
The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
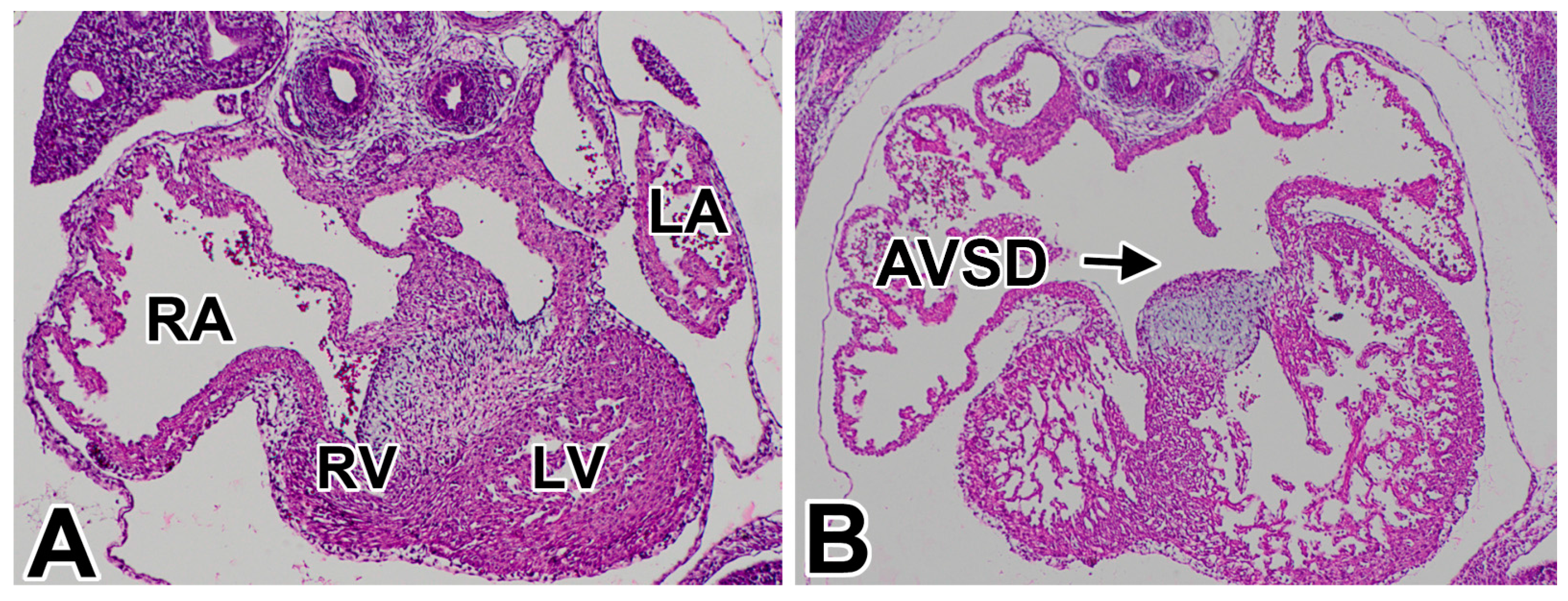
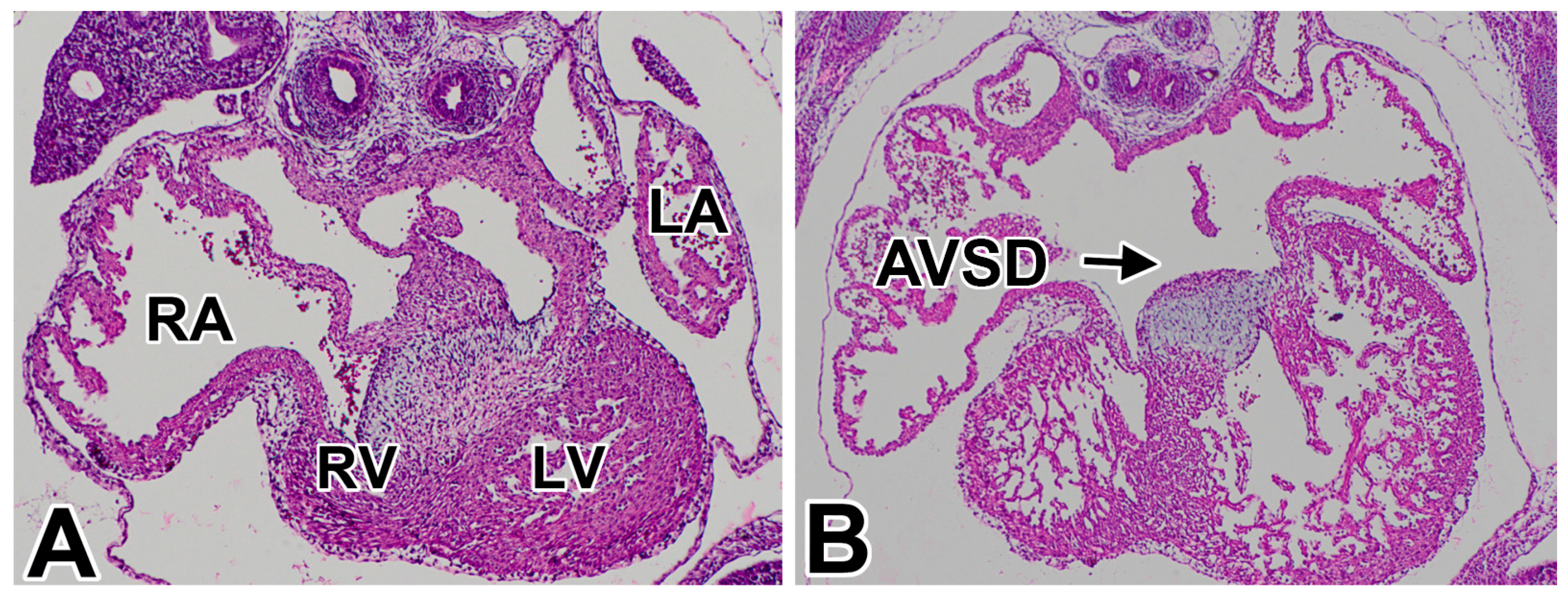
1.1. Atrioventricular Septal Defects in Humans
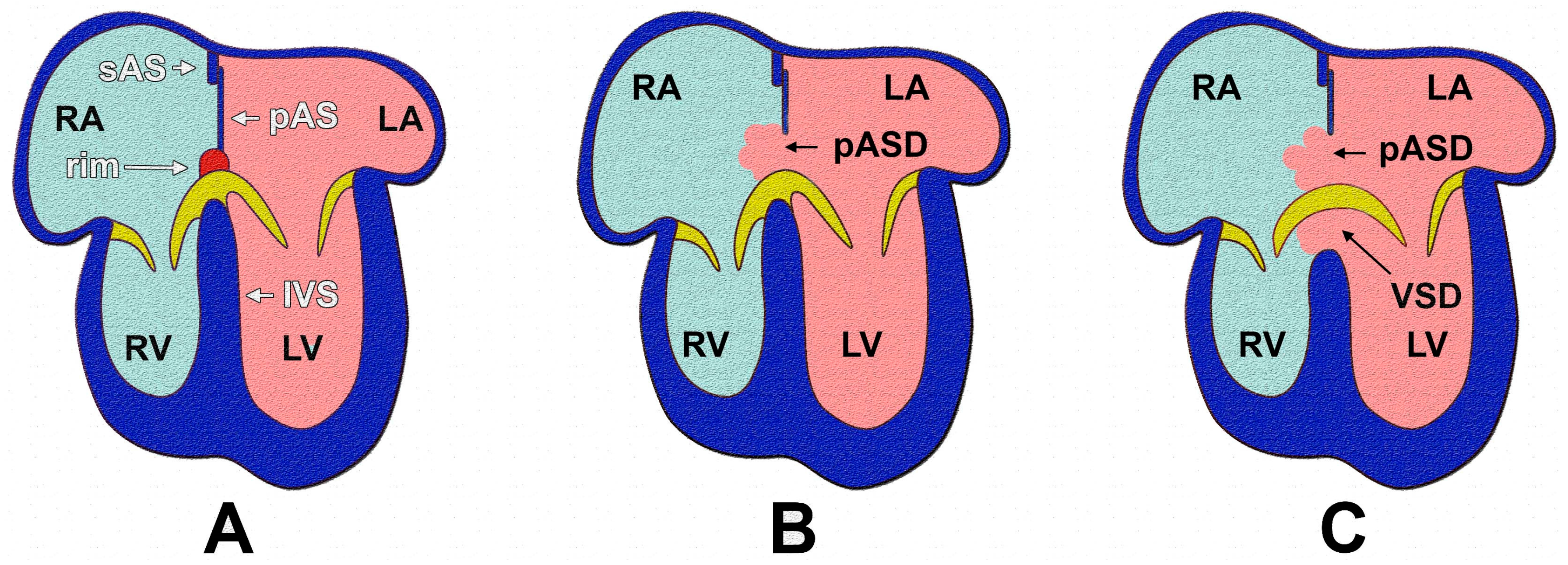
1.2. AVSDs—Subtypes
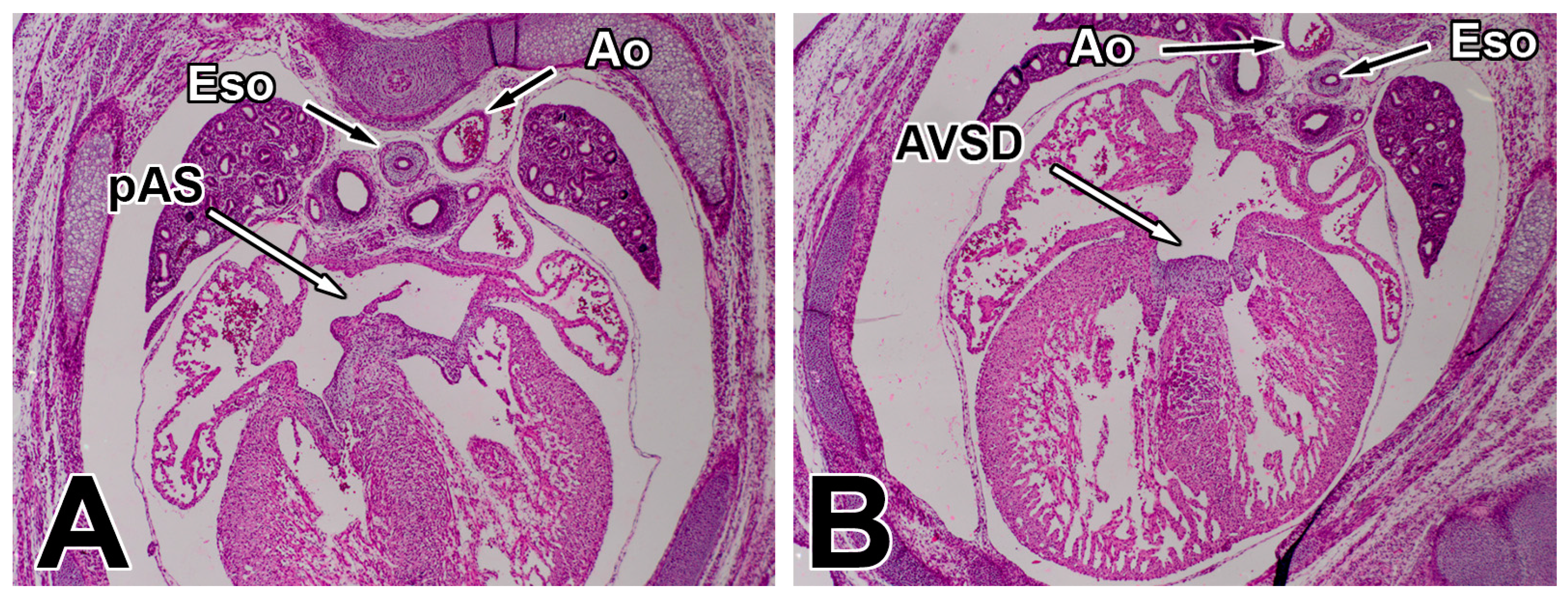
1.3. Pathogenesis of AVSDs—AV Cushions vs. DMP
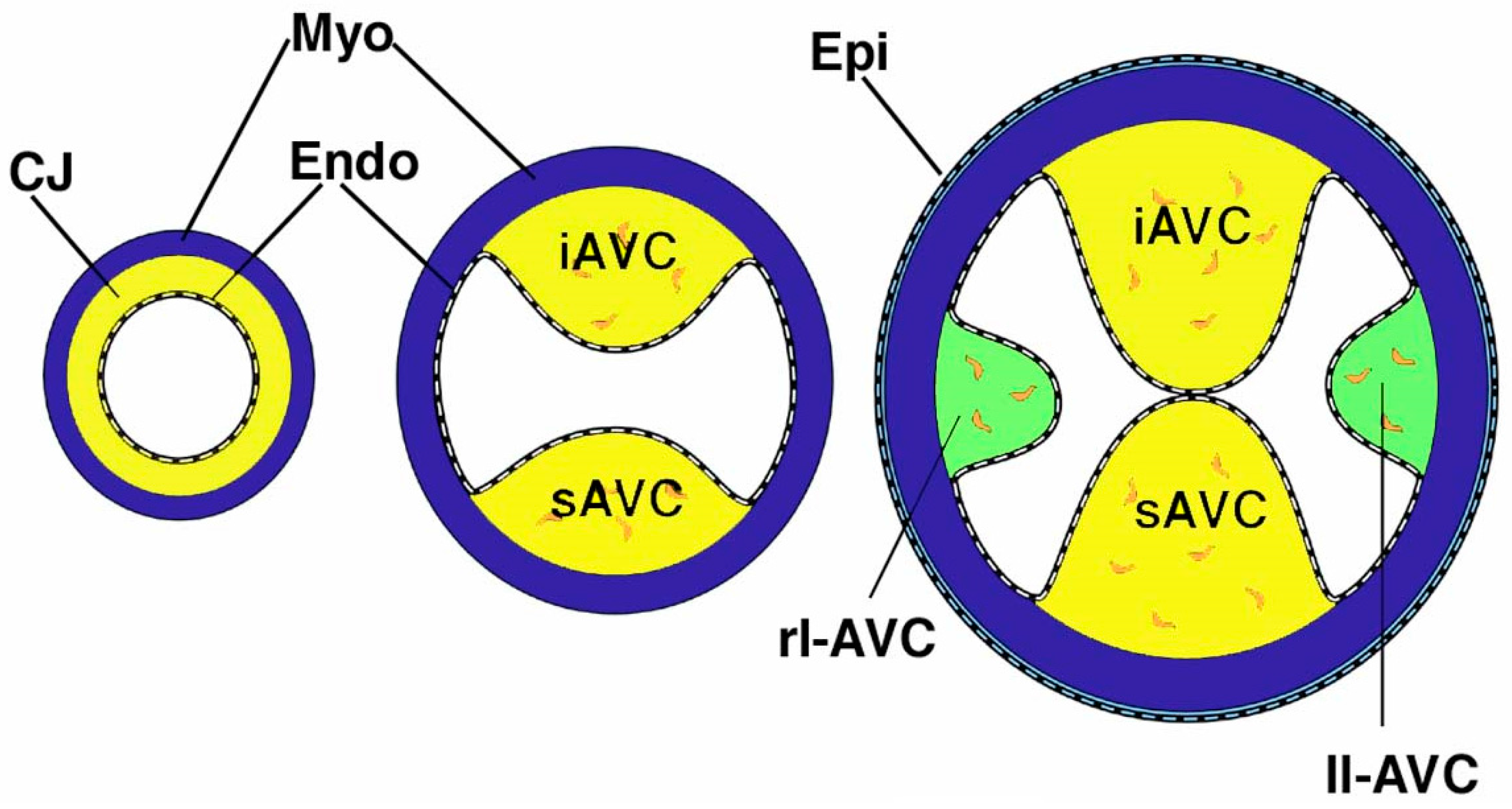
2. The Atrioventricular Mesenchymal Complex
2.1. The Mesenchymal AV Cushions
2.2. The Mesenchymal Cap on the Primary Atrial Septum
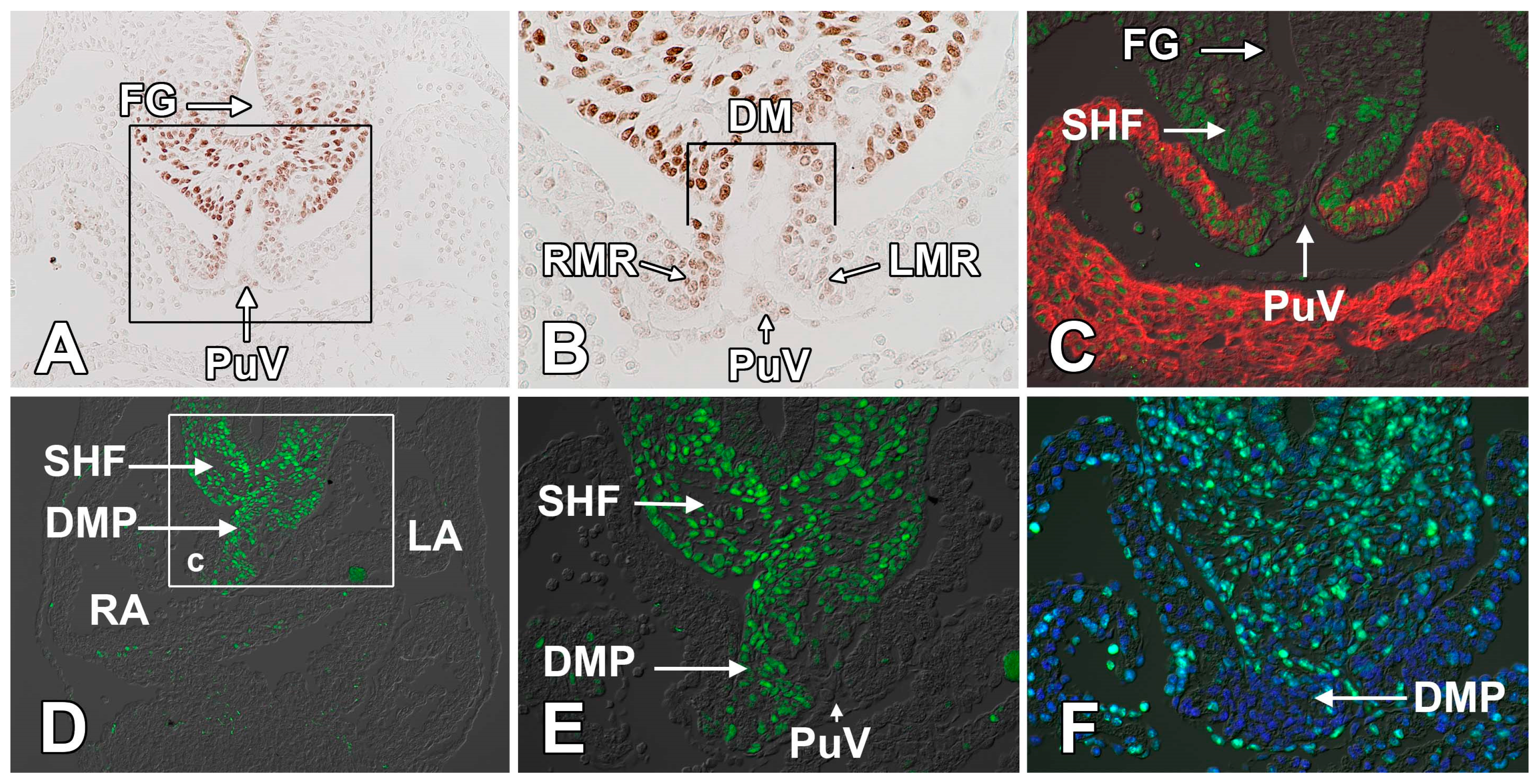
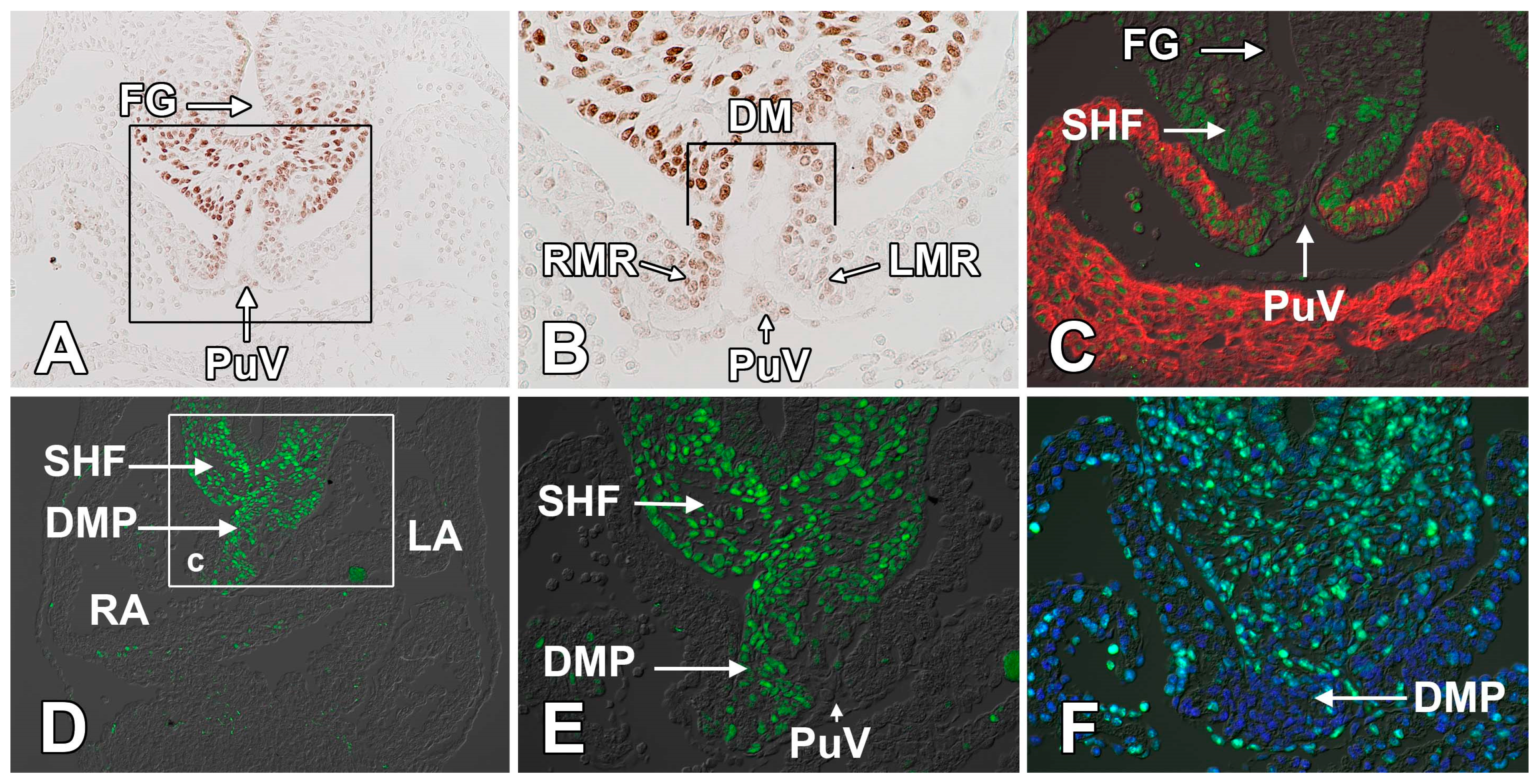
2.3. The Dorsal Mesenchymal Protrusion
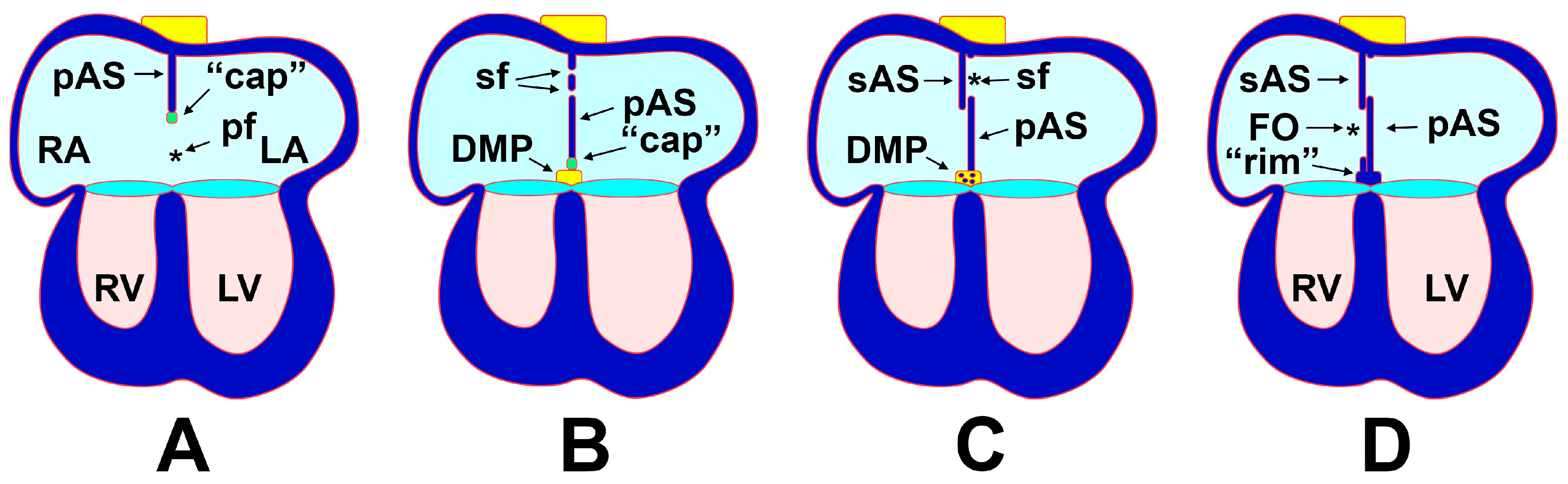
3. Atrial Septation
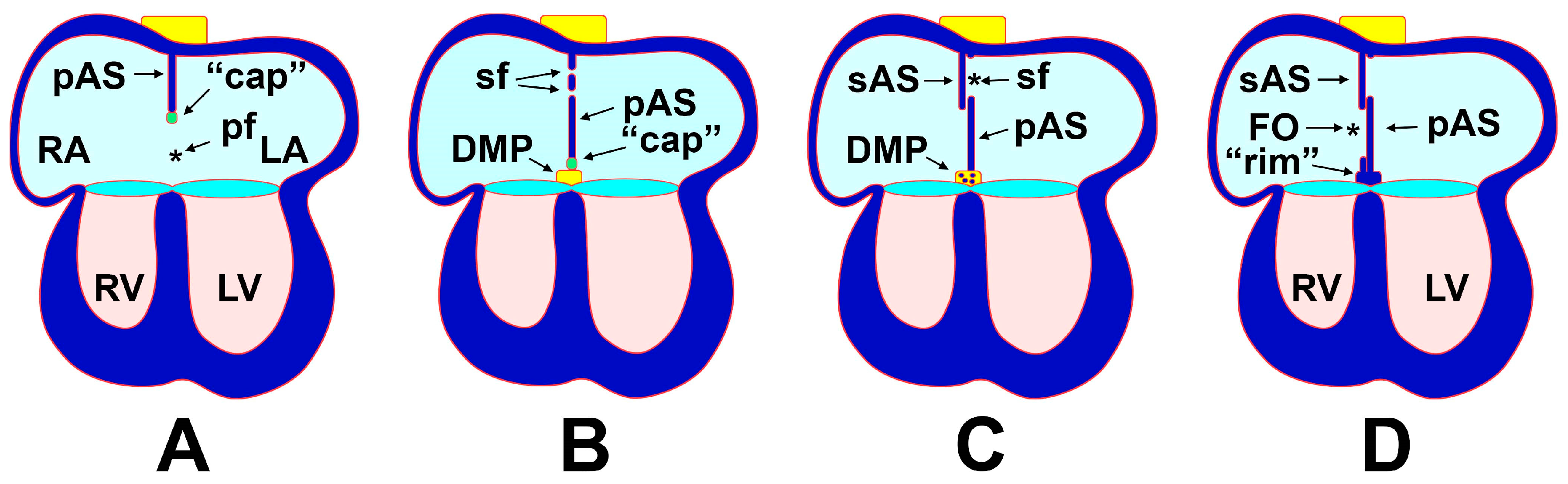
3.1. The Formation of the Atrial Septal Complex—The Primary Atrial Septum
3.2. The Formation of the Atrial Septal Complex–The Secondary Atrial Septum
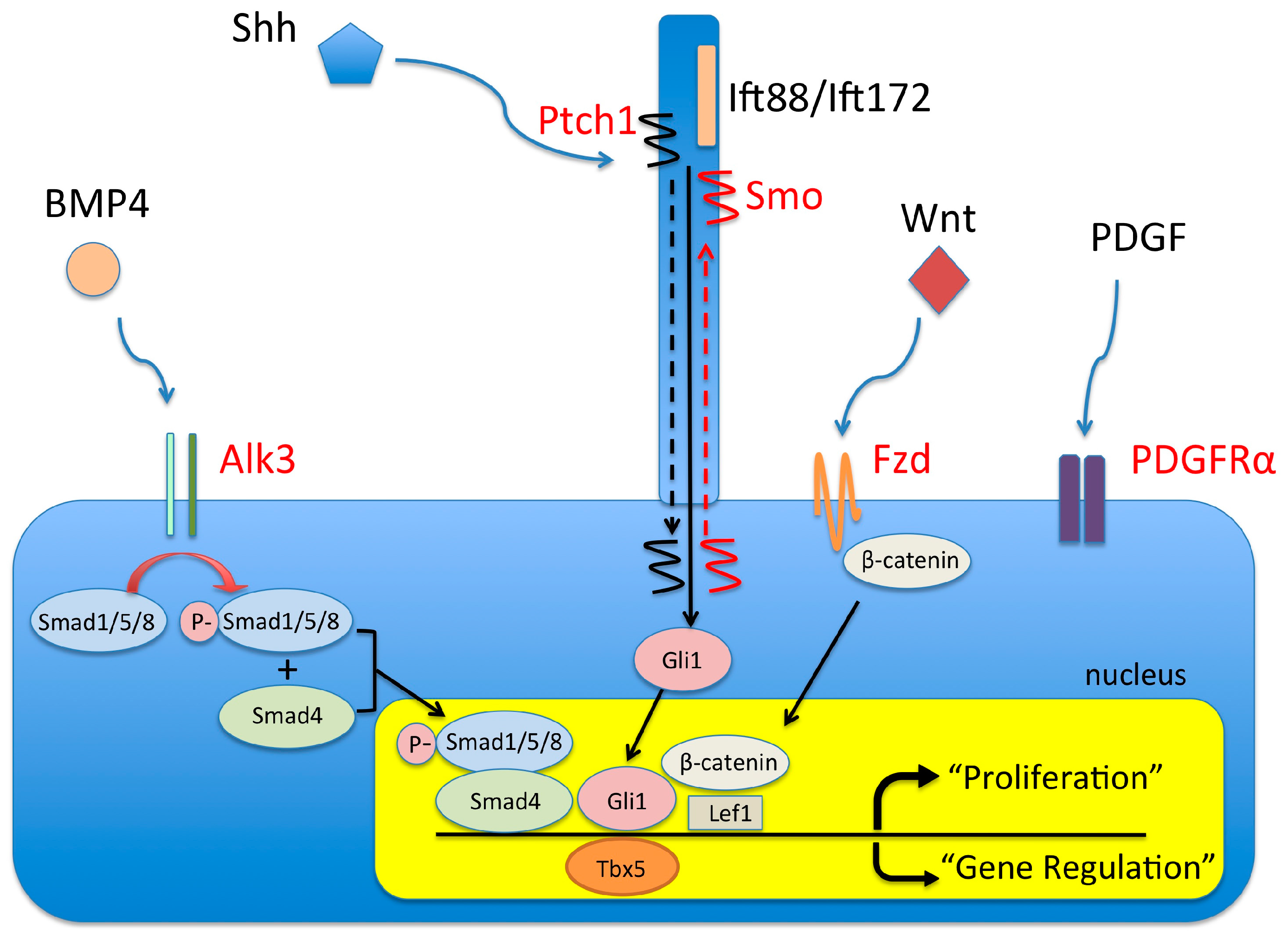
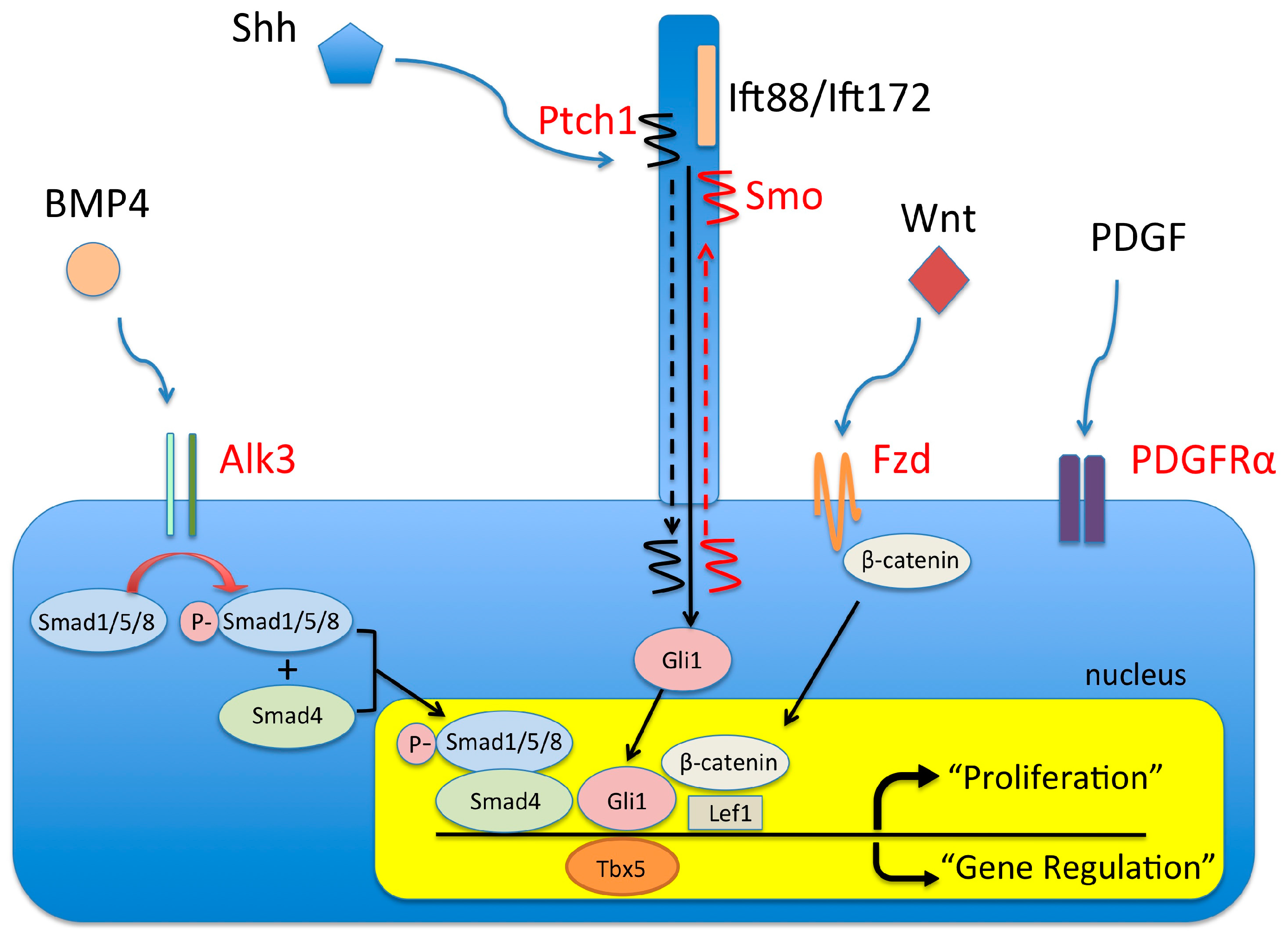
4. Molecular Regulation of DMP Development
4.1. The BMP Signaling Pathway and AVSDs
4.2. The Wnt/β-Catenin Signaling Pathway and AVSDs
4.3. The (Sonic) Hedgehog Signaling Pathway and AVSDs
4.4. Pdgf and Pdgf Receptors and AVSDs
4.5. Tbx5 and AVSD
4.6. Primary Cilia and AVSDs
4.7. AVSDs and Heterotaxy Syndrome
5. Etiology and Pathogenesis of AVSDs—Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Parker, S.E.; Mai, C.T.; Canfield, M.A.; Rickard, R.; Wang, Y.; Meyer, R.E.; Anderson, P.; Mason, C.A.; Collins, J.S.; Kirby, R.S.; et al. Updated national birth prevalence estimates for selected birth defects in the united states, 2004–2006. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Calkoen, E.E.; Hazekamp, M.G.; Blom, N.A.; Elders, B.B.; Gittenberger-de Groot, A.C.; Haak, M.C.; Bartelings, M.M.; Roest, A.A.; Jongbloed, M.R. Atrioventricular septal defect: From embryonic development to long-term follow-up. Int. J. Cardiol. 2016, 202, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Craig, B. Atrioventricular septal defect: From fetus to adult. Heart 2006, 92, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.A.; Joziasse, I.C.; Chocron, S.; van Dinther, M.; Guryev, V.; Verhoeven, M.C.; Rehmann, H.; van der Smagt, J.J.; Doevendans, P.A.; Cuppen, E.; et al. Dominant-negative alk2 allele associates with congenital heart defects. Circulation 2009, 119, 3062–3069. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, L.C.; Al Turki, S.; Manickaraj, A.K.; Manase, D.; Mulder, B.J.; Bergin, L.; Rosenberg, H.C.; Mondal, T.; Gordon, E.; Lougheed, J.; et al. Exome sequencing identifies rare variants in multiple genes in atrioventricular septal defect. Genet. Med. 2016, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.G.; Perrot, A.; Schmitt, K.; Mittelhaus, S.; Esenwein, E.M.; Stiller, B.; Geier, C.; Dietz, R.; Gessner, R.; Ozcelik, C.; et al. Mutations in GATA4, Nkx2.5, CRELD1, and bmp4 are infrequently found in patients with congenital cardiac septal defects. Am. J. Med. Genet. A 2008, 146A, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.W.; Morris, C.D.; Goldmuntz, E.; Reller, M.D.; Jones, M.A.; Steiner, R.D.; Maslen, C.L. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am. J. Hum. Genet. 2003, 72, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Zatyka, M.; Priestley, M.; Ladusans, E.J.; Fryer, A.E.; Mason, J.; Latif, F.; Maher, E.R. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2). Clin. Genet. 2005, 67, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Borlak, J. GATA4 zinc finger mutations as a molecular rationale for septation defects of the human heart. J. Med. Genet. 2005, 42, e32. [Google Scholar] [CrossRef] [PubMed]
- Inga, A.; Reamon-Buettner, S.M.; Borlak, J.; Resnick, M.A. Functional dissection of sequence-specific Nkx2-5 DNA binding domain mutations associated with human heart septation defects using a yeast-based system. Hum. Mol. Genet. 2005, 14, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Borlak, J. Somatic Nkx2-5 mutations as a novel mechanism of disease in complex congenital heart disease. J. Med. Genet. 2004, 41, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Reamon-Buettner, S.M.; Borlak, J. TBX5 mutations in Non-Holt-Oram Syndrome (HOS) malformed hearts. Hum. Mutat. 2004, 24, 104. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, C.A.; Hirata, J.; Wilson, P.D.; Ferencz, C.; Lurie, I.W. Atrioventricular septal defects: Possible etiologic differences between complete and partial defects. Teratology 2001, 63, 87–93. [Google Scholar] [CrossRef]
- Loffredo, C.A.; Wilson, P.D.; Ferencz, C. Maternal diabetes: An independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology 2001, 64, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Cleves, M.A.; Malik, S.; Yang, S.; Carter, T.C.; Hobbs, C.A. Maternal urinary tract infections and selected cardiovascular malformations. Birth Defects Res. A Clin. Mol. Teratol. 2008, 82, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Ferencz, C.; Correa-Villasenor, A.; Loffredo, C. Genetic and Environmental Risk Factors of Major Cardiovascular Malformations: The Baltimore-Washington Infant Study: 1981–1989; Futura Publishing Co., Inc.: Armonk, NY, 1997; pp. 1–463. [Google Scholar]
