
DNA and RNA Extraction and Quantitative Real-Time PCR-Based Assays for Biogas Biocenoses in an Interlaboratory Comparison


Abstract
:1. Introduction
- Does a given kit-based DNA extraction protocol (with small modifications) yield similar qPCR results in a ring-trial in an interlaboratory comparison? Do modifications that were introduced by some partners impact the results, and which differences can be seen if a traditional chloroform-based routine is used?
- How efficient is the extraction considering that maximum assay sensitivity, i.e., lowest possible method detection limit should be provided?
- How suitable are the extracts for qPCR? If PCR inhibition is observed, which qualities do the extracts have?
- Do the different qPCR protocols that were applied in the ring-trial yield different results? Are there any peculiarities explaining the differences?
- Which are the most important process steps, control points and benchmarks that should be respected in the qPCR analysis pipeline, and are traditional absorption ratio thresholds helpful?
- Which methods of RNA preservation can be recommended for biogas fermenter sludge samples?
- Which pitfalls do the different RNA extraction protocols present for different purposes such as RT-qPCR and metatranscriptome analysis?
2. Experimental Section
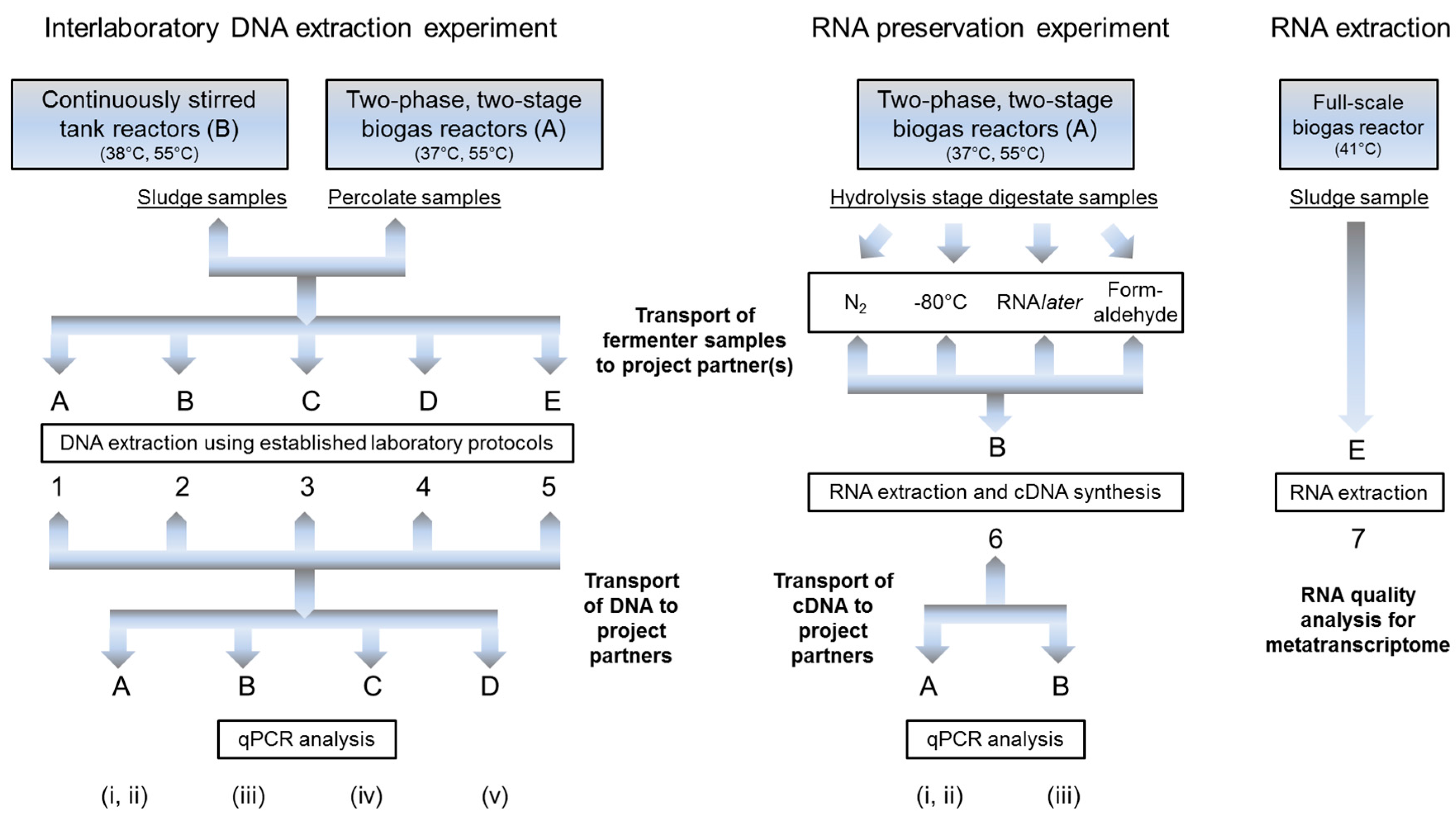
2.1. General Outline of the Experiments

2.2. Substrate, Reactor Set-Up and Sampling
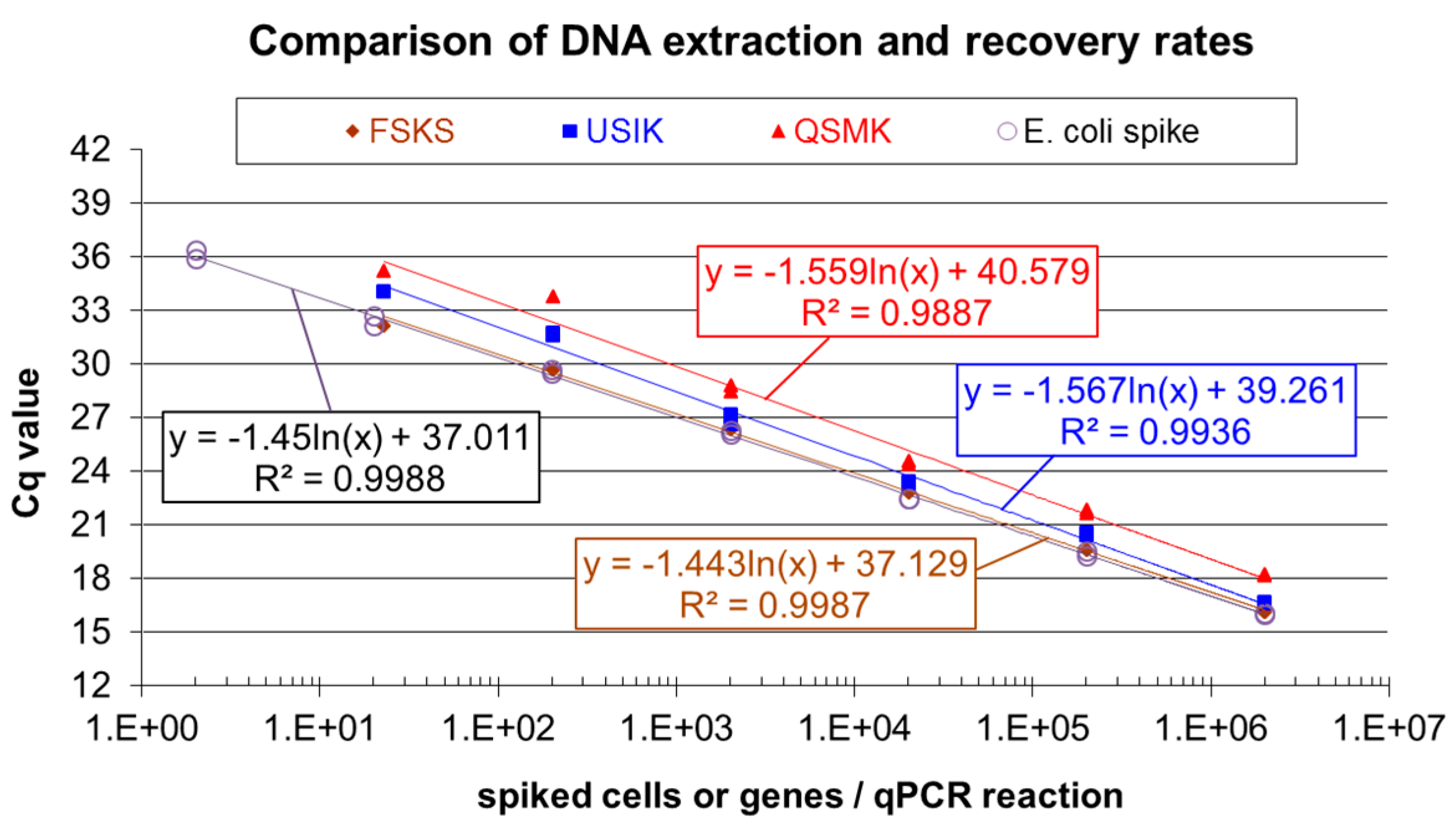
2.3. Comparison of DNA Extraction Kits and DNA Recovery Rates
2.4. DNA Extraction Ring-Trial

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol | 1 | 2 * | 3 * | 4 | 5 | |
|---|---|---|---|---|---|---|
