Adverse Effects of Fenofibrate in Mice Deficient in the Protein Quality Control Regulator, CHIP

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Metabolomic Analyses of Hearts
2.3. Ex Vivo Oxidation and ATP Assays
2.4. Fenofibrate Feeding
2.5. RNA Isolation and Quantitative Polymerase Chain Reaction (qPCR) Analysis of Gene Expression
2.6. Measurement of Blood Chemistry
2.7. Echocardiography
2.8. Morphological Analysis of Tissue by Histology and Transmission Electron Microscopy
3. Results
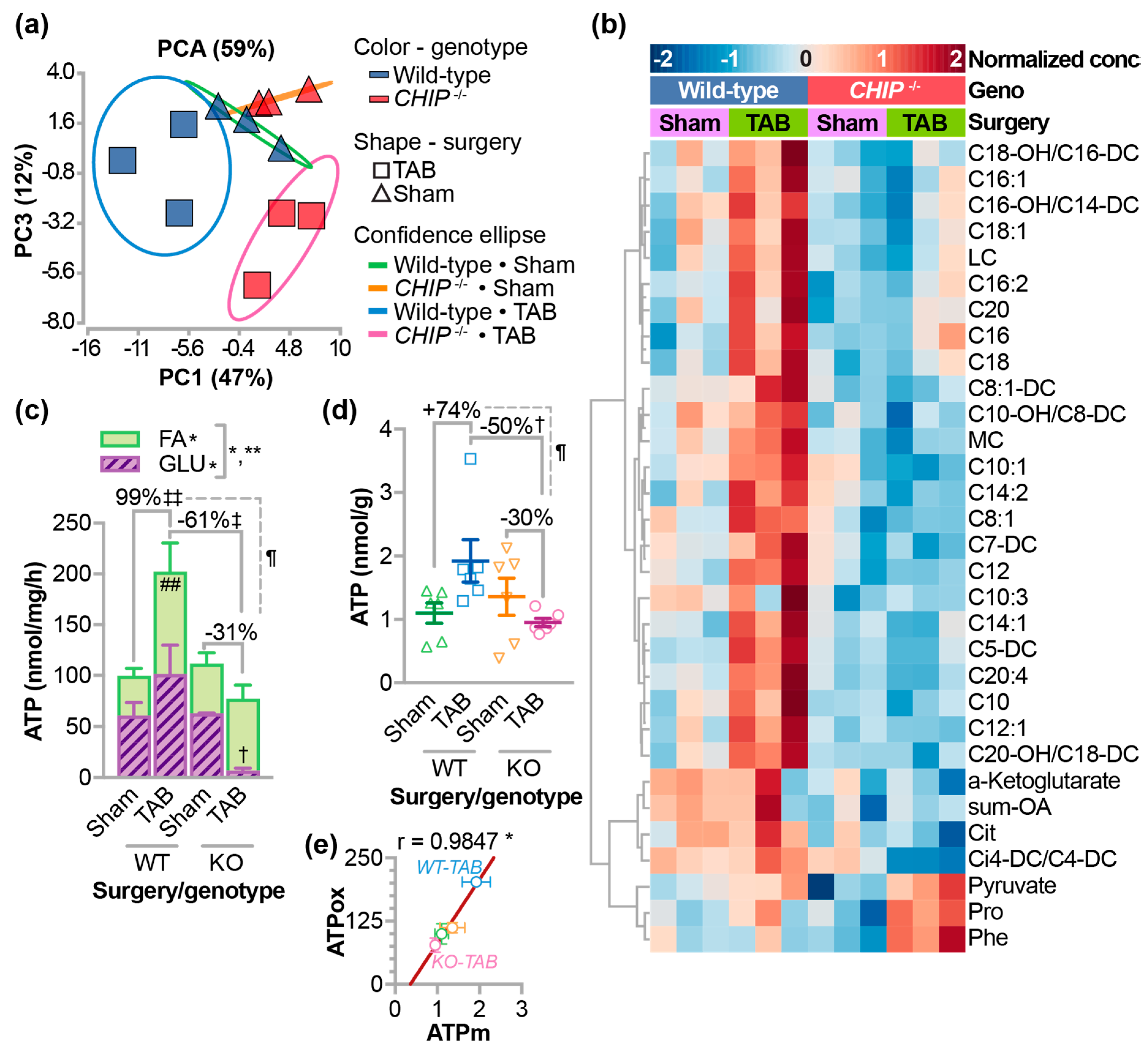
3.1. Role of CHIP in Fatty Acid Metabolism
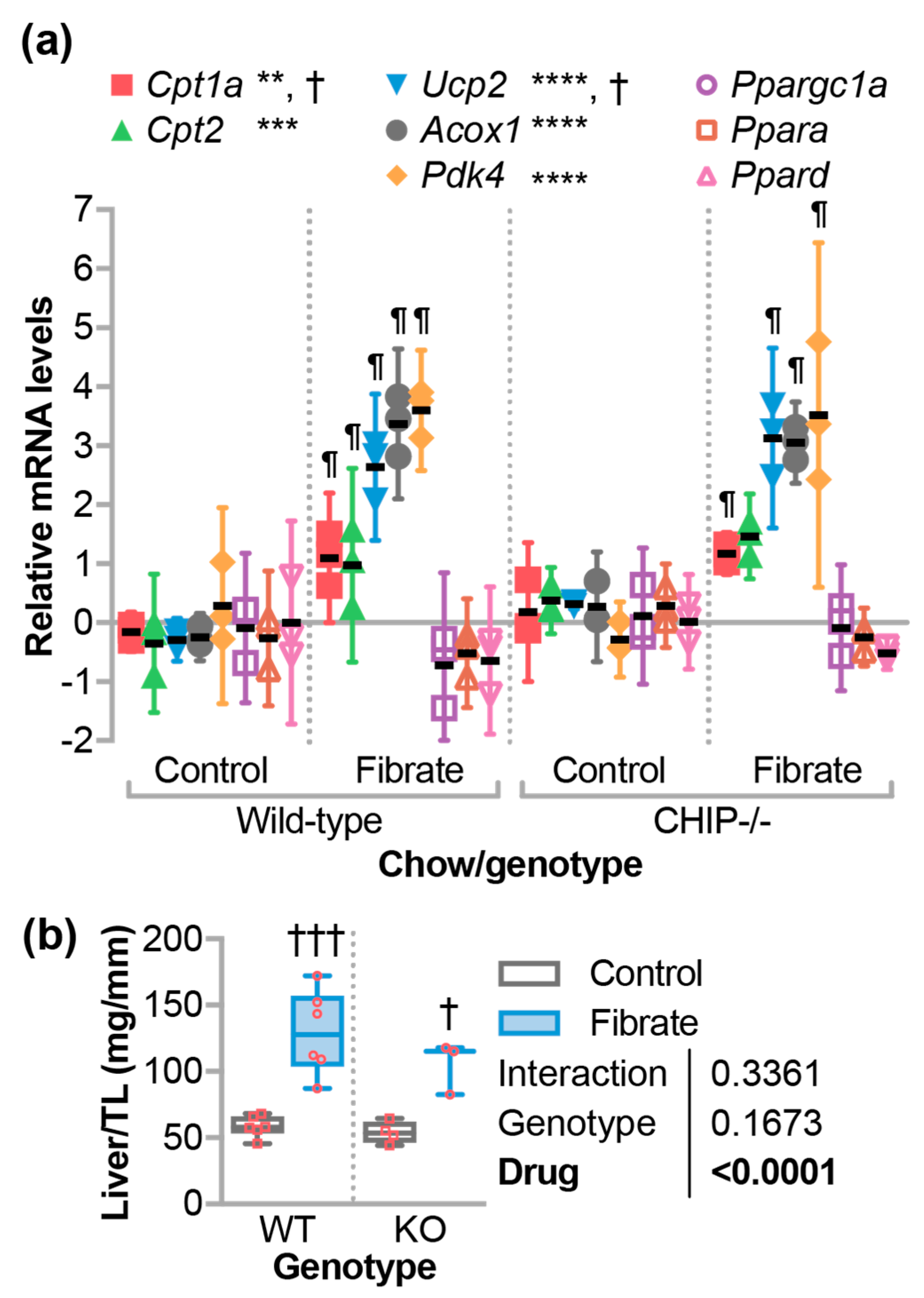
3.2. Fenofibrate-Activated PPARα Target Genes in the Liver
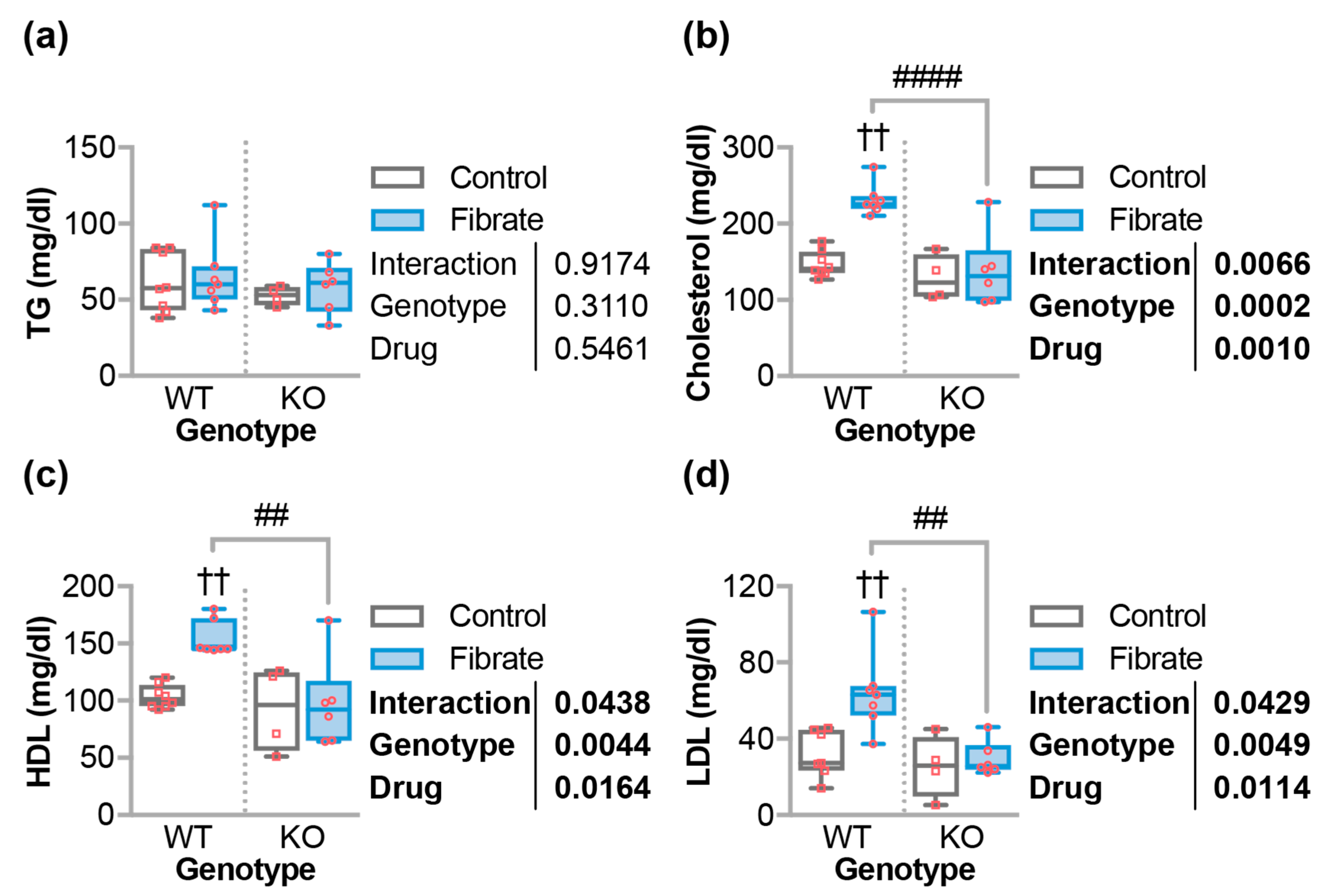
3.3. Fenofibrate Altered Circulating Cholesterol in Wild-Type But Not CHIP−/− Mice
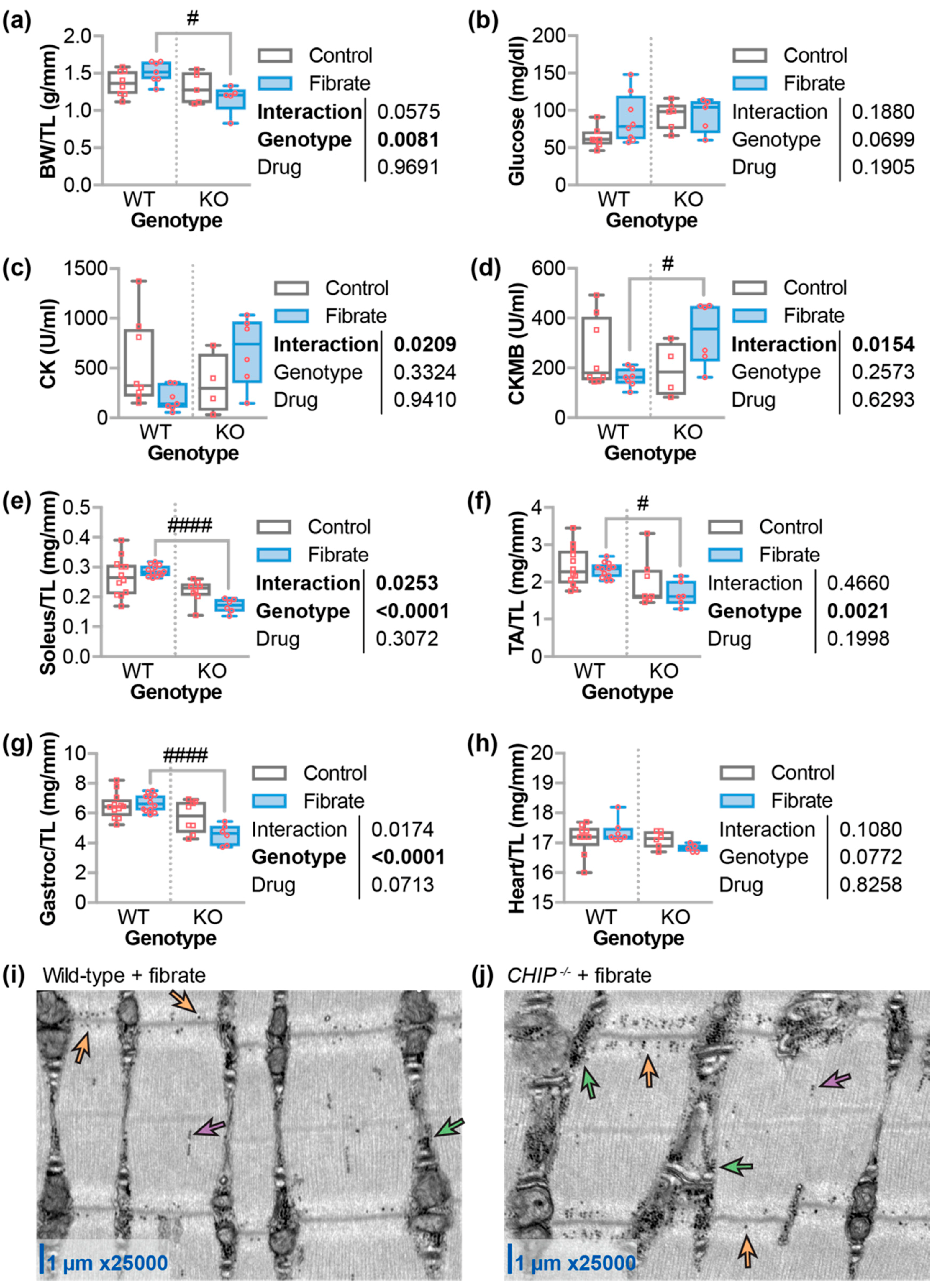
3.4. Differential Effects of Fenofibrate on Skeletal Muscle
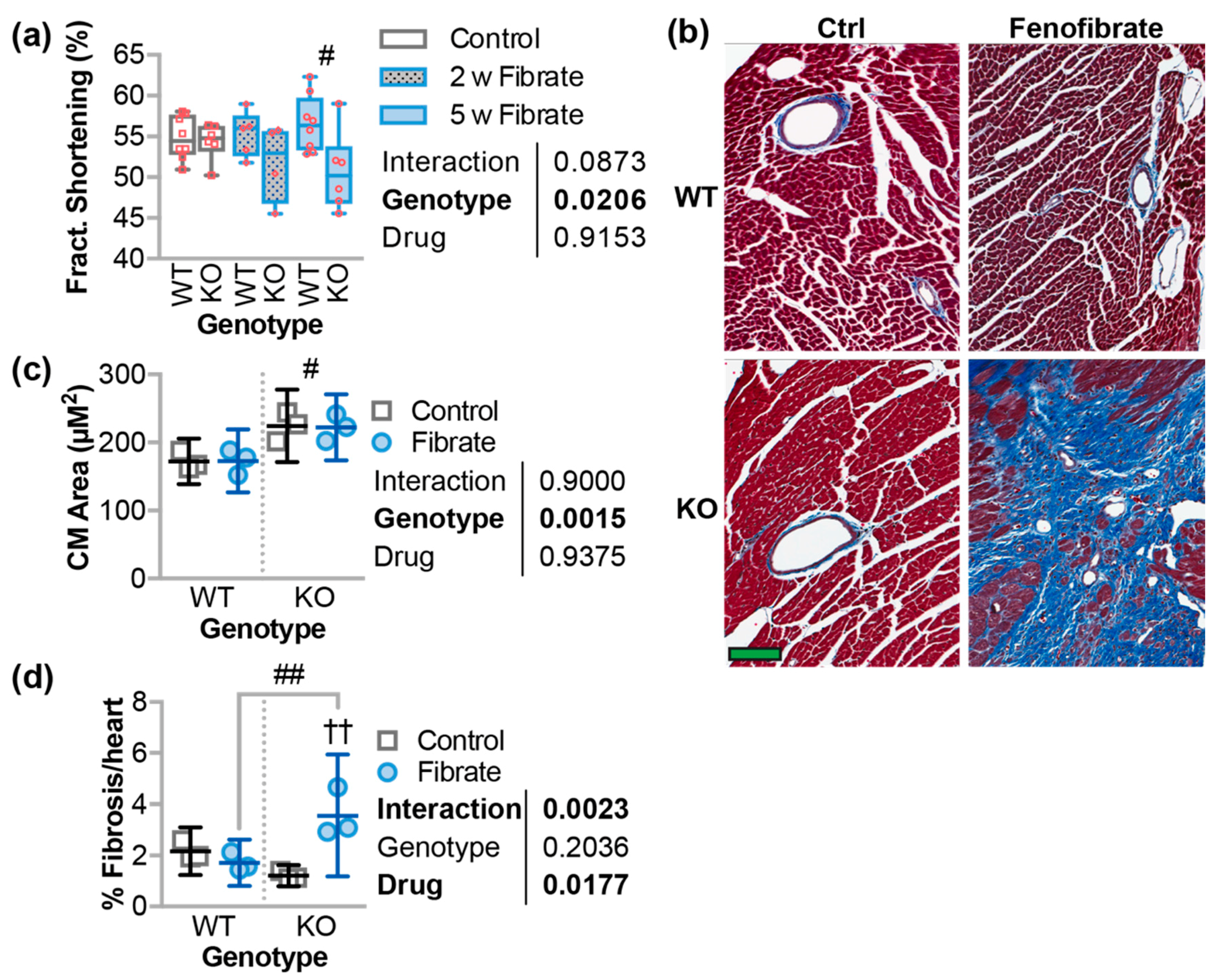
3.5. Fenofibrate Decreased Cardiac Function and Increased Fibrosis in CHIP−/− Mice
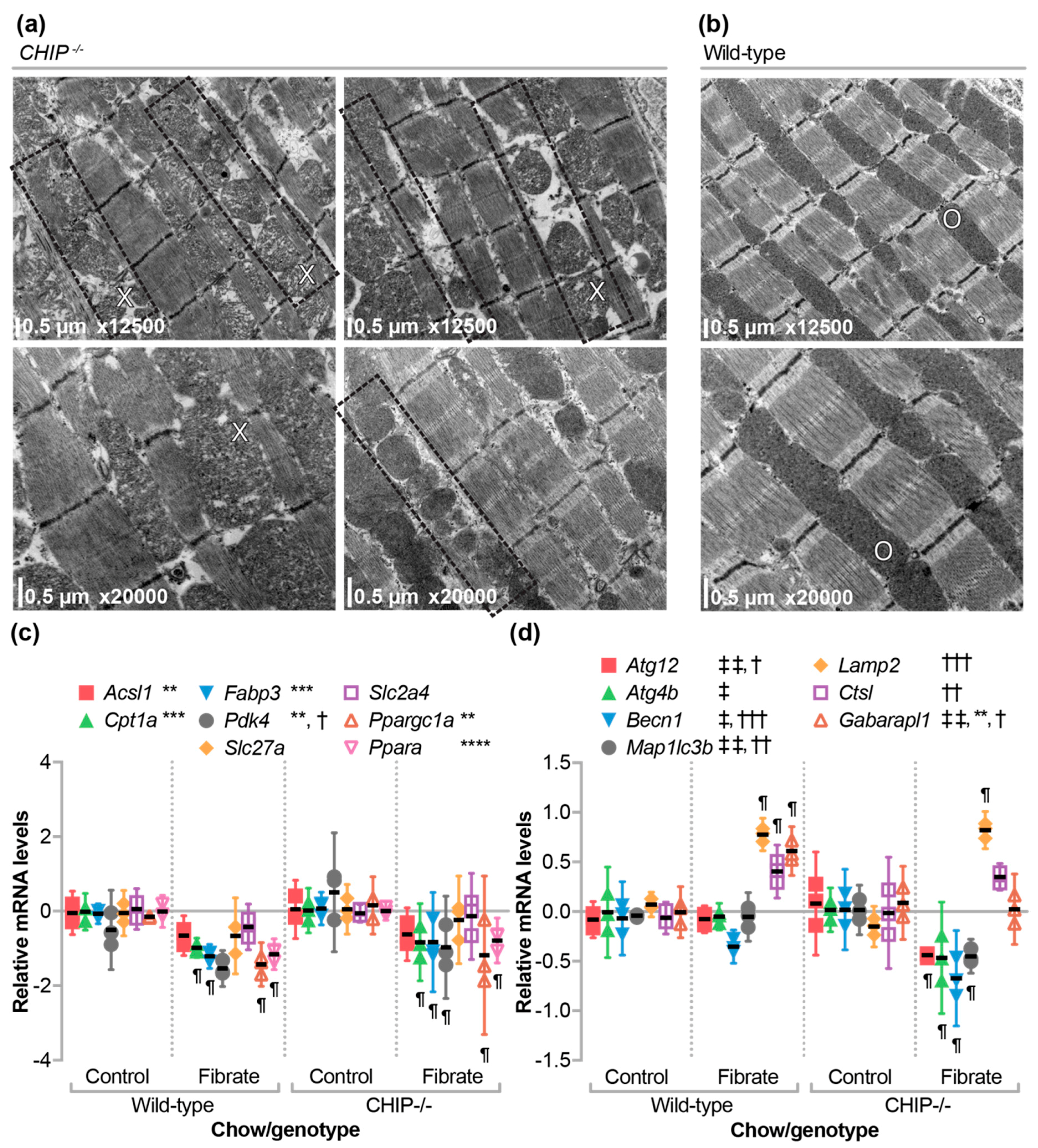
3.6. The Effect of Fenofibrate on Mitochondrial Ultrastructure in the Cardiac Sarcomere and on the Expression of Metabolic, Autophagy, and Mitophagy Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paul, I.; Ghosh, M.K. A CHIPotle in physiology and disease. Int. J. Biochem. Cell Biol. 2015, 58, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-H.; Schisler, J.C.; Rubel, C.E.; Tan, S.; Song, B.; McDonough, H.; Xu, L.; Portbury, A.L.; Mao, C.-Y.; True, C.; et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 2014, 23, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, S.M.; Patterson, C.; Schisler, J.C. Emerging evidence of coding mutations in the ubiquitin–proteasome system associated with cerebellar ataxias. Hum. Genome Var. 2014, 1, 14018. [Google Scholar] [CrossRef] [PubMed]
- Kanack, A.J.; Newsom, O.J.; Scaglione, K.M. Most mutations that cause spinocerebellar ataxia autosomal recessive type 16 (SCAR16) destabilize the protein quality-control E3 ligase CHIP. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pakdaman, Y.; Sanchez-Guixé, M.; Kleppe, R.; Erdal, S.; Bustad, H.J.; Bjørkhaug, L.; Haugarvoll, K.; Tzoulis, C.; Heimdal, K.; Knappskog, P.M.; et al. In vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, S.M.; Patterson, C.; Schisler, J.C. Minireview: Hey U(PS): Metabolic and Proteolytic Homeostasis Linked via AMPK and the Ubiquitin Proteasome System. Mol. Endocrinol. 2014, 28, 1602–1615. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.T.; Okada, S.; Minamino, T.; Iwanaga, K.; Liu, M.-L.; Sumida, T.; Nomura, S.; Sahara, N.; Mizoroki, T.; Takashima, A.; et al. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ. Res. 2010, 106, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Le, N.-T.; Takei, Y.; Shishido, T.; Woo, C.-H.; Chang, E.; Heo, K.-S.; Lee, H.; Lu, Y.; Morrell, C.; Oikawa, M.; et al. p90RSK targets the ERK5-CHIP ubiquitin E3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ. Res. 2012, 110, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, Z.; He, X.-R.; Michael, L.H.; Patterson, C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2836–H2842. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, T.-P.; Tian, C.; Jia, L.-X.; Du, J.; Li, H.-H. Carboxyl terminus of heat shock protein 70-interacting protein inhibits angiotensin II-induced cardiac remodeling. Am. J. Hypertens. 2012, 25, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Geng, Y.-J.; Bolli, R.; Rokosh, G.; Ferdinandy, P.; Patterson, C.; De Caterina, R. Co-activation of nuclear factor-κB and myocardin/serum response factor conveys the hypertrophy signal of high insulin levels in cardiac myoblasts. J. Biol. Chem. 2014, 289, 19585–19598. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Min, J.-N.; Wang, S.; McDonough, H.; Lockyer, P.; Wadosky, K.M.; Patterson, C. Carboxyl terminus of Hsp70-interacting protein (CHIP) is required to modulate cardiac hypertrophy and attenuate autophagy during exercise. Cell Biochem. Funct. 2013, 31, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Schisler, J.C.; Rubel, C.E.; Zhang, C.; Lockyer, P.; Cyr, D.M.; Patterson, C. CHIP protects against cardiac pressure overload through regulation of AMPK. J. Clin. Investig. 2013, 123, 3588–3599. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, B.; Tao, W.; Hao, Z.; Liu, M. Fibrates for secondary prevention of cardiovascular disease and stroke. Cochrane Database Syst. Rev. 2015, CD009580. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Harvengt, C. Effects of clofibrate, bezafibrate, fenofibrate and probucol on plasma lipolytic enzymes in normolipaemic subjects. Eur. J. Clin. Pharmacol. 1983, 25, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Malmendier, C.L.; Lontie, J.F.; Delcroix, C.; Dubois, D.Y.; Magot, T.; De Roy, L. Apolipoproteins C-II and C-III metabolism in hypertriglyceridemic patients. Effect of a drastic triglyceride reduction by combined diet restriction and fenofibrate administration. Atherosclerosis 1989, 77, 139–149. [Google Scholar] [CrossRef]
- Martin, G.; Schoonjans, K.; Lefebvre, A.M.; Staels, B.; Auwerx, J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARalpha and PPARgamma activators. J. Biol. Chem. 1997, 272, 28210–28217. [Google Scholar] [CrossRef] [PubMed]
