
The Comparative Genomics of Botryosphaeriaceae Suggests Gene Families of Botryosphaeria dothidea Related to Pathogenicity on Chinese Hickory Tree

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Growth Conditions, DNA Isolation, and Genomic Sequencing
2.2. Genome Assembly, Gene Prediction, and Functional Annotation
2.3. Orthologs Identification and Species Phylogenetic Tree Construction
2.4. Identification of Rapidly Evolving Ortholog Families
2.5. Positive Selection Analysis
2.6. Detection of Horizontal Gene Transfer (HGT)
2.7. Synteny Analysis
2.8. Prediction of Secretome and Small Cysteine-Rich Proteins (SCPs)
2.9. Plant Materials, Fungal Materials, and Inoculation Assays
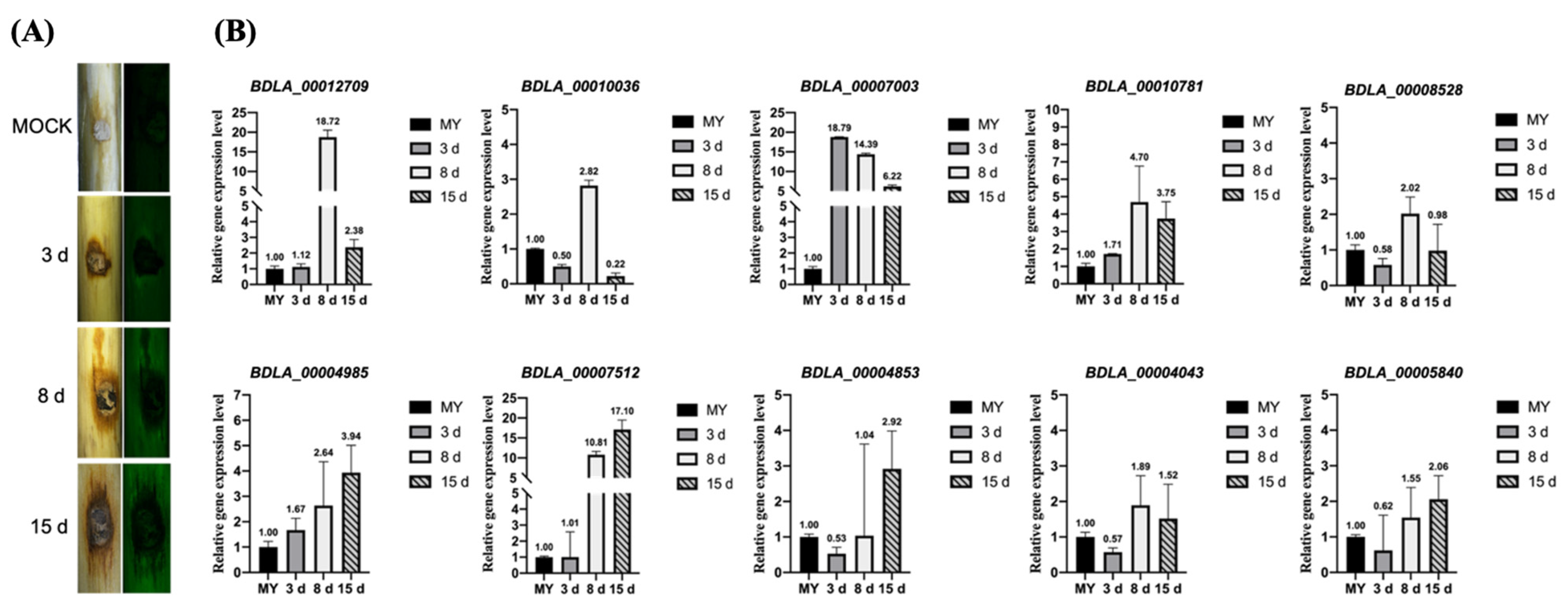
2.10. Validation of Gene Expression Using Quantitative qRT-PCR
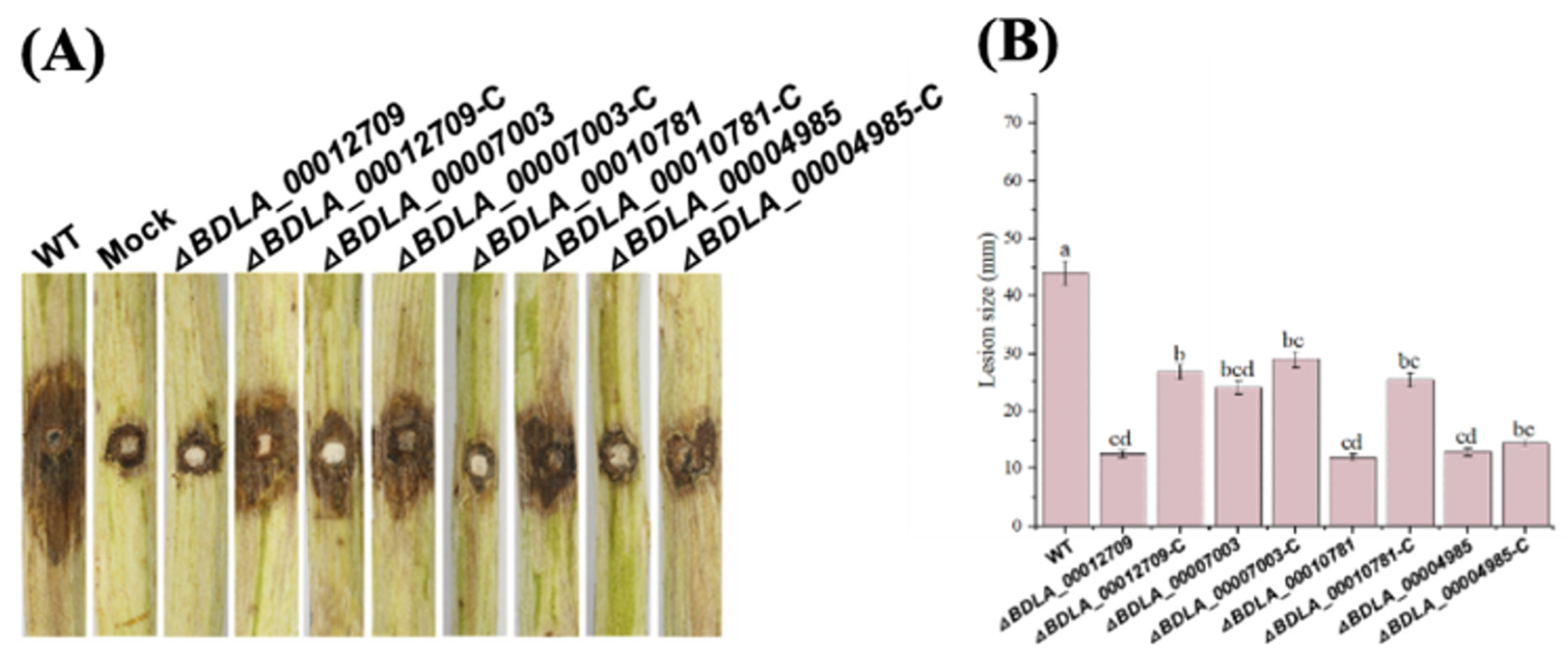
2.11. Construction of Deletion Mutants and Complemented Strains
3. Results
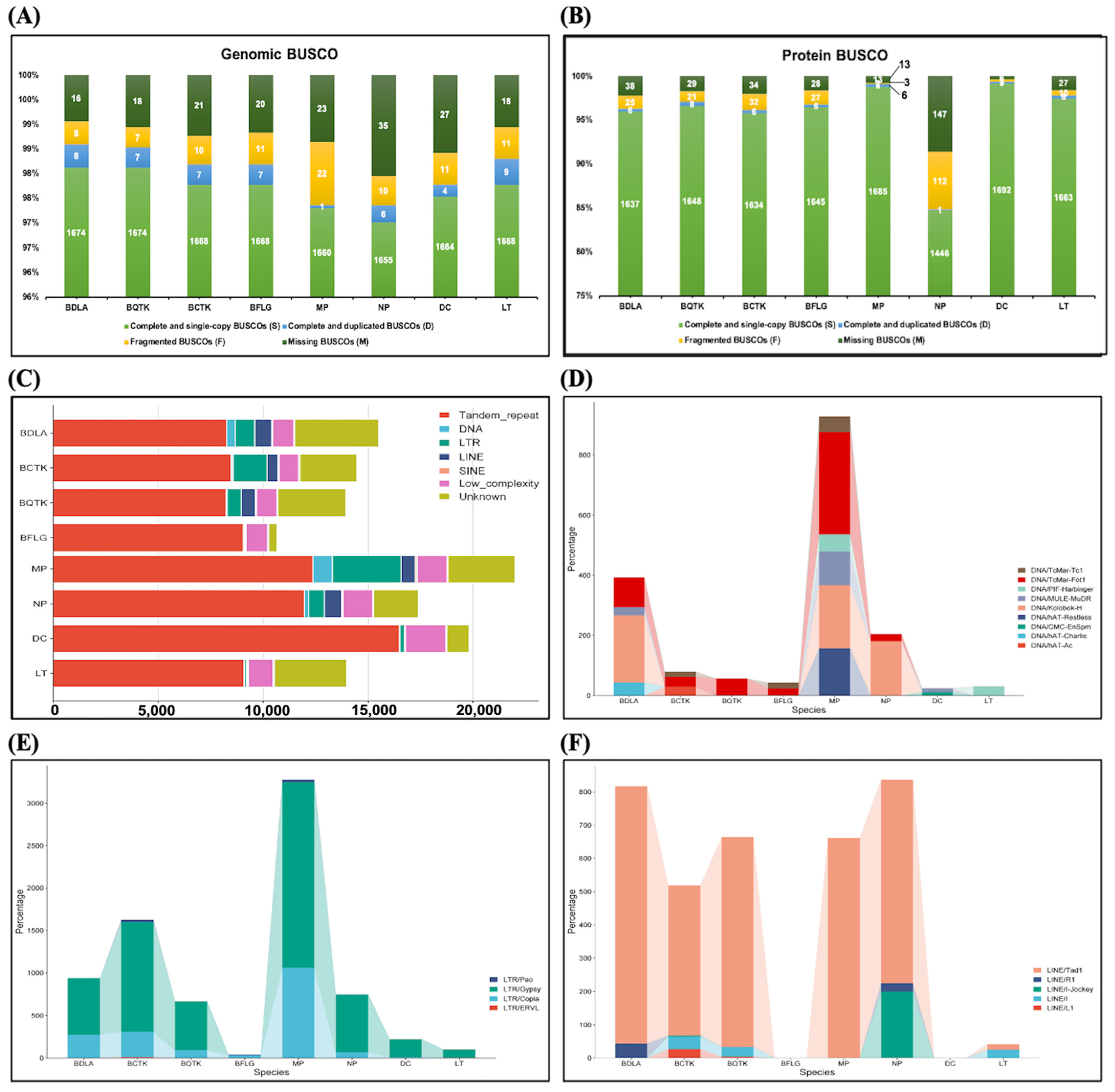
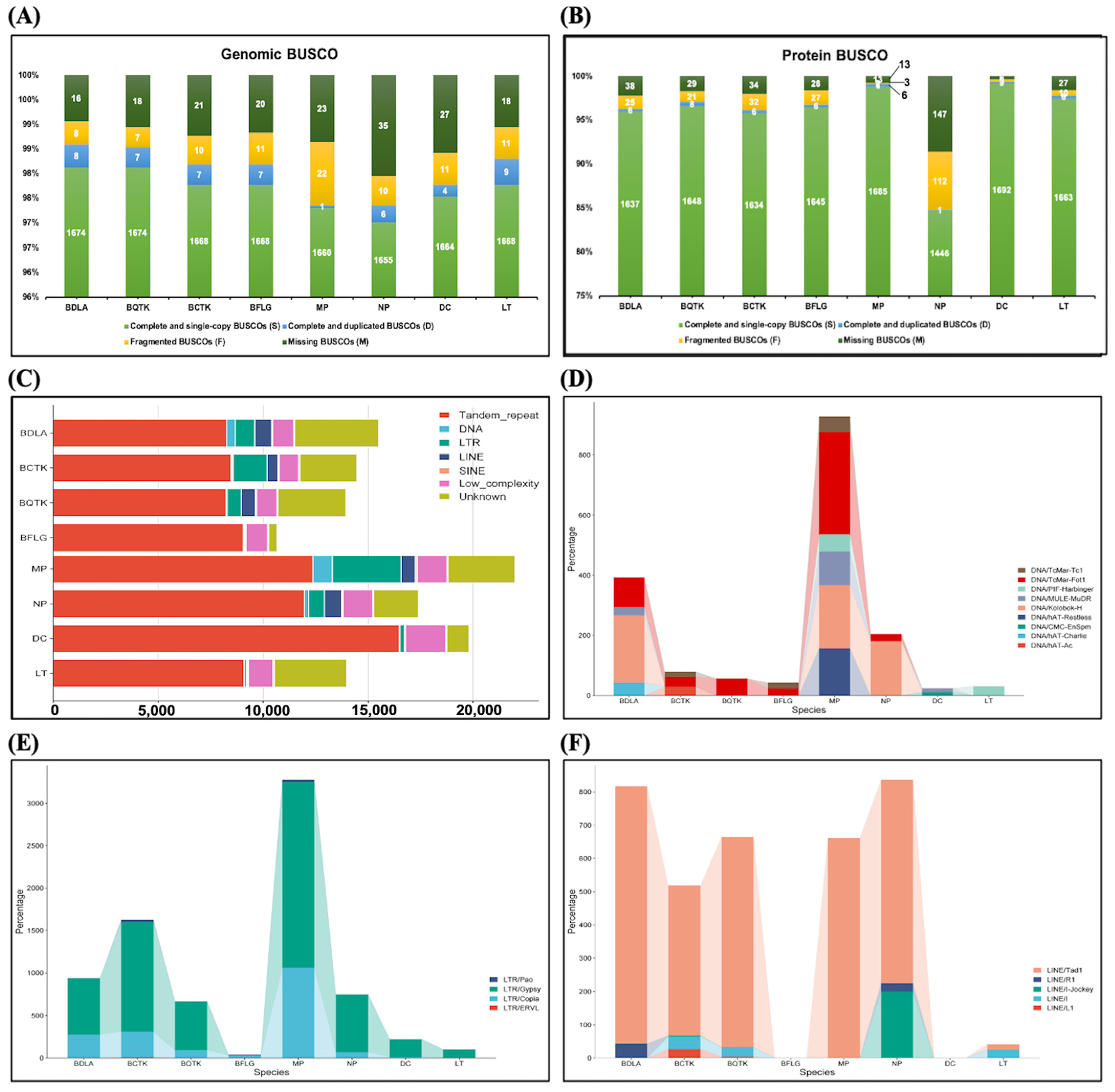
3.1. Genome Assemblies of Eight Botryosphaeriaceae Species and Features of Note
3.2. Identification of Repeat Sequences and Protein-Coding Genes
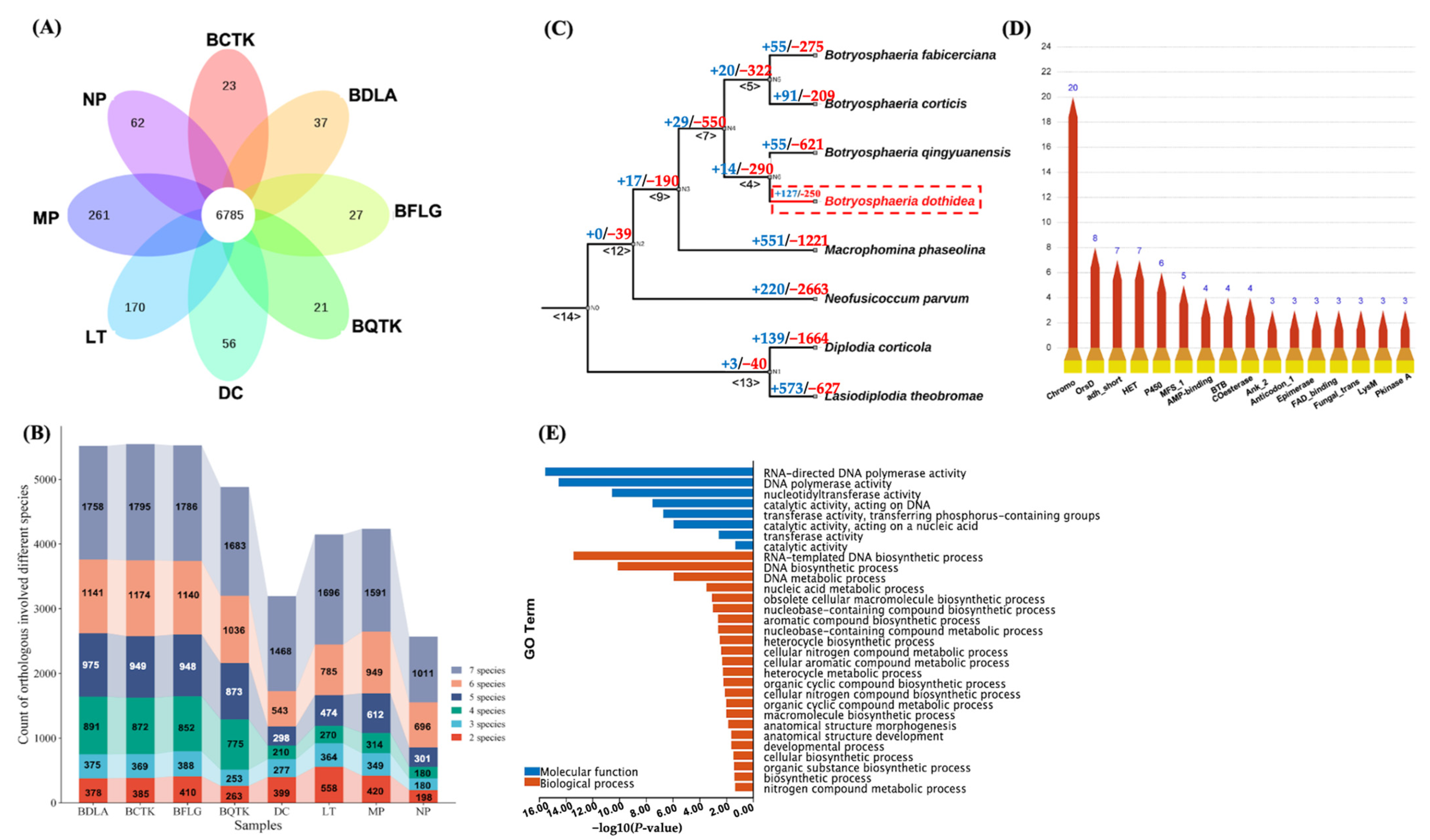
3.3. Synteny Analysis of BDLA Genome Reveals Two Accessory Contigs and Lineage-Specific (LS) Genes
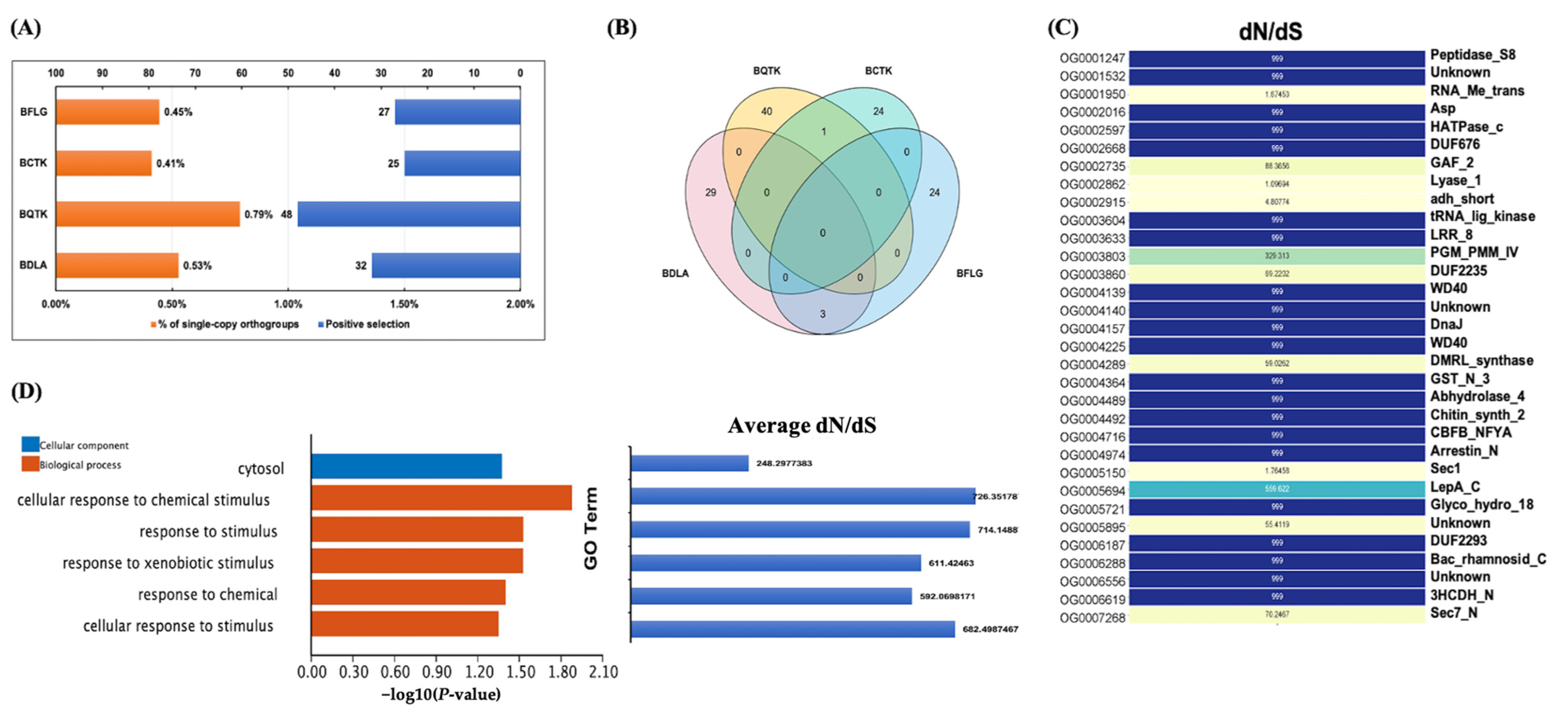
3.4. Ortholog Construction of Botryosphaeriaceae Species Uncovered Rapid Expanded Ortholog in BDLA
3.5. Positively Selected Genes (PSGs) of BDLA may Be Involved in Response to Xenobiotic Stimulus
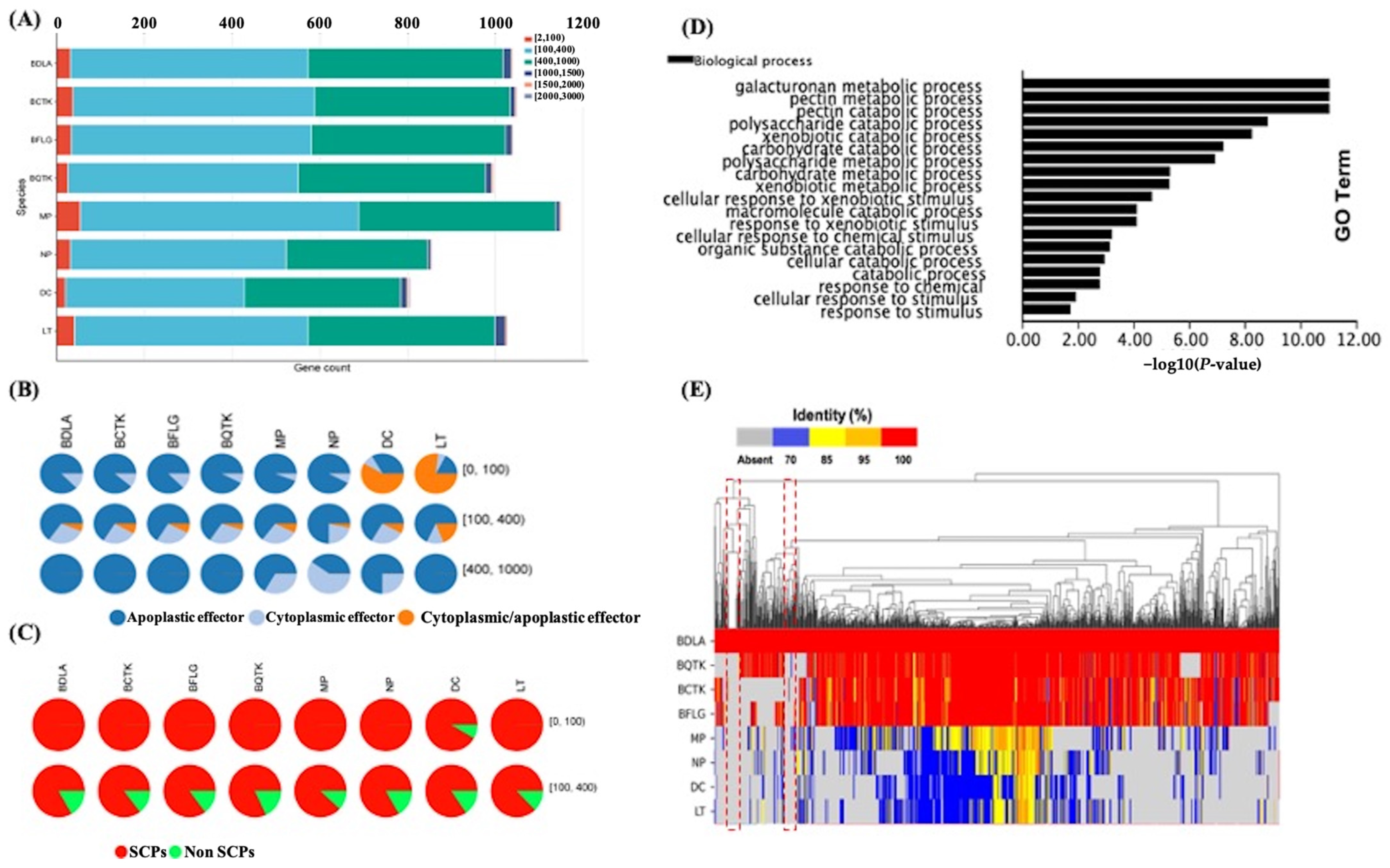
3.6. Predicted Secretome Comparison Gives Insight into BDLA-Specific Small Secret Cysteine-Rich Proteins (Effectors)
3.7. Identification of Horizontal Gene Transfer (HGT) Events in BDLA Genome
3.8. Secreted Glucanase, Orsellinic Acid Biosynthesis Enzyme, and MFS Transporters Play an Important Role in Pathogenicity of B. dothidea
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phillips, A.J.L.; Alves, A.; Abdollahzadeh, J.; Slippers, B.; Wingfield, M.J.; Groenewald, J.Z.; Crous, P.W. The Botryosphaeriaceae: Genera and species known from culture. Stud. Mycol. 2013, 76, 51–167. [Google Scholar] [CrossRef] [PubMed]
- Trakunyingcharoen, T.; Lombard, L.; Groenewald, J.Z.; Cheewangkoon, R.; To-Anun, C.; Crous, P.W. Caulicolous Botryosphaeriales from Thailand. Persoonia-Mol. Phylogeny Evol. Fungi 2015, 34, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Belair, M.; Restrepo-Leal, J.D.; Praz, C.; Fontaine, F.; Rémond, C.; Fernandez, O.; Besaury, L. Botryosphaeriaceae gene machinery: Correlation between diversity and virulence. Fungal Biol. 2023, 127, 1010–1031. [Google Scholar] [CrossRef] [PubMed]
- Slippers, B.; Wingfield, M.J. Botryosphaeriaceae as endophytes and latent pathogens of woody plants: Diversity, ecology and impact. Fungal Biol. Rev. 2007, 21, 90–106. [Google Scholar] [CrossRef]
- Dissanayake, A. Botryosphaeriaceae: Current status of genera and species. Mycosphere 2016, 7, 1001–1073. [Google Scholar] [CrossRef]