| extracted sample volume # | percolate (HR) | 2 mL | 100 μL | 40 μL | 2 mL | 26 g |
| sludge (CSTR) | 50 μL | 40 μL | 50 μL | 26 g | ||
| pretreatment of samples | 60 s Ultra-Turrax, centrifuged, 2× 0.85% KCl-washed | centrifuged, resuspended in ultrapure H2O | washed with PBS | |||
| cell lysis | 2× 20 s @ 5.0 bead beater (MP Biome-dicals, Lysing Matrix E Tubes) | 40 s @ 6.0 bead beater (MP Bio-medicals, Lysing Matrix E Tubes) | disruptor (Scientif. industr.); MP Bio-medicals Lysing Matrix E Tubes | CTAB/pronase ε/SDS/65 °C | ||
| involvement of a kit | FastDNA Spin kit for Soil | no | ||||
| DNA-purification | kit components, silica matrix based, no post-purification | chloroform:iso-amylalcohole, isopropanol | ||||
| elution volume # | 100 μL | 75 μL | 100 μL | |||
2.5. Preservation Methods for RNA Stability
2.6. RNA Extraction and Reverse Transcription
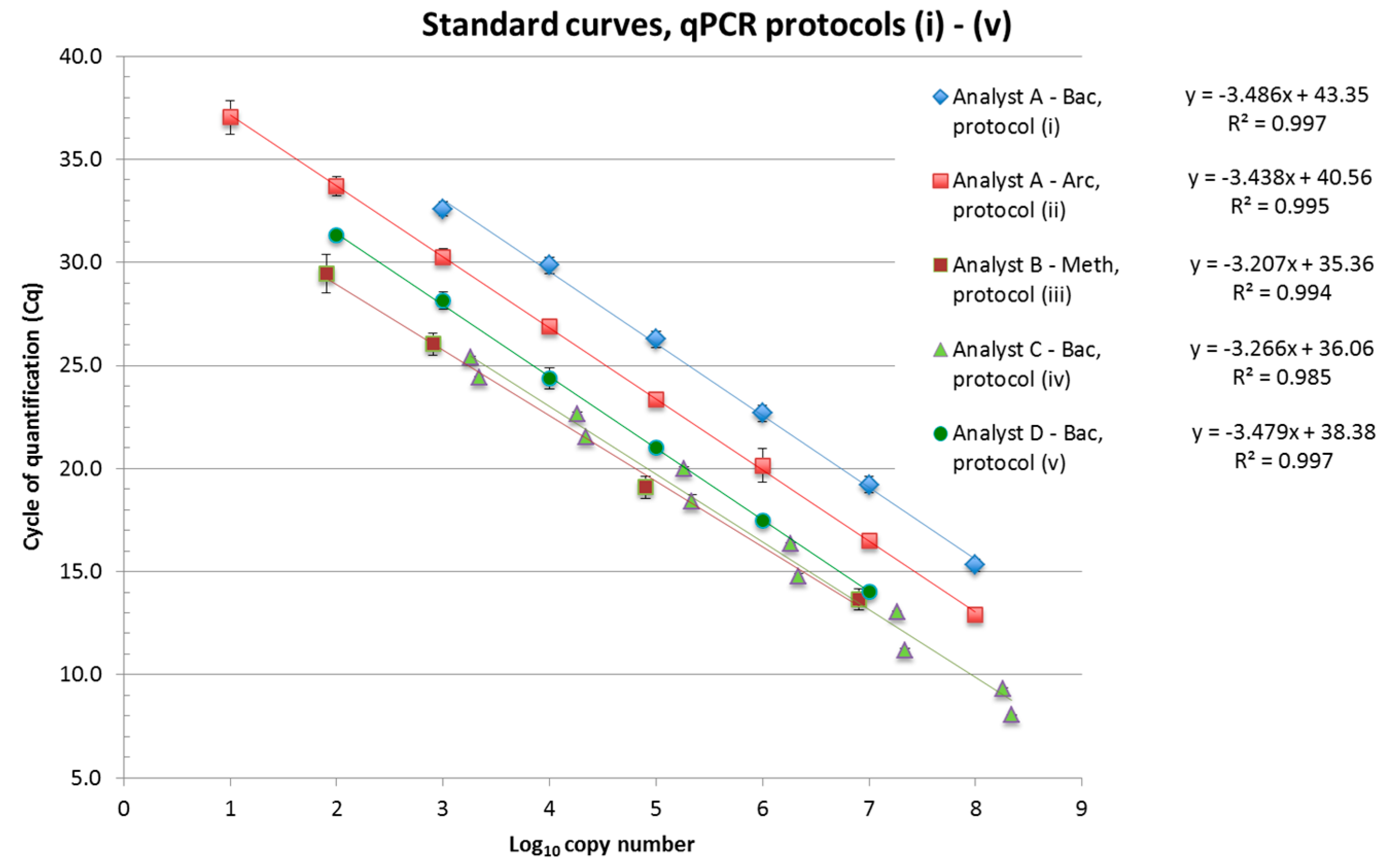
2.7. Real-Time Quantitative PCR Assays

3. Results and Discussion
3.1. Comparison of DNA Extraction Kits and DNA Recovery Rates

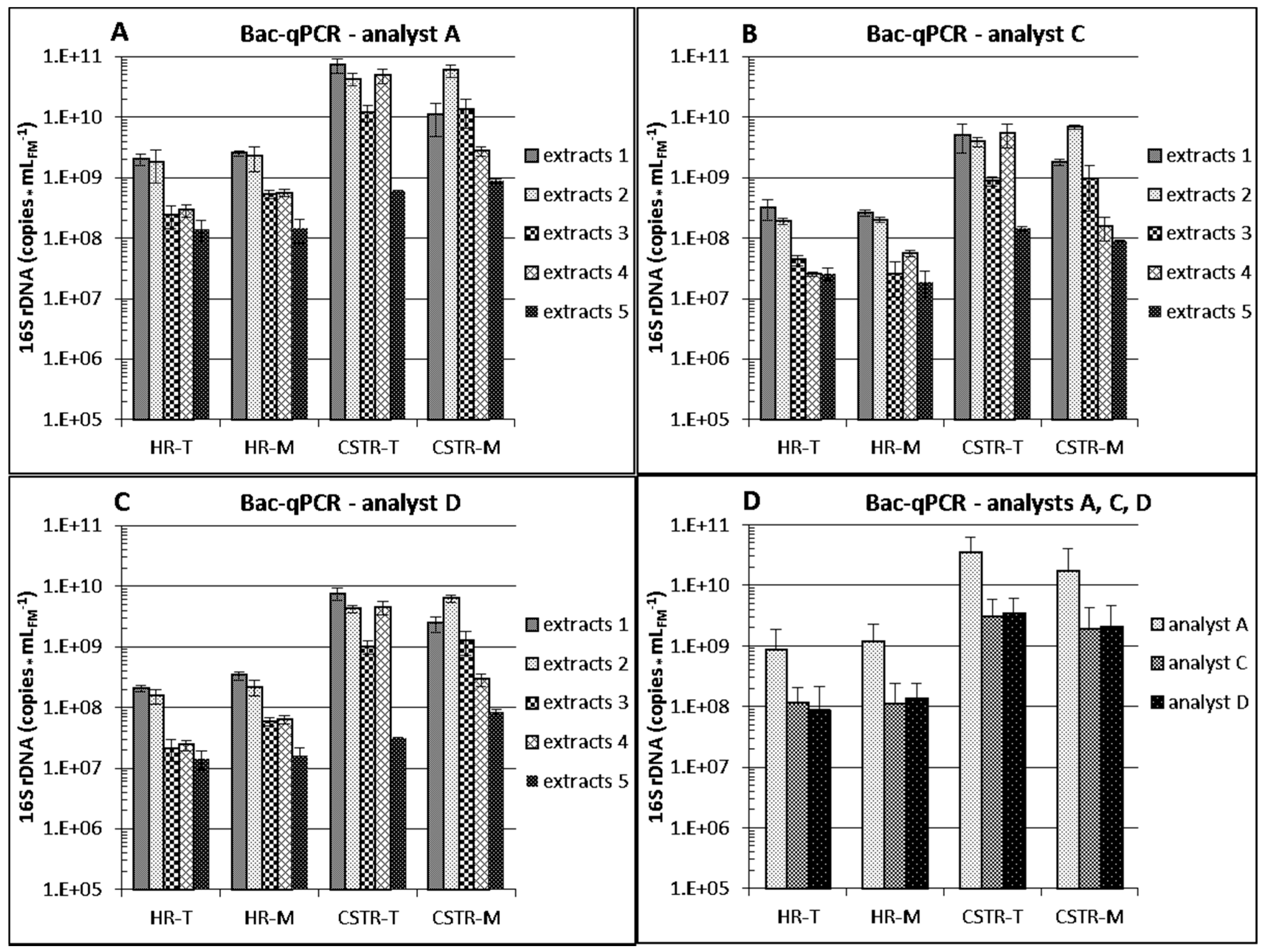
3.2. Interlaboratory Comparison of DNA Extraction
| qPCR Assay, Partner/Analyst | Bac (i), A | Arc (ii), A | Meth (iii), B | Bac (iv), C | Bac (v), D |
|---|---|---|---|---|---|
| CV (%) | |||||
| analytical | 7.56 | 7.20 | 7.47 | 10.10 | 13.38 |
| technical | 20.92 | 20.80 | 24.85 | n.d. | 16.33 |
| biological | 28.49 | 28.70 | 32.24 | 25.45 | 23.27 |
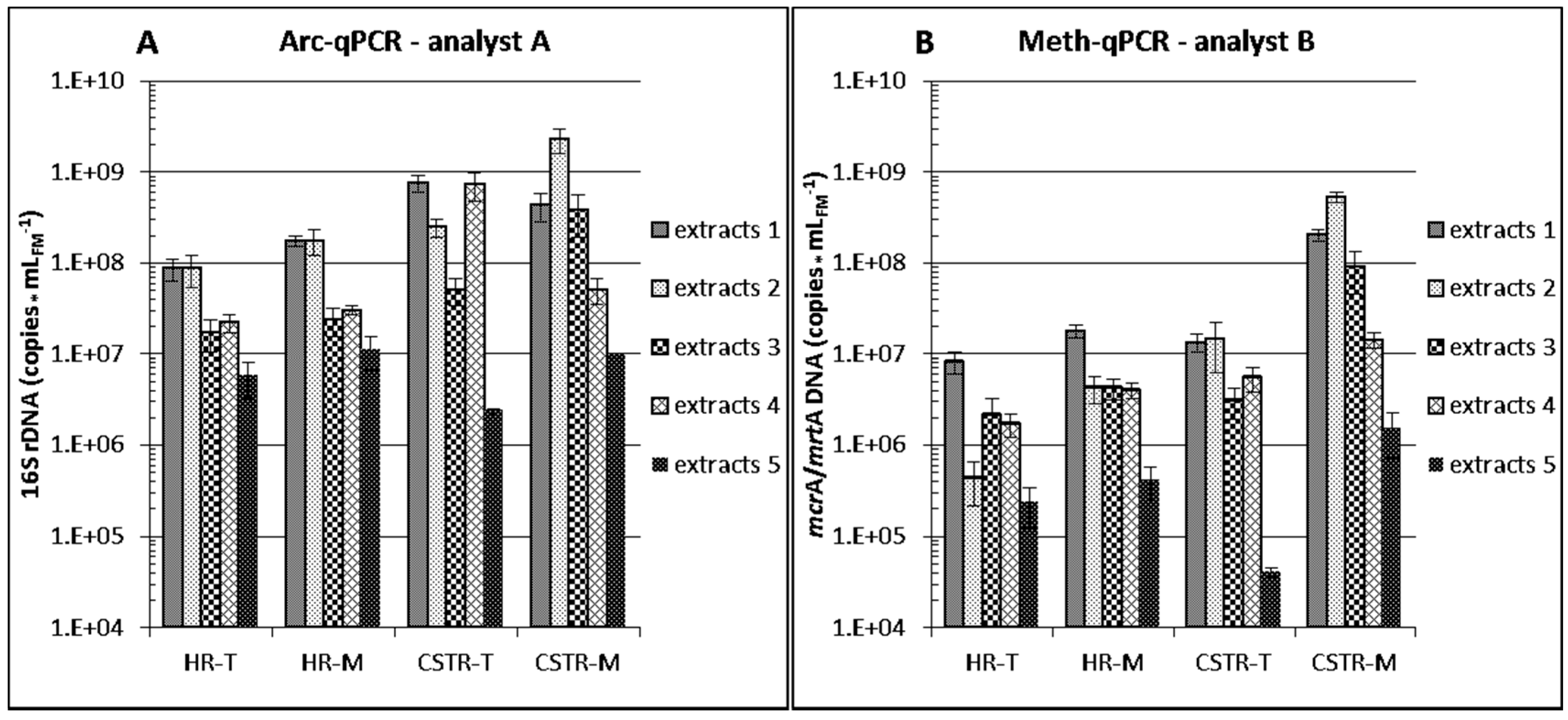
3.2.1. Differences between Reactor Types and Operation


3.2.2. Differences between Analytical Routines
3.2.3. Differences between DNA Extraction Protocols
| DNA Extracts Obtained with | Bac-qPCR (i, iv, v) | Arc-qPCR (ii) | Meth-qPCR (iii) |
|---|---|---|---|