- Vu-Dac, N.; Schoonjans, K.; Kosykh, V.; Dallongeville, J.; Fruchart, J.C.; Staels, B.; Auwerx, J. Fibrates increase human apolipoprotein A-II expression through activation of the peroxisome proliferator-activated receptor. J. Clin. Investig. 1995, 96, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, T.; Milionis, H.J. Treatment of hyperlipidaemia with fenofibrate and related fibrates. Expert Opin. Investig. Drugs 2008, 17, 1599–1614. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Muoio, D.M.; Shiota, M.; Fujimoto, Y.; Cline, G.W.; Shulman, G.I.; Koves, T.R.; Stevens, R.; Millington, D.; Newgard, C.B. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat. Med. 2004, 10, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-Y.; Kao, H.-J.; Li, S.-C.; Stevens, R.; Hillman, S.; Millington, D.; Chen, Y.-T. ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J. Clin. Investig. 2004, 113, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.V.; Joseph, J.W.; Ilkayeva, O.; Burgess, S.; Lu, D.; Ronnebaum, S.M.; Odegaard, M.; Becker, T.C.; Sherry, A.D.; Newgard, C.B. Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 22342–22351. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Veerkamp, J.H. Postnatal development of palmitate oxidation and mitochondrial enzyme activities in rat cardiac and skeletal muscle. Biochim. Biophys. Acta 1982, 711, 327–335. [Google Scholar] [CrossRef]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator–activated receptor-α during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.R.; Lopaschuk, G.D. Glucose oxidation rates in fatty acid-perfused isolated working hearts from diabetic rats. Biochim. Biophys. Acta 1989, 1006, 97–103. [Google Scholar] [CrossRef]
- Veerkamp, J.H.; van Moerkerk, T.B.; Glatz, J.F.; Zuurveld, J.G.; Jacobs, A.E.; Wagenmakers, A.J. 14CO2 production is no adequate measure of [14C]fatty acid oxidation. Biochem. Med. Metab. Biol. 1986, 35, 248–259. [Google Scholar] [CrossRef]
- Rodríguez, J.E.; Liao, J.-Y.; He, J.; Schisler, J.C.; Newgard, C.B.; Drujan, D.; Glass, D.L.; Frederick, C.B.; Yoder, B.C.; Lalush, D.S.; et al. The ubiquitin ligase MuRF1 regulates PPARα activity in the heart by enhancing nuclear export via monoubiquitination. Mol. Cell. Endocrinol. 2015, 413, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Parry, T.L.; Desai, G.; Schisler, J.C.; Li, L.; Quintana, M.T.; Stanley, N.; Lockyer, P.; Patterson, C.; Willis, M.S. Fenofibrate unexpectedly induces cardiac hypertrophy in mice lacking MuRF1. Cardiovasc. Pathol. 2016, 25, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Rojas, M.; Li, L.; Selzman, C.H.; Tang, R.