- Yan, J.Y.; Zhao, W.S.; Chen, Z.; Xing, Q.K.; Zhang, W.; Chethana, K.T.; Xue, M.F.; Xu, J.P.; Phillips, A.J.; Wang, Y. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae. DNA Res. 2018, 25, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Ashnaei, S.P. Grapevine, esca complex, and environment: The disease triangle. Phytopathol. Mediterr. 2019, 58, 17–37. [Google Scholar]
- Songy, A.; Fernandez, O.; Clément, C.; Larignon, P.; Fontaine, F. Grapevine trunk diseases under thermal and water stresses. Planta 2019, 249, 1655–1679. [Google Scholar] [CrossRef] [PubMed]
- Marsberg, A.; Kemler, M.; Jami, F.; Nagel, J.H.; Postma-Smidt, A.; Naidoo, S.; Wingfield, M.J.; Crous, P.W.; Spatafora, J.W.; Hesse, C.N.; et al. Botryosphaeria dothidea: A latent pathogen of global importance to woody plant health. Mol. Plant Pathol. 2017, 18, 477–488. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, P.; Chen, W.; Yang, G.; Gu, Y.; Wang, J.; Wang, J. First Report of Leaf Spot on Malania oleifera Caused by Botryosphaeria fabicerciana in China. Plant Dis. 2022, 106, 1759. [Google Scholar] [CrossRef]
- Feng, W.; Li, M.; Li, L.; He, Y.; Yang, J.; Sun, J.; Gao, Z.; Hu, M. First Report of Stem-End Rot Caused by Botryosphaeria fabicerciana on Mango Fruits in China. Plant Dis. 2023, 107, 2880. [Google Scholar] [CrossRef] [PubMed]
- Marquez, N.; Giachero, M.L.; Declerck, S.; Ducasse, D.A. Macrophomina Phaseolina: General Characteristics of Pathogenicity and Methods of Control. Front. Plant Sci. 2021, 12, 634397. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Haque, M.S.; Islam, M.M.; Emdad, E.M.; Halim, A.; Hossen, Q.M.M.; Hossain, M.Z.; Ahmed, B.; Rahim, S.; Rahman, M.S.; et al. Tools to Kill: Genome of One of the Most Destructive Plant Pathogenic Fungi Macrophomina Phaseolina. BMC Genom. 2012, 13, 493. [Google Scholar] [CrossRef] [PubMed]
- Abou-Mansour, E.; Débieux, J.-L.; Ramírez-Suero, M.; Bénard-Gellon, M.; Magnin-Robert, M.; Spagnolo, A.; Chong, J.; Farine, S.; Bertsch, C.; L’Haridon, F.; et al. Phytotoxic Metabolites from Neofusicoccum parvum, a Pathogen of Botryosphaeria Dieback of Grapevine. Phytochemistry 2015, 115, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Zlatković, M.; Keča, N.; Wingfield, M.J.; Jami, F.; Slippers, B. Botryosphaeriaceae Associated with the Die-Back of Ornamental Trees in the Western Balkans. Antonie Van Leeuwenhoek 2016, 109, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.L.; Stauder, C.M.; Martin, D.K.H.; Kasson, M.T. Morphological and Phylogenetic Resolution of Diplodia corticola and D. quercivora, Emerging Canker Pathogens of Oak (Quercus spp.), in the United States. Plant Dis. 2021, 105, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Asman, A.; Shao, J.; Balidion, J.F.; Strem, M.D.; Puig, A.S.; Meinhardt, L.W.; Bailey, B.A. Genome and Transcriptome Analysis of the Latent Pathogen Lasiodiplodia theobromae, an Emerging Threat to the Cacao Industry. Genome 2020, 63, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ozbudak, E.; Liu, G.; Hosmani, P.S.; Saha, S.; Flores-Gonzalez, M.; Mueller, L.A.; Rodrigues-Stuart, K.; Dewdney, M.M.; Lin, Y.; et al. Draft Genome Sequence Resource of the Citrus Stem-End Rot Fungal Pathogen Lasiodiplodia theobromae CITRA15. Phytopathology® 2021, 111, 761–764. [Google Scholar] [CrossRef]