| Mean over Analysts and Fermenters (copies · mLFM−1) | |||
| protocol 1 | 8.79 × 109 | 3.64 × 108 | 6.09 × 107 |
| protocol 2 | 1.07 × 1010 | 6.99 × 108 | 1,38 × 108 |
| protocol 3 | 2.54 × 109 | 1.18 × 108 | 2.47 × 107 |
| protocol 4 | 5.26 × 109 | 2.08 × 108 | 6.38 × 106 |
| protocol 5 | 3.52 × 108 | 7.17 × 106 | 5.42 × 105 |
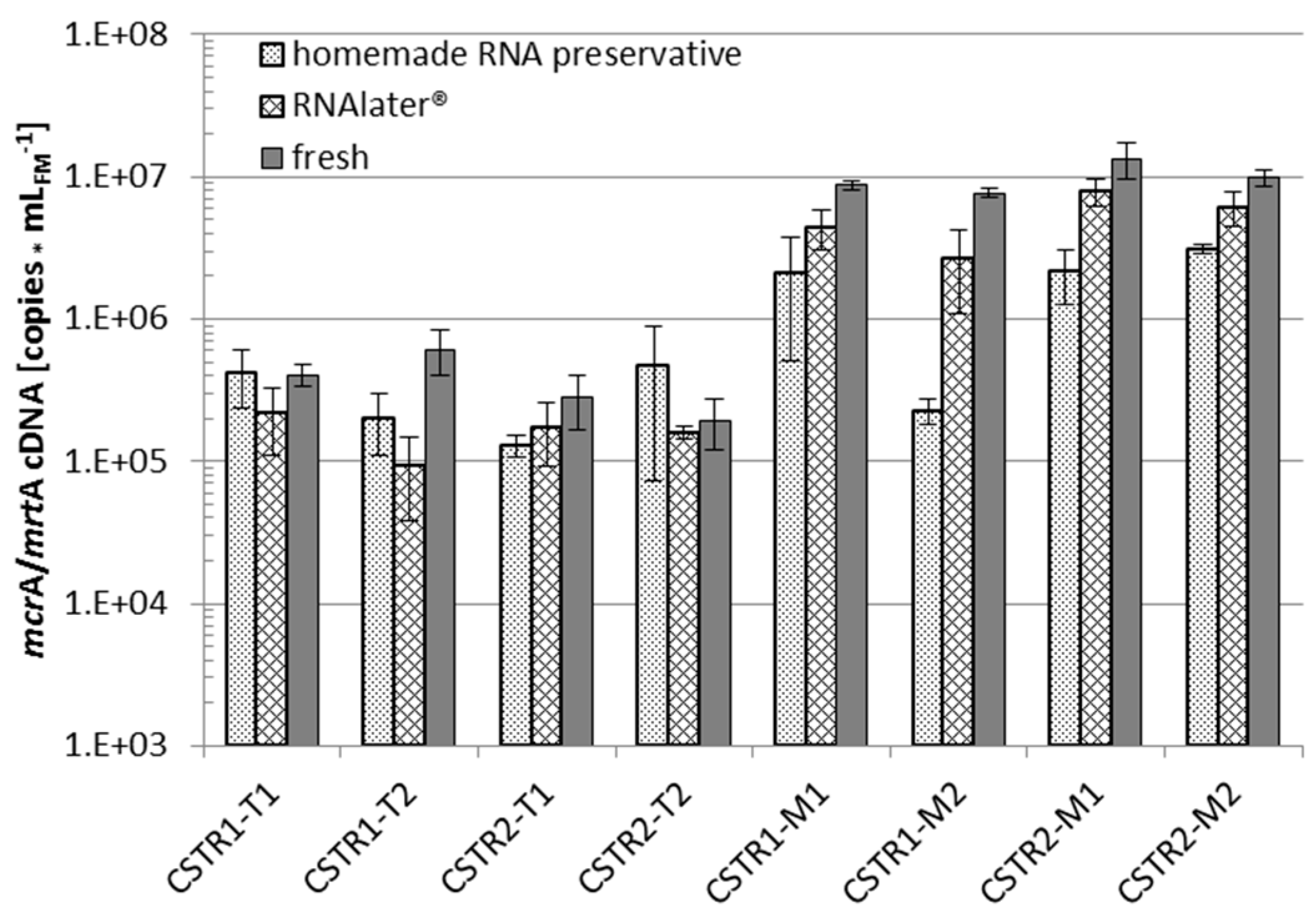
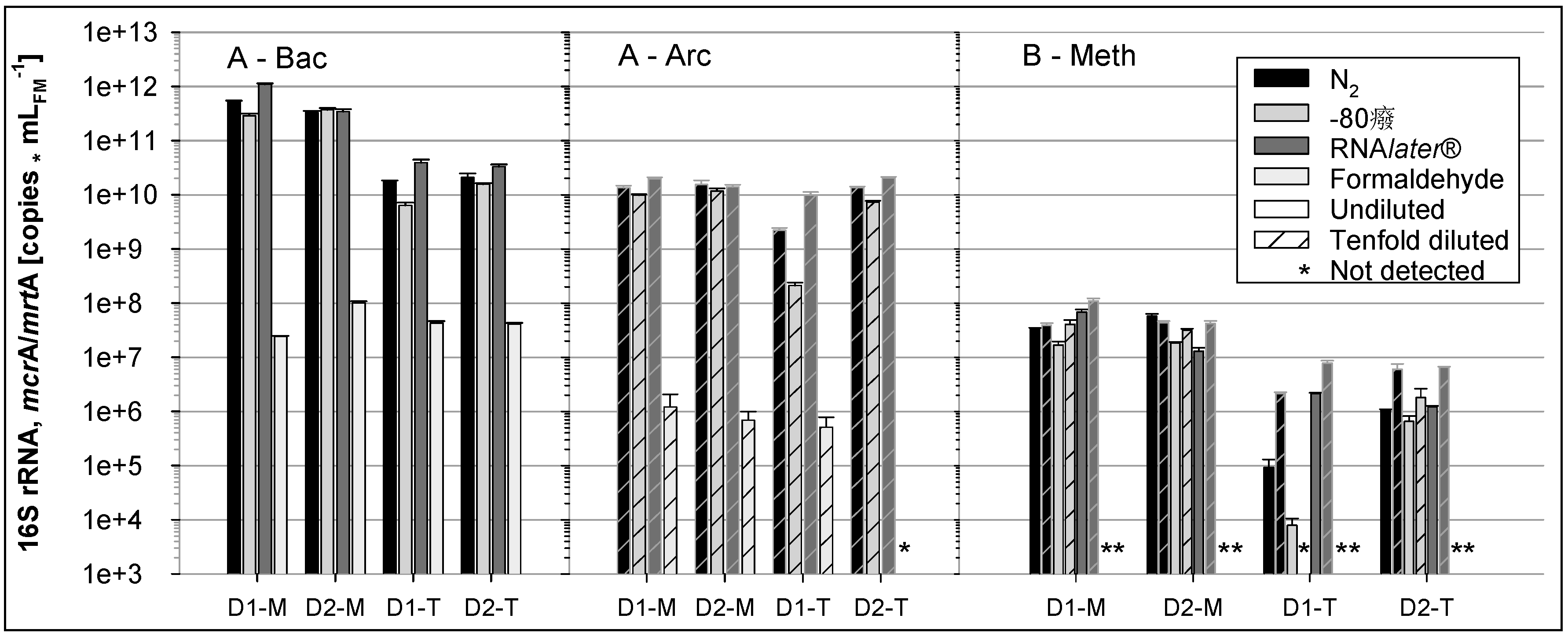
3.3. RNA Experiments
3.3.1. RNA Preservation


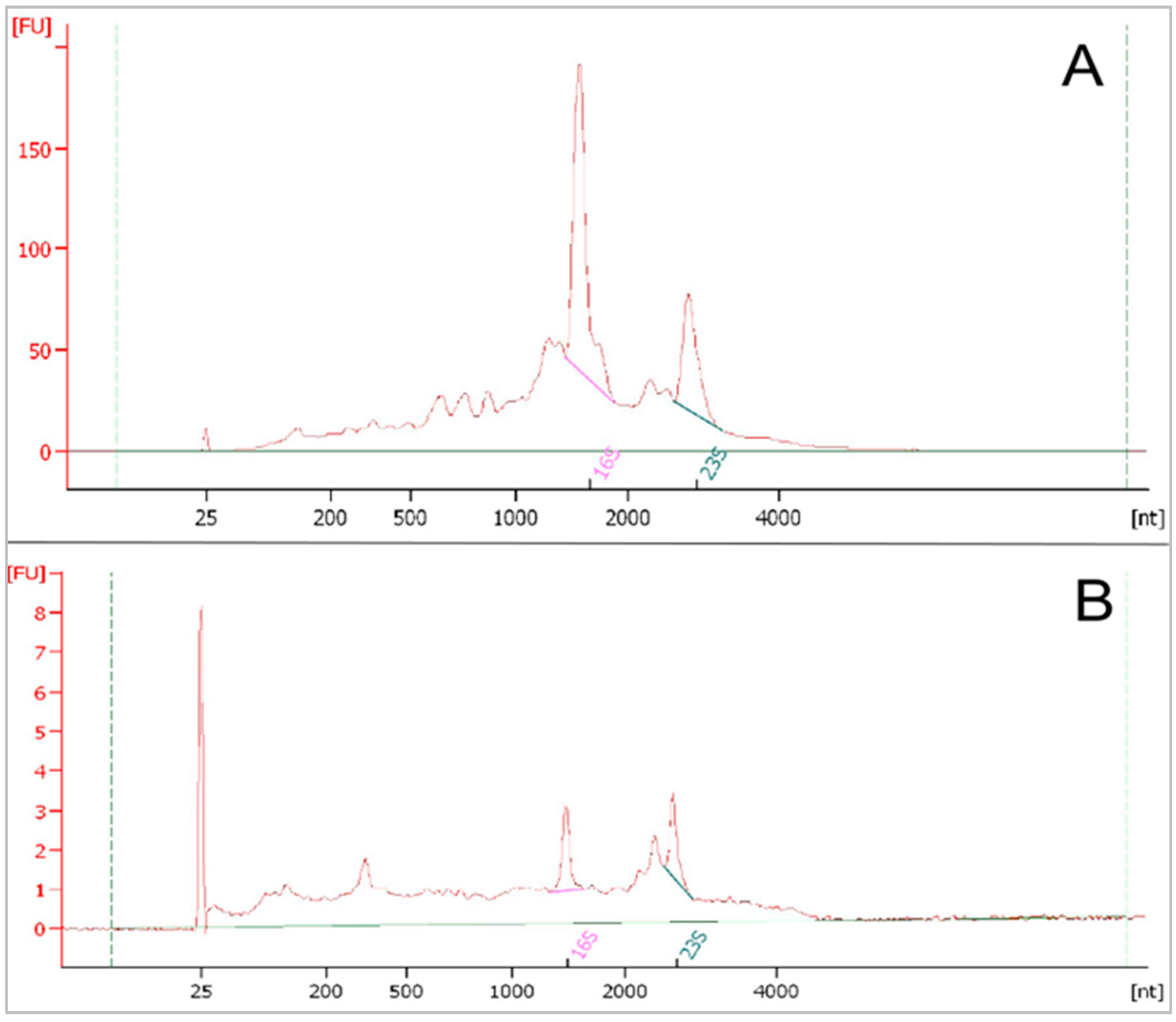
3.3.2. RNA Qualification and Quantification for Metatranscriptome Sequencing

4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Josefsen, M.H.; Löfström, C.; Hansen, T.; Reynisson, E.; Hoorfar, J. Instrumentation and Fluorescent Chemistries Used in qPCR. In Quantitative Real-Time PCR in Applied Microbiology; Filion, M., Ed.; Caister Academic Press: Norfolk, UK, 2012; pp. 27–52. [Google Scholar]
- Lebuhn, M.; Munk, B.; Effenberger, M. Agricultural biogas production in Germany—From practice to microbiology basics. Energy Sustain. Soc. 2014, 4. [Google Scholar] [CrossRef]
- Munk, B.; Lebuhn, M. Process diagnosis using methanogenic Archaea in maize-fed, trace element depleted fermenters. Anaerobe 2014, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Lebuhn, M.; Effenberger, M.; Gronauer, A.; Wilderer, P.A.; Wuertz, S. Using quantitative real-time PCR to determine the hygienic status of cattle manure. Water Sci. Technol. 2003, 48, 97–103. [Google Scholar] [PubMed]