- Malik, S.; Cleves, M.A.; Honein, M.A.; Romitti, P.A.; Botto, L.D.; Yang, S.; Hobbs, C.A. Maternal smoking and congenital heart defects. Pediatrics 2008, 121, e810–e816. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.E.; Kakarla, J.; Wessels, A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation 2012, 84, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.M.; Peng, L.Y.; Li, L.; Liu, X.Y.; Wang, J.; Zhang, X.L.; Yuan, F.; Li, R.G.; Qiu, X.B.; Yang, Y.Q. PITX2 loss-of-function mutation contributes to congenital endocardial cushion defect and axenfeld-rieger syndrome. PLoS ONE 2015, 10, e0124409. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Missen, G.A. Endocardial cushion defects; common atrio-ventricular canal and ostium primum. Br. Heart J. 1957, 19, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Sharratt, G.P.; Webb, S.; Anderson, R.H. The vestibular defect: An interatrial communication due to a deficiency in the atrial septal component derived from the vestibular spine. Cardiol. Young 2003, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.A.; Ottenkamp, J.; Wenink, A.G.; Gittenberger-de Groot, A.C. Deficiency of the vestibular spine in atrioventricular septal defects in human fetuses with down syndrome. Am. J. Cardiol. 2003, 91, 180–184. [Google Scholar] [CrossRef]
- Briggs, L.E.; Phelps, A.L.; Brown, E.; Kakarla, J.; Anderson, R.H.; van den Hoff, M.J.; Wessels, A. Expression of the bmp receptor alk3 in the second heart field is essential for development of the dorsal mesenchymal protrusion and atrioventricular septation. Circ. Res. 2013, 112, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.M.; Rho, S.; Petiet, A.; Davenport, C.L.; Johnson, G.A.; Meyers, E.N.; Klingensmith, J. Intracardiac septation requires hedgehog-dependent cellular contributions from outside the heart. Development 2008, 135, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yuan, L.; Goss, A.M.; Wang, T.; Yang, J.; Lepore, J.J.; Zhou, D.; Schwartz, R.J.; Patel, V.; Cohen, E.D.; et al. Characterization and in vivo pharmacological rescue of a Wnt2-Gata6 pathway required for cardiac inflow tract development. Dev. Cell 2010, 18, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.E.; Burns, T.A.; Lockhart, M.M.; Phelps, A.L.; Van den Hoff, M.J.; Wessels, A. Wnt/β-catenin and sonic hedgehog pathways interact in the regulation of the development of the dorsal mesenchymal protrusion. Dev. Dyn. 2016, 245, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Abu-Issa, R.; Kirby, M.L. Heart field: From mesoderm to heart tube. Annu. Rev. Cell Dev. Biol. 2007, 23, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.F.; Christoffels, V.M.; Anderson, R.H.; van den Hoff, M.J. The heart-forming fields: One or multiple? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, L.M.; Markwald, R.R. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 1995, 77, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Linask, K.K.; Vanauker, M. A role for the cytoskeleton in heart looping. ScientificWorldJournal 2007, 7, 280–298. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.; Habets, P.; Franco, D.; Campione, M.; de Jong, F.; Lamers, W.; Bao, Z.; Palmer, S.; Biben, C.; Harvey, R.; et al. Chamber formation and morphogenesis in the developing mammalian heart. Dev. Biol. 2000, 223, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Markwald, R.R.; Fitzharris, T.P.; Manasek, F.J. Structural development of endocardial cushions. Am. J. Anat. 1977, 148, 85–119. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.S.; Kern, C.B.; Wessels, A. Origin and fate of cardiac mesenchyme. Dev. Dyn. 2008, 237, 2804–2819. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Sedmera, D. Developmental anatomy of the heart: A tale of mice and man. Physiol. Genomics 2003, 15, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.S.; Wirrig, E.E.; Phelps, A.L.; Trusk, T.C.; Wessels, A. A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev. Dyn. 2007, 236, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Anderson, R.H.; Markwald, R.R.; Webb, S.; Brown, N.A.; Viragh, S.; Moorman, A.F.; Lamers, W.H. Atrial development in the human heart: An immunohistochemical study with emphasis on the role of mesenchymal tissues. Anat. Rec. 2000, 259, 288–300. [Google Scholar] [CrossRef]
- Mommersteeg, M.T.; Soufan, A.T.; de Lange, F.J.; van den Hoff, M.J.; Anderson, R.H.; Christoffels, V.M.; Moorman, A.F. Two distinct pools of mesenchyme contribute to the development of the atrial septum. Circ. Res. 2006, 99, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Gerety, M.; Watanabe, M. Polysialylated ncam expression on endocardial cells of the chick primary atrial septum. Anat. Rec. 1997, 247, 71–84. [Google Scholar] [CrossRef]
- Hoffmann, A.D.; Peterson, M.A.; Friedland-Little, J.M.; Anderson, S.A.; Moskowitz, I.P. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development 2009, 136, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Snarr, B.S.; O’Neal, J.L.; Chintalapudi, M.R.; Wirrig, E.E.; Phelps, A.L.; Kubalak, S.W.; Wessels, A. Isl1 expression at the venous pole identifies a novel role for the second heart field in cardiac development. Circ. Res. 2007, 101, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.; Brown, N.A.; Wessels, A.; Anderson, R.H. Development of the murine pulmonary vein and its relationship to the embryonic venous sinus. Anat. Rec. 1998, 250, 325–334. [Google Scholar] [CrossRef]