- Huang, Y.; Xiao, L.; Zhang, Z.; Zhang, R.; Wang, Z.; Huang, C.; Huang, R.; Luan, Y.; Fan, T.; Wang, J. The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition. GigaScience 2019, 8, giz036. [Google Scholar] [CrossRef]
- Zhang, C.Q.; Xu, B.C. First report of canker on pecan (Carya cathayensis) caused by Botryosphaeria dothidea in China. Plant Dis. 2011, 95, 1319. [Google Scholar] [CrossRef]
- Zhuang, C.J.; Wang, Q.W.; Wu, Q.Q.; Qiu, Z.L.; Xu, B.C.; Zhang, C.Q. Diversity of Botryosphaeriaceae species associated with Chinese hickory tree (Carya cathayensis) trunk cankers. Plant Dis. 2021, 105, 3869–3879. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liang, X.; Gleason, M.L.; Zhang, R.; Sun, G. Comparative genomics of Botryosphaeria dothidea and B. kuwatsukai, causal agents of apple ring rot, reveals both species expansion of pathogenicity-related genes and variations in virulence gene content during speciation. IMA Fungus 2018, 9, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wu, Q.; Huang, J.; Zhang, C.-Q. High-Quality Genome Assembly and Annotation Resource of Botryosphaeria dothidea Strain BDLA16-7, Causing Trunk Canker Disease on Chinese Hickory. Plant Dis. 2022, 106, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Koutsovoulos, G.D.; Granjeon Noriot, S.; Bailly-Bechet, M.; Danchin, E.G.; Rancurel, C. AvP: A software package for automatic phylogenetic detection of candidate horizontal gene transfers. PLoS Comput. Biol. 2022, 18, e1010686. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tian, L.; Zhang, D.-D.; Song, J.; Song, S.-S.; Yin, C.-M.; Zhou, L.; Liu, Y.; Wang, B.-L.; Kong, Z.-Q. Functional analyses of small secreted cysteine-rich proteins identified candidate effectors in Verticillium dahliae. Mol. Plant Pathol. 2020, 21, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Y.; Hu, S.; Huang, J.; Zhang, C. High-Quality Genome Assembly and Annotation Resource of Three Botry-osphaeria Pathogens Causing Chinese Hickory Canker. Mol. Plant-Microbe Interact. 2022, 35, 941–943. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Bae, H. The diversity of fungal genome. Biol. Proced. Online 2015, 17, 8. [Google Scholar] [CrossRef]