- Bergmann, I.; Mundt, K.; Sontag, M.; Baumstark, I.; Nettmann, E.; Klocke, M. Influence of DNA isolation on Q-PCR-based quantification of methanogenic Archaea in biogas fermenters. Syst. Appl. Microbiol. 2010, 33, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Cox, F.; Kittelmann, S.; Miri, V.H.; Zethof, M.; Noel, S.J.; Waghorn, G.C.; Janssen, P.H. Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Li, A.; Chu, Y.N.; Wang, X.; Ren, L.; Yu, J.; Liu, X.; Yan, J.; Zhang, L.; Wu, S.; Li, S. A pyrosequencing-based metagenomic study of methane-producing microbial community in solid-state biogas reactor. Biotechnol. Biofuels 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.; Nolan, T.; Bustin, S.A. MIQE: Guidelines for the Design and Publication of a Reliable Real-time PCR Assay. In Real-Time PCR: Advanced Technologies and Applications; Caister Acad. Press: Norfolk, UK, 2013; pp. 247–258. [Google Scholar]
- Johnson, G.; Nour, A.A.; Nolan, T.; Huggett, J.; Bustin, S. Minimum information necessary for quantitative real-time PCR experiments. In Quantitative Real-Time PCR; Springer: New York, NY, USA, 2014; pp. 5–17. [Google Scholar]
- Quantitative Real-Time PCR: Methods and Protocols. Methods in Molecular Biology; Biassoni, R., Raso, A., Eds.; Springer Science & Business Media: New York, NY, USA, 2014; Volume 1160. [Google Scholar]
- Sambrook, J.; Russel, D.W. Molecular Cloning. A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press, Cold Spring Harbor: New York, NY, USA, 2001; Volume 3. [Google Scholar]
- Munk, B.; Bauer, C.; Gronauer, A.; Lebuhn, M. Population dynamics of methanogens during acidification of biogas fermenters fed with maize silage. Eng. Life Sci. 2010, 10, 496–508. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Bachoon, D.S.; Chen, F.; Hodson, R.E. RNA recovery and detection of mRNA by RT-PCR from preserved prokaryotic samples. FEMS Microbiol. Lett. 2001, 201, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Van Eijsden, R.G.E.; Stassen, C.; Daenen, L.; van Mulders, S.E.; Bapat, P.M.; Siewers, V.; Goossens, K.V.Y.; Nielsen, J.; Delvaux, F.R.; van Hummelen, P.; et al. A universal fixation method based on quaternary ammonium salts (RNAlater) for omics-technologies: Saccharomyces cerevisiae as a case study. Biotechnol. Lett. 2013, 35, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, A.J.; Kurhela, E.; Aho, T.; Oittinen, T.; Tiisola, M. Storage of environmental samples for guaranteeing nucleic acid yields for molecular microbiological studies. Appl. Microbiol. Biotechnol. 2010, 88, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Lebuhn, M.; Effenberger, M.; Garces, G.; Gronauer, A.; Wilderer, P.A. Evaluating real-time PCR for the quantification of distinct pathogens and indicator organisms in environmental samples. Water Sci. Technol. 2004, 50, 263–270. [Google Scholar] [PubMed]
- Scott Adams, P. Data analysis and reporting. In Real-time PCR; Dorak, M.T., Ed.; Taylor & Francis Group: Abingdon, UK, 2006; pp. 39–62. [Google Scholar]
- Schönberg, M.; Linke, B. The influence of the temperature regime on the formation of methane in a two-phase anaerobic digestion process. Eng. Life Sci. 2012, 12, 279–286. [Google Scholar] [CrossRef]
- Munk, B.; Bauer, C.; Gronauer, A.; Lebuhn, M. A metabolic quotient for methanogenic Archaea. Water Sci. Technol. 2012, 66, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Nettmann, E.; Bergmann, I.; Mundt, K.; Linke, B.; Klocke, M. Archaea diversity within a commercial biogas plant utilizing herbal biomass determined by 16S rDNA and mcrA analysis. J. Appl. Microbiol. 2008, 105, 1835–1850. [Google Scholar] [CrossRef] [PubMed]
