashi, N.; Kim, C.S.; Kang, J.; Yu, R.; Kawada, T. Involvement of mast cells in adipose tissue fibrosis. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E247–E255. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Gan, P.Y.; Dewage, L.; Ma, F.T.; Ooi, J.D.; O’Sullivan, K.M.; Nikolic-Paterson, D.J.; Kitching, A.R.; Holdsworth, S.R. Mast cell activation and degranulation promotes renal fibrosis in experimental unilateral ureteric obstruction. Kidney Int. 2012, 82, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Erjefalt, J.S. Mast cells in human airways: The culprit? Eur. Respir. Rev. 2014, 23, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Ningyan, G.; Xu, Y.; Hongfei, S.; Jingjing, C.; Min, C. The role of macrophage migration inhibitory factor in mast cell-stimulated fibroblast proliferation and collagen production. PLoS ONE 2015, 10, e0122482. [Google Scholar] [CrossRef] [PubMed]
- Hugle, T. Beyond allergy: The role of mast cells in fibrosis. Swiss Med. Wkly. 2014, 144. [Google Scholar] [CrossRef] [PubMed]
- Gailit, J.; Marchese, M.J.; Kew, R.R.; Gruber, B.L. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J. Invest. Dermatol. 2001, 117, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Hatamochi, A.; Fujiwara, K.; Ueki, H. Effects of histamine on collagen synthesis by cultured fibroblasts derived from guinea pig skin. Arch. Dermatol. Res. 1985, 277, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Kupietzky, A.; Levi-Schaffer, F. The role of mast cell-derived histamine in the closure of an in vitro wound. Inflamm. Res. 1996, 45, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Gruber, B.L.; Kew, R.R.; Jelaska, A.; Marchese, M.J.; Garlick, J.; Ren, S.; Schwartz, L.B.; Korn, J.H. Human mast cells activate fibroblasts: Tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J. Immunol. 1997, 158, 2310–2317. [Google Scholar] [PubMed]
- Albrecht, M.; Frungieri, M.B.; Kunz, L.; Ramsch, R.; Meineke, V.; Kohn, F.M.; Mayerhofer, A. Divergent effects of the major mast cell products histamine, tryptase and TNF-alpha on human fibroblast behaviour. Cell. Mol. Life Sci. 2005, 62, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Hermes, B.; Feldmann-Boddeker, I.; Welker, P.; Algermissen, B.; Steckelings, M.U.; Grabbe, J.; Henz, B.M. Altered expression of mast cell chymase and tryptase and of c-Kit in human cutaneous scar tissue. J. Investig. Dermatol. 2000, 114, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Kofford, M.W.; Schwartz, L.B.; Schechter, N.M.; Yager, D.R.; Diegelmann, R.F.; Graham, M.F. Cleavage of type I procollagen by human mast cell chymase initiates collagen fibril formation and generates a unique carboxyl-terminal propeptide. J. Biol. Chem. 1997, 272, 7127–7131. [Google Scholar] [CrossRef] [PubMed]
- Pistorio, A.L.; Ehrlich, H.P. Modulatory effects of connexin-43 expression on gap junction intercellular communications with mast cells and fibroblasts. J. Cell. Biochem. 2011, 112, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Au, S.R.; Au, K.; Saggers, G.C.; Karne, N.; Ehrlich, H.P. Rat mast cells communicate with fibroblasts via gap junction intercellular communications. J. Cell. Biochem. 2007, 100, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Moyer, K.E.; Saggers, G.C.; Ehrlich, H.P. Mast cells promote fibroblast populated collagen lattice contraction through gap junction intercellular communication. Wound Repair Regen. 2004, 12, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.T.; Saggers, G.C.; Moyer, K.E.; Ehrlich, H.P. Rat mast cells enhance fibroblast proliferation and fibroblast-populated collagen lattice contraction through gap junctional intercellular communications. Plast. Reconstr. Surg. 2011, 127, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ness, V.P.; Yang, X.; Chen, H.; Luo, J.; Brown, E.B.; Zhang, X. Spatiotemporal analyses of osteogenesis and angiogenesis via intravital imaging in cranial bone defect repair. J. Bone Miner. Res. 2015, 30, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Scholten, J.; Hartmann, K.; Gerbaulet, A.; Krieg, T.; Muller, W.; Testa, G.; Roers, A. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 2008, 17, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.B.; Gilsenan, A.W.; Midkiff, K.; Sherrill, B.; Wu, Y.; Mann, B.H.; Masica, D. The us postmarketing surveillance study of adult osteosarcoma and teriparatide: Study design and findings from the first 7 years. J. Bone Miner. Res. 2012, 27, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Aspenberg, P.; Genant, H.K.; Johansson, T.; Nino, A.J.; See, K.; Krohn, K.; Garcia-Hernandez, P.A.; Recknor, C.P.; Einhorn, T.A.; Dalsky, G.P.; et al. Teriparatide for acceleration of fracture repair in humans: A prospective, randomized, double-blind study of 102 postmenopausal women with distal radial fractures. J. Bone Miner. Res. 2009, 25, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Aspenberg, P.; Johansson, T. Teriparatide improves early callus formation in distal radial fractures. Acta Orthop. 2010, 81, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Peichl, P.; Holzer, L.A.; Maier, R.; Holzer, G. Parathyroid hormone 1–84 accelerates fracture-healing in pubic bones of elderly osteoporotic women. J. Bone Jt. Surg. Am. 2011, 93, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Bashutski, J.D.; Eber, R.M.; Kinney, J.S.; Benavides, E.; Maitra, S.; Braun, T.M.; Giannobile, W.V.; McCauley, L.K. Teriparatide and osseous regeneration in the oral cavity. N. Engl. J. Med. 2010, 363, 2396–2405. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antebi, B.; Zhang, L.; Sheyn, D.; Pelled, G.; Zhang, X.; Gazit, Z.; Schwarz, E.M.; Gazit, D. Controlling Arteriogenesis and Mast Cells Are Central to Bioengineering Solutions for Critical Bone Defect Repair Using Allografts. Bioengineering 2016, 3, 6. https://doi.org/10.3390/bioengineering3010006
Antebi B, Zhang L, Sheyn D, Pelled G, Zhang X, Gazit Z, Schwarz EM, Gazit D. Controlling Arteriogenesis and Mast Cells Are Central to Bioengineering Solutions for Critical Bone Defect Repair Using Allografts. Bioengineering. 2016; 3(1):6. https://doi.org/10.3390/bioengineering3010006
Chicago/Turabian StyleAntebi, Ben, Longze Zhang, Dmitriy Sheyn, Gadi Pelled, Xinping Zhang, Zulma Gazit, Edward M. Schwarz, and Dan Gazit. 2016. "Controlling Arteriogenesis and Mast Cells Are Central to Bioengineering Solutions for Critical Bone Defect Repair Using Allografts" Bioengineering 3, no. 1: 6. https://doi.org/10.3390/bioengineering3010006