-H.; Stansfield, W.E.; Rodriguez, J.E.; Glass, D.J.; Patterson, C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H997–H1006. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Ren, R.; Cruz-Topete, D.; Bird, G.S.; Myers, P.H.; Boyle, M.C.; Schneider, M.D.; Willis, M.S.; Cidlowski, J.A. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. USA 2013, 110, 17035–17040. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Dyer, L.A.; Ren, R.; Lockyer, P.; Moreno-Miralles, I.; Schisler, J.C.; Patterson, C. BMPER regulates cardiomyocyte size and vessel density in vivo. Cardiovasc. Pathol. 2013, 22, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Li, L.; Rodríguez, J.E.; Hilliard, E.G.; Charles, P.C.; Patterson, C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ. Res. 2009, 105, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Grimaldi, P.; Auwerx, J. Acyl-CoA synthetase mRNA expression is controlled by fibric-acid derivatives, feeding and liver proliferation. Eur. J. Biochem. 1993, 216, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, E.; de Lange, P.; Moreno, M.; Lombardi, A.; Ragni, M.; Feola, A.; Schiavo, L.; Goglia, F.; Lanni, A. Fenofibrate activates the biochemical pathways and the de novo expression of genes related to lipid handling and uncoupling protein-3 functions in liver of normal rats. Biochim. Biophys. Acta 2006, 1757, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.-J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [PubMed]
- Cattley, R.C. Regulation of cell proliferation and cell death by peroxisome proliferators. Microsc. Res. Tech. 2003, 61, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Declercq, V.; Yeganeh, B.; Moshtaghi-Kashanian, G.-R.; Khademi, H.; Bahadori, B.; Moghadasian, M.H. Paradoxical effects of fenofibrate and nicotinic acid in apo E-deficient mice. J. Cardiovasc. Pharmacol. 2005, 46, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Schisler, J.C.; Patterson, C.; Willis, M.S. Skeletal muscle mitochondrial alterations in carboxyl terminun of HSC70 interacting protein (CHIP)−/− mice. Afr. J. Cell. Pathol. 2016, 28–36. [Google Scholar]
- Gottlieb, R.A.; Mentzer, R.M. Autophagy: An affair of the heart. Heart Fail. Rev. 2013, 18, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Y.; Gu, J.; Wang, S.; Zhou, S.; Wang, Y.; Tan, Y.; Feng, W.; Fu, Y.; Mellen, N.; et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin. Sci. 2016, 130, 625–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pan, Y.; Xiong, R.; Zheng, J.; Li, Q.; Zhang, S.; Li, X.; Pan, X.; Yang, S. FGF21 mediates the protective effect of fenofibrate against acetaminophen -induced hepatotoxicity via activating autophagy in mice. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Song, K.D.; Lee, H.-K.; Yi, S.; Lee, Y.S.; Heo, T.-H.; Jun, H.S.; Kim, S.-J. Fibrates inhibit the apoptosis of Batten disease lymphoblast cells via autophagy recovery and regulation of mitochondrial membrane potential. In Vitro Cell. Dev. Biol. Anim. 2016, 52, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Lizama, B.N.; Palubinsky, A.M.; Raveendran, V.A.; Moore, A.M.; Federspiel, J.D.; Codreanu, S.G.; Liebler, D.C.; McLaughlin, B. Neuronal Preconditioning Requires the Mitophagic Activity of C-terminus of HSC70-Interacting Protein. J. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Sha, Y.; Eissa, N.T. The E3 ubiquitin ligase STUB1 regulates autophagy and mitochondrial biogenesis by modulating TFEB activity. Mol. Cell. Oncol. 2017, 4, e1372867. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, J.; Noguchi, Y.; Hattori, Y.; Yamaguchi, M.; Yamazaki, Y.; Ikari, A. Threonine-408 Regulates the Stability of Human Pregnane X Receptor through Its Phosphorylation and the CHIP/Chaperone-Autophagy Pathway. Drug Metab. Dispos. 2016, 44, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Ying, Z.; Wang, H.; Chen, D.; Gao, F.; Ren, H.; Wang, G. Regulation of autophagic flux by CHIP. Neurosci. Bull. 2015, 31, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.V.; Fôfo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girão, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.E.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M.A. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [PubMed]
- Pascual, F.; Schisler, J.C.; Grevengoed, T.J.; Willis, M.S.; Coleman, R.A. Modeling the Transition From Decompensated to Pathological Hypertrophy. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Schisler, J.C.; Coleman, R.A. mTORtuous effect on the elastic heart. Oncotarget 2015, 6, 16810–16811. [Google Scholar] [CrossRef] [PubMed]
- Schisler, J.C.; Grevengoed, T.J.; Pascual, F.; Cooper, D.E.; Ellis, J.M.; Paul, D.S.; Willis, M.S.; Patterson, C.; Jia, W.; Coleman, R.A. Cardiac energy dependence on glucose increases metabolites related to glutathione and activates metabolic genes controlled by mechanistic target of rapamycin. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Gariot, P.; Barrat, E.; Drouin, P.; Genton, P.; Pointel, J.P.; Foliguet, B.; Kolopp, M.; Debry, G. Morphometric study of human hepatic cell modifications induced by fenofibrate. Metab. Clin. Exp. 1987, 36, 203–210. [Google Scholar] [CrossRef]