- Stolze, Y.; Zakrzewski, M.; Maus, I.; Eikmeyer, F.; Jaenicke, S.; Rottmann, N.; Siebner, C.; Pühler, A.; Schlüter, A. Comparative metagenomics of biogas-producing microbial communities from production-scale biogas plants operating under wet or dry fermentation conditions. Biotechnol. Biofuels 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Booijink, C.C.; Klaassens, E.S.; Heilig, H.G.; Kleerebezem, M.; Smidt, H.; de Vos, W.M. Isolation of RNA from bacterial samples of the human gastrointestinal tract. Nat. Protoc. 2006, 1, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Urich, T.; Lanzén, A.; Qi, J.; Huson, D.H.; Schleper, C.; Schuster, S.C. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Klocke, M.; Nettmann, E.; Bergmann, I.; Mundt, K.; Souidi, K.; Mumme, J.; Linke, B. Characterization of the methanogenic Archaea within two-phase biogas reactor systems operated with plant biomass. Syst. Appl. Microbiol. 2008, 31, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Fey, A.; Eichler, S.; Flavier, S.; Christen, R.; Höfle, M.G.; Guzmán, C.A. Establishment of a real-time PCR-based approach for accurate quantification of bacterial RNA targets in water, using Salmonella as a model organism. Appl. Environ. Microbiol. 2004, 70, 3618–3623. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Korthals, M.; Gronauer, A.; Lebuhn, M. Methanogens in biogas production from renewable resources—A novel molecular population analysis approach. Water Sci. Technol. 2008, 58, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, J.; Vilgalys, R.; Jackson, R.B. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 2005, 71, 4117–4120. [Google Scholar] [CrossRef] [PubMed]