- Auer, J. The development of the human pulmonary vein and its major variations. Anat. Rec. 1948, 101, 581–594. [Google Scholar] [CrossRef] [PubMed]
- DeRuiter, M.C.; Gittenberger-De Groot, A.C.; Wenink, A.C.; Poelmann, R.E.; Mentink, M.M. In normal development pulmonary veins are connected to the sinus venosus segment in the left atrium. Anat. Rec. 1995, 243, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, W.; Palie, J.; Lu, M.F.; Brown, N.A.; Martin, J.F. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development 2002, 129, 5081–5091. [Google Scholar] [PubMed]
- Anderson, R.H.; Brown, N.A.; Webb, S. Development and structure of the atrial septum. Heart 2002, 88, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Campione, M.; Kelly, R.; Zammit, P.S.; Buckingham, M.; Lamers, W.H.; Moorman, A.F. Multiple transcriptional domains, with distinct left and right components, in the atrial chambers of the developing heart. Circ. Res. 2000, 87, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahid, E.; Rice, D.; Pelliniemi, L.J.; Jokinen, E. Overlapping and differential localization of Bmp-2, Bmp-4, Msx-2 and apoptosis in the endocardial cushion and adjacent tissues of the developing mouse heart. Cell Tissue Res. 2001, 305, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Somi, S.; Buffing, A.A.; Moorman, A.F.; Van Den Hoff, M.J. Dynamic patterns of expression of BMP isoforms 2, 4, 5, 6, and 7 during chicken heart development. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2004, 279, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Greene, S.B.; Martin, J.F. Bmp signaling in congenital heart disease: New developments and future directions. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, S.; Fukuda, K. Multiple roles for bmp signaling in cardiac development. Drug Discov. Today Ther. Strateg. 2008, 5, 209–214. [Google Scholar] [CrossRef]
- Sugi, Y.; Yamamura, H.; Okagawa, H.; Markwald, R.R. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev. Biol. 2004, 269, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lu, M.F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Selever, J.; Wang, D.; Lu, M.F.; Moses, K.A.; Schwartz, R.J.; Martin, J.F. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc. Natl. Acad. Sci. USA 2004, 101, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- McCulley, D.J.; Kang, J.O.; Martin, J.F.; Black, B.L. Bmp4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dyn. 2008, 237, 3200–3209. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Komatsu, Y.; Tanaka, M.; McCann, K.L.; Mishina, Y. Bmp2 and Bmp4 genetically interact to support multiple aspects of mouse development including functional heart development. Genesis 2009, 47, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Dehart, D.B.; Sulik, K.K.; Hogan, B.L. Distinct requirements for extra-embryonic and embryonic bone morphogenetic protein 4 in the formation of the node and primitive streak and coordination of left-right asymmetry in the mouse. Development 2002, 129, 4685–4696. [Google Scholar] [PubMed]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B.L. An essential role of bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [PubMed]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Fu, X.; Wang, J.; Lu, M.F.; Chen, L.; Baldini, A.; Klein, W.H.; Martin, J.F. Canonical wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. USA 2007, 104, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.D.; Wang, Z.; Lepore, J.J.; Lu, M.M.; Taketo, M.M.; Epstein, D.J.; Morrisey, E.E. Wnt/β-catenin signaling promotes expansion of isl-1-positive cardiac progenitor cells through regulation of fgf signaling. J. Clin. Investig. 2007, 117, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cui, L.; Zhou, W.; Dufort, D.; Zhang, X.; Cai, C.L.; Bu, L.; Yang, L.; Martin, J.; Kemler, R.; et al. β-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 9313–9318. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kokubo, H.; Miyagawa-Tomita, S.; Endo, M.; Igarashi, K.; Aisaki, K.; Kanno, J.; Saga, Y. Activation of Notch1 signaling in cardiogenic mesoderm induces abnormal heart morphogenesis in mouse. Development 2006, 133, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Herzog, K.; Willnow, T.E. LRP2, an auxiliary receptor that controls sonic hedgehog signaling in development and disease. Dev. Dyn. 2016, 245, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.M.; Schwartz, R.; Klingensmith, J.; Meyers, E.N. Independent requirements for hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Washington Smoak, I.; Byrd, N.A.; Abu-Issa, R.; Goddeeris, M.M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E.N. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Hildreth, V.; Webb, S.; Chaudhry, B.; Peat, J.D.; Phillips, H.M.; Brown, N.; Anderson, R.H.; Henderson, D.J. Left cardiac isomerism in the sonic hedgehog null mouse. J. Anat. 2009, 214, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, N.M.; Winkel, L.C.; Nisancioglu, M.H.; Maas, S.; Wisse, L.J.; Armulik, A.; Poelmann, R.E.; Lie-Venema, H.; Betsholtz, C.; Gittenberger-de Groot, A.C. PDGF-B signaling is important for murine cardiac development: Its role in developing atrioventricular valves, coronaries, and cardiac innervation. Dev. Dyn. 2008, 237, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. The PDGF α receptor is required for neural crest cell development and for normal patterning of the somites. Development 1997, 124, 2691–2700. [Google Scholar] [PubMed]