- Guo, Y.; Jolly, R.A.; Halstead, B.W.; Baker, T.K.; Stutz, J.P.; Huffman, M.; Calley, J.N.; West, A.; Gao, H.; Searfoss, G.H.; et al. Underlying mechanisms of pharmacology and toxicity of a novel PPAR agonist revealed using rodent and canine hepatocytes. Toxicol. Sci. 2007, 96, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Das, S.K.; Jha, P.; Al Zoughbi, W.; Schauer, S.; Claudel, T.; Sexl, V.; Vesely, P.; Birner-Gruenberger, R.; Kratky, D.; et al. The PPARα agonist fenofibrate suppresses B-cell lymphoma in mice by modulating lipid metabolism. Biochim. Biophys. Acta 2013, 1831, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Duhaney, T.-A.S.; Silva, D.S.D.; Cui, L.; Ip, P.C.; Joseph, L.; Sam, F. Effects of Fenofibrate on Cardiac Remodeling in Aldosterone-Induced Hypertension. Hypertension 2007, 50, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Miyauchi, T.; Sakai, S.; Takanashi, M.; Irukayama-Tomobe, Y.; Yamaguchi, I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J. Am. Coll. Cardiol. 2004, 43, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Dhahri, W.; Couet, J.; Roussel, É.; Drolet, M.-C.; Arsenault, M. Fenofibrate reduces cardiac remodeling and improves cardiac function in a rat model of severe left ventricle volume overload. Life Sci. 2013, 92, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Labinskyy, V.; Bellomo, M.; Chandler, M.P.; Young, M.E.; Lionetti, V.; Qanud, K.; Bigazzi, F.; Sampietro, T.; Stanley, W.C.; Recchia, F.A. Chronic activation of peroxisome proliferator-activated receptor-alpha with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. J. Pharmacol. Exp. Ther. 2007, 321, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-B.; Li, X.-X.; Chen, Y.-G.; Zhang, C.; Zhang, M.-X.; Zhao, X.-Q.; Hao, M.-X.; Hou, X.-Y.; Gong, M.-L.; Zhao, Y.-X.; et al. Effects and mechanisms of PPARalpha activator fenofibrate on myocardial remodelling in hypertension. J. Cell. Mol. Med. 2009, 13, 4444–4452. [Google Scholar] [CrossRef] [PubMed]
- Duhaney, T.-A.S.; Cui, L.; Rude, M.K.; Lebrasseur, N.K.; Ngoy, S.; De Silva, D.S.; Siwik, D.A.; Liao, R.; Sam, F. Peroxisome proliferator-activated receptor alpha-independent actions of fenofibrate exacerbates left ventricular dilation and fibrosis in chronic pressure overload. Hypertension 2007, 49, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Punthakee, Z.; Scully, L.J.; Guindi, M.M.; Ooi, T.C. Liver fibrosis attributed to lipid lowering medications: Two cases. J. Intern. Med. 2001, 250, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Attridge, R.L.; Frei, C.R.; Ryan, L.; Koeller, J.; Linn, W.D. Fenofibrate-associated nephrotoxicity: A review of current evidence. Am. J. Health Syst. Pharm. 