- Karakashev, D.; Batstone, D.J.; Angelidaki, I. Influence of environmental conditions on methanogenic compositions in anaerobic biogas reactors. Appl. Environ. Microbiol. 2005, 71, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Pervin, H.M.; Dennis, P.G.; Lim, H.J.; Tyson, G.W.; Batstone, D.J.; Bond, P.L. Drivers of microbial community composition in mesophilic and thermophilic temperature-phased anaerobic digestion pre-treatment reactors. Water Res. 2013, 47, 7098–7108. [Google Scholar] [CrossRef] [PubMed]
- Pap, B.; Györkei, Á.; Boboescu, I.Z.; Nagy, I.K.; Bíró, T.; Kondorosi, É.; Maróti, G. Temperature-dependent transformation of biogas-producing microbial communities points to the increased importance of hydrogenotrophic methanogenesis under thermophilic operation. Bioresour. Technol. 2015, 177, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Lebuhn, M.; Hanreich, A.; Klocke, M.; Schlüter, A.; Bauer, C.; Marín Pérez, C. Towards molecular biomarkers for biogas production from lignocellulose-rich substrates. Anaerobe 2014, 29, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Hospodsky, D.; Yamamoto, N.; Peccia, J. Accuracy, precision, and method detection limits of quantitative PCR for airborne bacteria and fungi. Appl. Environ. Microbiol. 2010, 76, 7004–7012. [Google Scholar] [CrossRef] [PubMed]
- Garcés, G.; Effenberger, M.; Najdrowski, M.; Gronauer, A.; Wilderer, P.A.; Lebuhn, M. Quantitative real-time PCR for detecting Cryptosporidium parvum in cattle manure and anaerobic digester samples—Methodological advances in DNA extraction. In Proceedings of the 8th Latin American Workshop and Symposium on Anaerobic Digestion, Punta del Este, Uruguay, 2–5 October 2005; pp. 68–73.