- Chen, J.; Liu, C.; Gui, Y.; Si, K.; Zhang, D.; Wang, J.; Short, D.P.G.; Huang, J.; Li, N.; Liang, Y.; et al. Comparative genomics reveals cotton-specific virulence factors in flexible genomic regions in Verticillium dahliae and evidence of horizontal gene transfer from Fusarium. New Phytol. 2018, 217, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; Ling, C.; Chaguza, C.; Salter, S.J.; Hinfonthong, P.; Nikolaou, E.; Tate, N.; Pastusiak, A.; Turner, C.; Chewapreecha, C. Pneumococcal within-host diversity during colonization, transmission and treatment. Nat. Microbiol. 2022, 7, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, P.; Li, C.; Han, S.; Zhao, C.; Xia, H.; Bi, Y.; Guo, B.; Zhang, X.; Wang, X. Comparative analysis of NBS-LRR genes and their response to Aspergillus flavus in Arachis. PLoS ONE 2017, 12, e0171181. [Google Scholar] [CrossRef] [PubMed]
- Shanks, S.G.; Carpp, L.N.; Struthers, M.S.; McCann, R.K.; Bryant, N.J. The Sec1/Munc18 protein Vps45 regulates cellular levels of its SNARE binding partners Tlg2 and Snc2 in Saccharomyces cerevisiae. PLoS ONE 2012, 7, e49628. [Google Scholar] [CrossRef] [PubMed]
- Diefenbacher, M.; Thorsteinsdottir, H.; Spang, A. The Dsl1 tethering complex actively participates in soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptor (SNARE) complex assembly at the endoplasmic reticulum in Saccharomyces Cerevisiae. J. Biol. Chem. 2011, 286, 25027–25038. [Google Scholar] [CrossRef]
- Wang, Y.; Pruitt, R.N.; Nürnberger, T.; Wang, Y. Evasion of plant immunity by microbial pathogens. Nat. Rev. Microbiol. 2022, 20, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Rep, M. Small proteins of plant-pathogenic fungi secreted during host colonization. FEMS Microbiol. Lett. 2005, 253, 19–27. [Google Scholar] [CrossRef]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Gladyshev, E.A.; Meselson, M.; Arkhipova, I.R. Massive horizontal gene transfer in bdelloid rotifers. Science 2008, 320, 1210–1213. [Google Scholar] [CrossRef]
- Bernardi, G. The neoselectionist theory of genome evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 8385–8390. [Google Scholar] [CrossRef]
- Kanzaki, H.; Yoshida, K.; Saitoh, H.; Fujisaki, K.; Hirabuchi, A.; Alaux, L.; Fournier, E.; Tharreau, D.; Terauchi, R. Arms race co-evolution of Magnaporthe oryzae AVR-Pik and rice Pik genes driven by their physical interactions. Plant J. 2012, 72, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Kamoun, S. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat. Rev. Microbiol. 2012, 10, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, S.-X.; He, Q.; Fan, R. Advances in the development of HIV integrase strand transfer inhibitors. Eur. J. Med. Chem. 2021, 225, 113787. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, J.; Wang, Y.; Hong, N.; Zhang, F.; Xu, W.; Wang, G. Hypovirulence of the phytopathogenic fungus Botry-osphaeria dothidea: Association with a coinfecting chrysovirus and a partitivirus. J. Virol. 2014, 88, 7517–7527. [Google Scholar] [CrossRef] [PubMed]
- Porquier, A.; Tisserant, C.; Salinas, F.; Glassl, C.; Wange, L.; Enard, W.; Hauser, A.; Hahn, M.; Weiberg, A. Retrotransposons as Pathogenicity Factors of the Plant Pathogenic Fungus Botrytis cinerea. Genome Biol. 2021, 22, 225. [Google Scholar] [CrossRef] [PubMed]