2013, 70, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Olivé, M.; Abdul-Hussein, S.; Oldfors, A.; González-Costello, J.; van der Ven, P.F.M.; Fürst, D.O.; González, L.; Moreno, D.; Torrejón-Escribano, B.; Alió, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Wang, J.; Kang, L.; Wang, Y.; Zou, Y.; Feng, X.; Wang, D.; Ahmad, F.; Zhou, X.; Hui, R.; et al. Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathy. Int. J. Mol. Sci. 2014, 15, 9302–9313. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | No Drug | 2 w | 5 w |
| Wild-type (N) | 8 | 5 | 8 |
| CHIP−/− (N) | 6 | 4 | 6 |
| Analysis | |||
| Echo | ✓ | ✓ | ✓ |
| Blood labs | ✓ | ✓ | |
| Histology/RNA | ✓ | ✓ |
| Gene | UPL Probe | Sense (5′–3′) | Anti-sense (5′–3′) |
|---|---|---|---|
| Acox1 | #45 | gcgccagtctgaaatcaag | actgctgcgtctgaaaatcc |
| Cpt1a | #109 | gctgtcaaagataccgtgagc | tctccctccttcatcagtgg |
| Cpt2 | #71 | ccaaagaagcagcgatgg | tagagctcaggcagggtga |
| Pdk4 | #22 | ctgcctgaccgcttagtga | cttctgggctcttctcatgg |
| Ucp2 | #2 | acagccttctgcactcctg | ggctgggagacgaaacact |
| Ppard | #11 | atgggggaccagaacacac | ggaggaattctgggagaggt |
| Ppargc1a | #6 | cagtcgcaacatgctcaag | tggggtcatttggtgactct |
| Ppara | #41 | cacgcatgtgaaggctgtaa | cagctccgatcacacttgtc |
| Acsl1 | #84 | cagcctcactgcccttttc | ggttggtggttctctatgcag |
| Fabp3 | #56 | ctttgtcggtacctggaagc | tggtcatgctagccacctg |
| Slc27a | #1 | gacaagctggatcaggcaag | gaggccacagaggctgttc |
| Slc2a4 | #5 | gacggacactccatctgttg | gccacgatggagacatagc |
| Atg12 | SYBR | ggcctcggaacagttgttta | cagcaccgaaatgtctctga |
| Atg4b | SYBR | attgctgtggggtttttctg | aaccccaggattttcagagg |
| Becn1 | SYBR | ggccaataagatgggtctga | cactgcctccagtgtcttca |
| Ctsl | SYBR | gtggactgttctcacgctcaag | tccgtccttcgcttcatagg |
| Gabarapl1 | SYBR | catcgtggagaaggctccta | atacagctggcccatggtag |
| Lamp2 | SYBR | tggctaatggctcagctttc | atgggcacaaggaagttgtc |
| Map1lc3b | SYBR | cgtcctggacaagaccaagt | attgctgtcccgaatgtctc |
| Genotype | Wild-Type | CHIP−/− | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | Effect | p | Drug | 0 | 2 w | 5 w | 0 | 2 w | 5 w |
| N | 8 | 5 | 8 | 6 | 4 | 6 | |||
| HR (bpm) | Inter | 0.915 | 600.7 ± 23.8 | 660.3 ± 17.9 | 640.5 ± 17.8 | 602.0 ± 28.0 | 645.3 ± 26.5 | 625.5 ± 17.5 | |
| Geno | 0.085 | ||||||||
| Drug | 0.604 | ||||||||
| IVS;d (mm) | Inter | 0.0004 | 1.07 ± 0.02 | 0.99 ± 0.02 | 1.11 ± 0.03 | 1.04 ± 0.02 | 1.3 ± 0.5 ***,### | 1.04 ± 0.06 ††† | |
| Geno | 0.0027 | ||||||||
| Drug | 0.146 | ||||||||
| LVID;d (mm) | Inter | 0.699 | 3.50 ± 0.16 | 3.09 ± 0.14 | 3.35 ± 0.10 | 3.26 ± 0.14 | 3.09 ± 0.24 | 3.3 ± 0.13 | |
| Geno | 0.137 | ||||||||
| Drug | 0.425 | ||||||||
| LVPW;d (mm) | Inter | 0.017 | 1.04 ± 0.03 | 1.02 ± 0.01 | 1.09 ± 0.04 | 1.00 ± 0.042 | 1.17 ± 0.07 | 0.99 ± 0.05 † | |
| Geno | 0.143 | ||||||||