- Tallquist, M.D.; Soriano, P. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development 2003, 130, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.A.; Lie-Venema, H.; Vicente-Steijn, R.; Bleyl, S.B.; van Den Akker, N.M.; Maas, S.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Platelet-derived growth factor is involved in the differentiation of second heart field-derived cardiac structures in chicken embryos. Dev. Dyn. 2009, 238, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Van Den Akker, N.M.; Lie-Venema, H.; Maas, S.; Eralp, I.; DeRuiter, M.C.; Poelmann, R.E.; Gittenberger-De Groot, A.C. Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Dev. Dyn. 2005, 233, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Bleyl, S.B.; Saijoh, Y.; Bax, N.A.; Gittenberger-de Groot, A.C.; Wisse, L.J.; Chapman, S.C.; Hunter, J.; Shiratori, H.; Hamada, H.; Yamada, S.; et al. Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: Integrating evidence from human genetics and model organisms. Hum. Mol. Genet. 2010, 19, 1286–1301. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.A.; Bleyl, S.B.; Gallini, R.; Wisse, L.J.; Hunter, J.; Van Oorschot, A.A.; Mahtab, E.A.; Lie-Venema, H.; Goumans, M.J.; Betsholtz, C.; et al. Cardiac malformations in Pdgfrα mutant embryos are associated with increased expression of WT1 and NKX2.5 in the second heart field. Dev. Dyn. 2010, 239, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Basson, C.T.; Bachinsky, D.R.; Lin, R.C.; Levi, T.; Elkins, J.A.; Soults, J.; Grayzel, D.; Kroumpouzou, E.; Traill, T.A.; Leblanc-Straceski, J.; et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat. Genet. 1997, 15, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Y.; Newbury-Ecob, R.A.; Terrett, J.A.; Wilson, D.I.; Curtis, A.R.; Yi, C.H.; Gebuhr, T.; Bullen, P.J.; Robson, S.C.; Strachan, T.; et al. Holt-oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat. Genet. 1997, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Hoffmann, A.D.; Burnicka-Turek, O.; Friedland-Little, J.M.; Zhang, K.; Moskowitz, I.P. Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 2012, 23, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Friedland-Little, J.M.; Hoffmann, A.D.; Ocbina, P.J.; Peterson, M.A.; Bosman, J.D.; Chen, Y.; Cheng, S.Y.; Anderson, K.V.; Moskowitz, I.P. A novel murine allele of intraflagellar transport protein 172 causes a syndrome including vacterl-like features with hydrocephalus. Hum. Mol. Genet. 2011, 20, 3725–3737. [Google Scholar] [CrossRef] [PubMed]