- Muszewska, A.; Hoffman-Sommer, M.; Grynberg, M. LTR Retrotransposons in Fungi. PLoS ONE 2011, 6, e29425. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, E.; Brown, S.; Tian, C.; Glass, N.L. Transcriptional profiling and functional analysis of heterokaryon incompatibility in Neurospora crassa reveals that reactive oxygen species, but not metacaspases, are associated with programmed cell death. Microbiology 2009, 155, 3957. [Google Scholar] [CrossRef] [PubMed]
- Biella, S.; Smith, M.L.; Aist, J.R.; Cortesi, P.; Milgroom, M.G. Programmed cell death correlates with virus transmission in a filamentous fungus. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2002, 269, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.A.; Stipanovic, R.D.; Crutcher, F.K.; Nichols, R.L.; Puckhaber, L.S.; Liu, J. FUBT, a Putative MFS Transporter, Promotes Secretion of Fusaric Acid in the Cotton Pathogen Fusarium oxysporum f. sp. vasinfectum. Microbiology 2015, 161, 875–883. [Google Scholar] [CrossRef]
- Vela-Corcía, D.; Aditya Srivastava, D.; Dafa-Berger, A.; Rotem, N.; Barda, O.; Levy, M. MFS Transporter from Botrytis cinerea Provides Tolerance to Glucosinolate-Breakdown Products and Is Required for Pathogenicity. Nat. Commun. 2019, 10, 2886. [Google Scholar] [CrossRef]
- Carroll, G. Fungal endophytes in stems and leaves: From latent pathogen to mutualistic symbiont. Ecology 1988, 69, 2–9. [Google Scholar] [CrossRef]
- Rana, K.L.; Kour, D.; Sheikh, I.; Yadav, N.; Yadav, A.N.; Kumar, V.; Singh, B.P.; Dhaliwal, H.S.; Saxena, A.K. Biodiversity of endophytic fungi from diverse niches and their biotechnological applications. In Advances in Endophytic Fungal Research: Present Status and Future Challenges; Springer: Berlin/Heidelberg, Germany, 2019; pp. 105–144. [Google Scholar]
- Hertweck, C. Hidden biosynthetic treasures brought to light. Nat. Chem. Biol. 2009, 5, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Schroeckh, V.; Scherlach, K.; Nützmann, H.-W.; Shelest, E.; Schmidt-Heck, W.; Schuemann, J.; Martin, K.; Hertweck, C.; Brakhage, A.A. Intimate bacterial–fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 2009, 106, 14558–14563. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.M. One-step functionalization of silver Nanoparticles using the orsellinic acid compound isolated from the endophytic fungus Epicoccum Nigrum: Characterization and antifungal activity. Int. J. Nanomater. Chem. 2015, 1, 103–110. [Google Scholar]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Pei, Y.; Shen, D.; Ji, P.; Xu, R.; Xue, X.; Peng, H.; Liang, X.; Dou, D. Phytophthora sojae leucine-rich repeat receptor-like kinas-es: Diverse and essential roles in development and pathogenicity. Iscience 2021, 24, 102725. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Z.; Yong, S.; Yu, S.; Feng, H.; Yin, M.; Ye, W.; Wang, Y.; Qiu, M. An oomycete-specific leucine-rich re-peat-containing protein is involved in zoospore flagellum development in Phytophthora sojae. Phytopathology® 2022, 112, 2351–2359. [Google Scholar] [CrossRef]