| Drug | 0.948 | ||||||||
| IVS;s (mm) | Inter | 0.0029 | 1.75 ± 0.06 | 1.69 ± 0.05 | 1.86 ± 0.05 | 1.74 ± 0.07 | 2.02 ± 0.06 ## | 1.69 ± 0.09 † | |
| Geno | 0.247 | ||||||||
| Drug | 0.398 | ||||||||
| LVID;s (mm) | Inter | 0.263 | 1.61 ± 0.08 | 1.39 ± 0.09 | 1.43 ± 0.05 | 1.49 ± 0.09 | 1.51 ± 0.20 | 1.63 ± 1.11 | |
| Geno | 0.572 | ||||||||
| Drug | 0.426 | ||||||||
| LVPW;s (mm) | Inter | 0.473 | 1.59 ± 0.04 | 1.57 ± 0.08 | 1.68 ± 0.07 | 1.53 ± 0.05 | 1.53 ± 0.06 | 1.5 ± 0.05 | |
| Geno | 0.782 | ||||||||
| Drug | 0.068 | ||||||||
| LV Vol;d (µL) | Inter | 0.617 | 51.92 ± 5.57 | 38.30 ± 4.02 | 46.04 ± 3.07 | 43.46 ± 4.40 | 38.84 ± 7.82 | 44.86 ± 4.39 | |
| Geno | 0.161 | ||||||||
| Drug | 0.447 | ||||||||
| LV Vol;s (µL) | Inter | 0.254 | 7.46 ± 0.91 | 5.13 ± 0.88 | 5.51 ± 0.51 | 6.16 ± 1.03 | 6.80 ± 2.42 | 7.89 ± 1.20 | |
| Geno | 0.723 | ||||||||
| Drug | 0.329 | ||||||||
| LV Mass (mg) | Inter | 0.0039 | 140.18 ± 7.13 | 109.95 ± 6.87 | 140.74 ± 8.32 | 120.86 ± 11.18 | 150.08 ± 8.21 # | 122.31 ± 11.08 | |
| Geno | 0.986 | ||||||||
| Drug | 0.917 | ||||||||
| EF (%) | Inter | 0.087 | 85.42 ± 0.75 | 86.89 ± 1.0 | 87.78 ± 1.12 | 86.05 ± 0.86 | 83.84 ± 2.40 | 82.7 ± 1.71 | |
| Geno | 0.915 | ||||||||
| Drug | 0.021 | ||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravi, S.; Parry, T.L.; Willis, M.S.; Lockyer, P.; Patterson, C.; Bain, J.R.; Stevens, R.D.; Ilkayeva, O.R.; Newgard, C.B.; Schisler, J.C. Adverse Effects of Fenofibrate in Mice Deficient in the Protein Quality Control Regulator, CHIP. J. Cardiovasc. Dev. Dis. 2018, 5, 43. https://doi.org/10.3390/jcdd5030043
Ravi S, Parry TL, Willis MS, Lockyer P, Patterson C, Bain JR, Stevens RD, Ilkayeva OR, Newgard CB, Schisler JC. Adverse Effects of Fenofibrate in Mice Deficient in the Protein Quality Control Regulator, CHIP. Journal of Cardiovascular Development and Disease. 2018; 5(3):43. https://doi.org/10.3390/jcdd5030043
Chicago/Turabian StyleRavi, Saranya, Traci L. Parry, Monte S. Willis, Pamela Lockyer, Cam Patterson, James R. Bain, Robert D. Stevens, Olga R. Ilkayeva, Christopher B. Newgard, and Jonathan C. Schisler. 2018. "Adverse Effects of Fenofibrate in Mice Deficient in the Protein Quality Control Regulator, CHIP" Journal of Cardiovascular Development and Disease 5, no. 3: 43. https://doi.org/10.3390/jcdd5030043
APA StyleRavi, S., Parry, T. L., Willis, M. S., Lockyer, P., Patterson, C., Bain, J. R., Stevens, R. D., Ilkayeva, O. R., Newgard, C. B., & Schisler, J. C. (2018). Adverse Effects of Fenofibrate in Mice Deficient in the Protein Quality Control Regulator, CHIP. Journal of Cardiovascular Development and Disease, 5(3), 43. https://doi.org/10.3390/jcdd5030043