- Willaredt, M.A.; Gorgas, K.; Gardner, H.A.; Tucker, K.L. Multiple essential roles for primary cilia in heart development. Cilia 2012, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, C.; Rivals, I.; Ait Yahya-Graison, E.; Dauphinot, L.; Paly, E.; Mircher, C.; Ravel, A.; Grattau, Y.; Blehaut, H.; Megarbane, A.; et al. Molecular signatures of cardiac defects in down syndrome lymphoblastoid cell lines suggest altered ciliome and hedgehog pathways. PLoS ONE 2012, 7, e41616. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Klena, N.T.; Gabriel, G.C.; Liu, X.; Kim, A.J.; Lemke, K.; Chen, Y.; Chatterjee, B.; Devine, W.; Damerla, R.R.; et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 2015, 521, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Belmont, J.W.; Ware, S.M. Genetics of human heterotaxias. Eur. J. Hum. Genet. 2006, 14, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.S.; Ahmed, M.M.; Spicer, D.E.; Backer, C.L.; Anderson, R.H. Manifestations of bodily isomerism. Cardiovasc. Pathol. 2016, 25, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; McCrindle, B.W.; Smallhorn, J.F.; Golding, F.; Caldarone, C.A.; Taketazu, M.; Jaeggi, E.T. Clinical features, management, and outcome of children with fetal and postnatal diagnoses of isomerism syndromes. Circulation 2005, 112, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Tanaka, Y.; Okada, Y.; Takeda, S.; Harada, A.; Kanai, Y.; Kido, M.; Hirokawa, N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking kif3b motor protein. Cell 1998, 95, 829–837. [Google Scholar] [CrossRef]
- Burnicka-Turek, O.; Steimle, J.D.; Huang, W.; Felker, L.; Kamp, A.; Kweon, J.; Peterson, M.; Reeves, R.H.; Maslen, C.L.; Gruber, P.J.; et al. Cilia gene mutations cause atrioventricular septal defects by multiple mechanisms. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burns, T.; Yang, Y.; Hiriart, E.; Wessels, A. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. J. Cardiovasc. Dev. Dis. 2016, 3, 29. https://doi.org/10.3390/jcdd3040029
Burns T, Yang Y, Hiriart E, Wessels A. The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. Journal of Cardiovascular Development and Disease. 2016; 3(4):29. https://doi.org/10.3390/jcdd3040029
Chicago/Turabian StyleBurns, Tara, Yanping Yang, Emilye Hiriart, and Andy Wessels. 2016. "The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects" Journal of Cardiovascular Development and Disease 3, no. 4: 29. https://doi.org/10.3390/jcdd3040029
APA StyleBurns, T., Yang, Y., Hiriart, E., & Wessels, A. (2016). The Dorsal Mesenchymal Protrusion and the Pathogenesis of Atrioventricular Septal Defects. Journal of Cardiovascular Development and Disease, 3(4), 29. https://doi.org/10.3390/jcdd3040029