- Krijger, J.-J.; Thon, M.R.; Deising, H.B.; Wirsel, S.G. Compositions of fungal secretomes indicate a greater impact of phylogenetic history than lifestyle adaptation. BMC Genom. 2014, 15, 1–19. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Contigs | N50 (Mb) | Assembly Length | Repeat Percent (%) | Predicted Genes | GC Content (%) |
|---|---|---|---|---|---|---|
| BDLA | 15 | 3.86 | 45.98 | 8.2 | 13,052 | 52.27 |
| BQTK | 14 | 3.93 | 44.31 | 6.25 | 12,141 | 52.81 |
| BCTK | 13 | 3.96 | 45.13 | 8.46 | 12,910 | 51.9 |
| BFLG | 13 | 3.92 | 44.87 | 6.51 | 12,863 | 51.99 |
| MP | 75 | 4.83 | 50.55 | 17.92 | 14,845 | 36.4 |
| NP | 18 | 2.54 | 43.99 | 6.89 | 10,366 | 48.42 |
| DC | 181 | 0.46 | 34.99 | 5.06 | 10,839 | 43.23 |
| LT | 296 | 0.88 | 43.69 | 2.72 | 13,054 | 42.08 |
| Annotation Statistic | BDLA | BQTK | BCTK | BFLG | MP | NP | DC | LT |
|---|---|---|---|---|---|---|---|---|
| Average gene length (bp) | 1660 | 1757 | 1601 | 1649 | 1750 | 1530 | 1747 | 1677 |
| Total length of CDS (Mb) | 18.51 | 17.71 | 18.1 | 18.26 | 20.15 | 13.21 | 17.23 | 19.5 |
| % of genome covered by genes | 47.13 | 48.14 | 45.79 | 47.27 | 51.38 | 36.06 | 54.13 | 50.11 |
| Gene density (gene/kb) | 0.28 | 0.27 | 0.29 | 0.29 | 0.29 | 0.24 | 0.31 | 0.3 |
| Exons per gene | 2.53 | 2.41 | 2.51 | 2.55 | 2.97 | 3.16 | 2.99 | 3.08 |
| Introns per gene | 1.53 | 1.42 | 1.51 | 1.55 | 1.96 | 2.16 | 1.99 | 2.08 |
| Gene ID | Protein Length | Cysteine Proportion | SCP Identification | EffectorP Prediction | PHI Annotation | Pfam Domain | Function Annotation |
|---|---|---|---|---|---|---|---|
| BDLA_00001985-RA | 111 | 6.25 | Y | Apoplastic effector | - | - | - |
| BDLA_00002075-RA | 227 | 0.88 | Y | - | effector_(plant_avirulence_determinant) | Glyco_hydro_61 | Fungal cellulose-binding domain-containing protein |
| BDLA_00002749-RA | 306 | 1.63 | Y | - | - | - | - |
| BDLA_00003112-RA | 189 | 1.05 | Y | - | - | - | - |
| BDLA_00003469-RA | 290 | 0.34 | Y | - | reduced_virulence | TB2_DP1_HVA22 | Belongs to the type-B carboxylesterase lipase family |
| BDLA_00003643-RA | 393 | 0.76 | Y | - | loss_of_pathogenicity | p450 | Belongs to the cytochrome P450 family |
| BDLA_00006476-RA | 292 | 3.07 | Y | - | - | - | - |
| BDLA_00007123-RA | 140 | 4.26 | Y | - | - | - | - |
| BDLA_00007170-RA | 234 | 6.38 | Y | Apoplastic effector | - | - | - |
| BDLA_00007782-RA | 102 | 0.97 | Y | Cytoplasmic effector | - | - | - |
| BDLA_00008471-RA | 318 | 0.94 | Y | - | - | - | - |
| BDLA_00008838-RA | 128 | 3.88 | Y | - | - | - | - |
| BDLA_00009176-RA | 284 | 3.51 | Y | - | - | - | - |
| BDLA_00009481-RA | 136 | 1.46 | Y | - | - | SnoaL_2 | Snoal-like polyketide cyclase family protein |
| BDLA_00009719-RA | 345 | 1.45 | Y | - | reduced_virulence | Glyco_hydro_10 | glycoside hydrolase family 10 protein |
| BDLA_00009878-RA | 124 | 1.6 | Y | - | reduced_virulence | Glycos_transf_1 | Starch synthase catalytic domain(ags1) |
| BDLA_00010018-RA | 90 | 8.79 | Y | Apoplastic effector | - | - | - |