- Garcés, G.; Effenberger, M.; Najdrowski, M.; Wackwitz, C.; Gronauer, A.; Wilderer, P.A.; Lebuhn, M. Quantification of Cryptosporidium parvum in anaerobic digesters treating manure by (reverse-transcription) quantitative real-time PCR, infectivity and excystation tests. Water Sci. Technol. 2006, 53, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Sherman, M.E.; Rader, J.S.; Carreon, J.; Schiffman, M.; Baker, C.C. Cervical Tissue collection Methods for RNA Preservation Comparison of Snap-frozen, Ethanol-fixed and RNAlater-fixation. Diagn. Mol. Pathol. 2006, 15, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Riesgo, A.; Pérez-Porro, A.R.; Carmona, S.; Leys, S.P.; Giribet, G. Optimization of preservation and storage time of sponge tissues to obtain quality mRNA for next-generation sequencing. Mol. Ecol. Res. 2012, 12, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Mutter, G.L.; Zahrieh, D.; Liu, C.; Neuberg, D.; Finkelstein, D.; Baker, H.E.; Warrington, J.A. Comparison of frozen and RNAlater solid tissue storage methods for use in RNA expression microarrays. BMC Genom. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallup, J.M. qPCR inhibition and amplification of difficult templates. In PCR Troubleshooting and Optimization, the Essential Guide; Kennedy, S., Oswald, N., Eds.; Caister Academic Press: Norfolk, UK, 2011; pp. 23–65. [Google Scholar]
- Lebuhn, M.; Weiß, S.; Munk, B.; Guebitz, G.M. Microbiology and Molecular Biology Tools for Biogas Process Analysis, Diagnosis and Control. In Biogas Science and Technology: Advances in Biochemical Engineering/Biotechnology; Guebitz, G.M., Ed.; Springer: Cham, Switzerland, 2015; Volume 151, pp. 1–40. [Google Scholar]
- Lebuhn, M.; Wilderer, P. Abschlussbericht des StMUGV-Projekts “Biogastechnologie zur umweltverträglichen Flüssigmistverwertung und Energiegewinnung in Wasserschutzgebieten: wasserwirtschaftliche und hygienische Begleituntersuchung, Projektteil: Mikrobiologische, parasitologische und virologische Untersuchungen“. Technische Universität München, Lehrstuhl für Siedlungswasserwirtschaft. 2006. Available online: http://www.sww.bgu.tum.de/fileadmin/w00bom/www/_migrated_content_uploads/StMUGV-Abschlussbericht_2006_Lebuhn.pdf (accessed on 28 December 2015).
- Su, C.; Lei, L.; Duan, Y.; Zhang, K.Q.; Yang, J. Culture-independent methods for studying environmental microorganisms: Methods, application, and perspective. Appl. Microbiol. Biotechnol. 2012, 93, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Filiatrault, M.J. Progress in prokaryotic transcriptomics. Curr. Opin. Microbiol. 2011, 14, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Cossart, P. Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010, 11, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pascault, N.; Loux, V.; Derozier, S.; Martin, V.; Debroas, D.; Maloufi, S.; Humbert, J.F.; Leloup, J. Technical challenges in metatranscriptomic studies applied to the bacterial communities of freshwater ecosystems. Genetica 2014, 143, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Mäder, U.; Nicolas, P.; Richard, H.; Bessières, P.; Aymerich, S. Comprehensive identification and quantification of microbial transcriptomes by genome-wide unbiased methods. Curr. Opin. Biotechnol. 2011, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.A. Metatranscriptomics: Eavesdropping on complex microbial communities. Microbe 2009, 4, 329–335. [Google Scholar]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebuhn, M.; Derenkó, J.; Rademacher, A.; Helbig, S.; Munk, B.; Pechtl, A.; Stolze, Y.; Prowe, S.; Schwarz, W.H.; Schlüter, A.; et al. DNA and RNA Extraction and Quantitative Real-Time PCR-Based Assays for Biogas Biocenoses in an Interlaboratory Comparison. Bioengineering 2016, 3, 7. https://doi.org/10.3390/bioengineering3010007
Lebuhn M, Derenkó J, Rademacher A, Helbig S, Munk B, Pechtl A, Stolze Y, Prowe S, Schwarz WH, Schlüter A, et al. DNA and RNA Extraction and Quantitative Real-Time PCR-Based Assays for Biogas Biocenoses in an Interlaboratory Comparison. Bioengineering. 2016; 3(1):7. https://doi.org/10.3390/bioengineering3010007
Chicago/Turabian StyleLebuhn, Michael, Jaqueline Derenkó, Antje Rademacher, Susanne Helbig, Bernhard Munk, Alexander Pechtl, Yvonne Stolze, Steffen Prowe, Wolfgang H. Schwarz, Andreas Schlüter, and et al. 2016. "DNA and RNA Extraction and Quantitative Real-Time PCR-Based Assays for Biogas Biocenoses in an Interlaboratory Comparison" Bioengineering 3, no. 1: 7. https://doi.org/10.3390/bioengineering3010007
APA StyleLebuhn, M., Derenkó, J., Rademacher, A., Helbig, S., Munk, B., Pechtl, A., Stolze, Y., Prowe, S., Schwarz, W. H., Schlüter, A., Liebl, W., & Klocke, M. (2016). DNA and RNA Extraction and Quantitative Real-Time PCR-Based Assays for Biogas Biocenoses in an Interlaboratory Comparison. Bioengineering, 3(1), 7. https://doi.org/10.3390/bioengineering3010007