| BDLA_00011085-RA | 73 | 8.11 | Y | Apoplastic effector | - | - | - |
| BDLA_00011913-RA | 209 | 0.48 | Y | - | reduced_virulence | Lipase_GDSL_2 | GDSL-like Lipase/Acylhydrolase family |
| BDLA_00012014-RA | 152 | 8.5 | Y | - | - | - | - |
| BDLA_00012024-RA | 287 | 0.69 | Y | - | - | Peptidase_S15 | X-Pro dipeptidyl-peptidase |
| BDLA_00012709-RA | 383 | 2.08 | Y | - | reduced_virulence | Glyco_hydro_16 | glycoside hydrolase family 16 protein |
| BDLA_00012987-RA | 233 | 2.56 | Y | Apoplastic effector | - | - | - |
| BDLA_00012997-RA | 304 | 2.62 | Y | - | - | - | - |
| BDLA_00013002-RA | 270 | 1.48 | Y | - | - | DUF3455 | Protein of unknown function (DUF3455) |
| BDLA_00013015-RA | 274 | 1.09 | Y | - | increased_virulence_(hypervirulence)_reduced_virulence | Glyco_hydro_18 | Belongs to the glycosyl hydrolase 18 family(CHT2) |
| Gene ID | HGT Event ID | Pfam Domain | Putative Function | Best Hit on PHI-Base (E-Value) | Most Likely Donor |
|---|---|---|---|---|---|
| BDLA_00000792-RA | HGT_1 | Cupin_3 | Protein of unknown function (DUF861) | - | Bacteria |
| BDLA_00005852-RA | HGT_2 | - | - | Q9HV44; heat shock protein; reduced virulence | Bacteria |
| BDLA_00004793-RA | HGT_3 | - | - | A0A098DK85; protease; unaffected pathogenicity | Eukaryota |
| BDLA_00003727-RA | HGT_4 | HRI1 | Protein HRI1 | - | Fungi |
| BDLA_00007369-RA | HGT_5 | Methyltransf_11 | ubiE/COQ5 methyltransferase family | - | Fungi |
| BDLA_00007786-RA | HGT_6 | Ctr | ctr copper transporter family protein(ctr4) | B0XUP5; copper transporters; unaffected pathogenicity | Fungi |
| BDLA_00010862-RA | HGT_7 | Peptidase_M43 | Pregnancy-associated plasma protein-A | C5P3X6; Metalloproteinase; reduced virulence | Fungi |
| BDLA_00001191-RA | HGT_8 | Ras | GTPase activity | A0A139Y2L7; small GTPase; reduced virulence | Fungi |
| BDLA_00003074-RA | HGT_9 | Acetyltransf_7 | Acetyltransferase (GNAT) domain | - | Fungi |
| BDLA_00010710-RA | HGT_10 | EHN | Epoxide hydrolase N terminus | - | Fungi |
| BDLA_00010289-RA | HGT_11 | Zn_clus | Oxidoreductase NAD-binding domain | Q4W9X3; flavohemoglobins; unaffected pathogenicity | Fungi |
| BDLA_00005439-RA | HGT_12 | Glyco_hydro_7 | Belongs to the glycosyl hydrolase 7 (cellulase C) family | G4ZRT3; conserved glycoside hydrolase family 7 cellobiohydrolase; reduced virulence | Fungi |
| BDLA_00011030-RA | HGT_13 | Abhydrolase_1 | Serine aminopeptidase, S33 | Q8PC98; effector protein; effector (plant avirulence determinant) | Fungi |
| BDLA_00007899-RA | HGT_14 | DAO | FAD dependent oxidoreductase | - | Fungi |
| BDLA_00010741-RA | HGT_15 | Jacalin | Jacalin-like lectin domain | P9WKQ1; extracellular nuclease; reduced virulence | Fungi |
| BDLA_00010678-RA | HGT_16 | DUF1349 | Protein of unknown function (DUF1349) | - | Fungi |
| BDLA_00007280-RA | HGT_17 | MFS_MOT1 | Molybdate transporter of MFS superfamily | - | Fungi |
| BDLA_00002083-RA | HGT_18 | Lipase_GDSL_2 | GDSL-like Lipase/Acylhydrolase family | - | Fungi |
| BDLA_00002724-RA | HGT_19 | Peptidase_S58 | Peptidase family S58 | - | Fungi |
| BDLA_00007942-RA | HGT_20 | - | - | - | HGT_complex |
| BDLA_00003219-RA | HGT_21 | RVT_2 | Mitochondrial protein | - | NM_Eukaryota_complex |
| BDLA_00005227-RA | HGT_22 | - | - | - | NM_Eukaryota_complex |
| BDLA_00012074-RA | HGT_23 | PALP | Pyridoxal-phosphate-dependent enzyme | G2XJR0;1-Aminocyclopropane-1-carboxylate deaminase; increased virulence (hypervirulence) | Oomycota |
| BDLA_00008930-RA | HGT_24 | NAD_binding_8 | FAD-binding domain | Q5GFD3; Mannitol 1-phosphate dehydrogenase; unaffected pathogenicity | Oomycota |
| BDLA_00008949-RA | HGT_25 | Ank_4 | spectrin binding | J9VLD1; Cyclin-dependent protein kinase inhibitor; reduced virulence | Bacteria |
| BDLA_00004977-RA | HGT_26 | - | Podospora anserina S mat genomic DNA chromosome | - | Bacteria |
| BDLA_00012548-RA | HGT_27 | PYNP_C | thymidine phosphorylase activity (TYMP) | - | Bacteria |
| BDLA_00003435-RA | HGT_28 | Lactamase_B | Alkyl sulfatase dimerization (MA20_17395) | U3M7S2; heat-sensitive alkyl sulphatase; unaffected pathogenicity | Bacteria |
| BDLA_00003351-RA | HGT_29 | - | - | Q8J286; copper transporting ATPase; loss of pathogenicity | Bacteria |
| BDLA_00006818-RA | HGT_30 | - | - | J9VLD1; cyclin-dependent protein kinase inhibitor; reduced virulence | Eukaryota |
| BDLA_00008015-RA | HGT_31 | UDPGT | Belongs to the OSBP family | - | Fungi |
| BDLA_00007233-RA | HGT_32 | - | - | - | Fungi |
| BDLA_00010519-RA | HGT_33 | - | - | - | Fungi |
| BDLA_00002024-RA | HGT_34 | - | - | - | Fungi |
| BDLA_00008349-RA | HGT_35 | - | - | - | Fungi |
| BDLA_00001002-RA | HGT_36 | - | - | - | Fungi |
| BDLA_00009177-RA | HGT_37 | - | - | - | Fungi |
| BDLA_00010857-RA | HGT_38 | - | - | - | Fungi |
| BDLA_00004645-RA | HGT_39 | - | - | - | Oomycota |
| BDLA_00012380-RA | HGT_40 | - | - | - | Oomycota |
| BDLA_00012910-RA | HGT_41 | - | - | - | Viridiplantae |
| BDLA_00002811-RA | HGT_42 | - | - | - | Viridiplantae |
| BDLA_00001009-RA | HGT_43 | - | - | - | Viridiplantae |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, D.; Jiang, Y.; Zhang, Y.; Mao, C.; Ma, T.; Zhang, C. The Comparative Genomics of Botryosphaeriaceae Suggests Gene Families of Botryosphaeria dothidea Related to Pathogenicity on Chinese Hickory Tree. J. Fungi 2024, 10, 299. https://doi.org/10.3390/jof10040299
Liang D, Jiang Y, Zhang Y, Mao C, Ma T, Zhang C. The Comparative Genomics of Botryosphaeriaceae Suggests Gene Families of Botryosphaeria dothidea Related to Pathogenicity on Chinese Hickory Tree. Journal of Fungi. 2024; 10(4):299. https://doi.org/10.3390/jof10040299
Chicago/Turabian StyleLiang, Dong, Yiru Jiang, Yu Zhang, Chengxing Mao, Tianlin Ma, and Chuanqing Zhang. 2024. "The Comparative Genomics of Botryosphaeriaceae Suggests Gene Families of Botryosphaeria dothidea Related to Pathogenicity on Chinese Hickory Tree" Journal of Fungi 10, no. 4: 299. https://doi.org/10.3390/jof10040299