

Topical Meloxicam Hydroxypropyl Guar Hydrogels Based on Low-Substituted Hydroxypropyl Cellulose Solid Dispersions
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Solubility Studies at pH 5.8
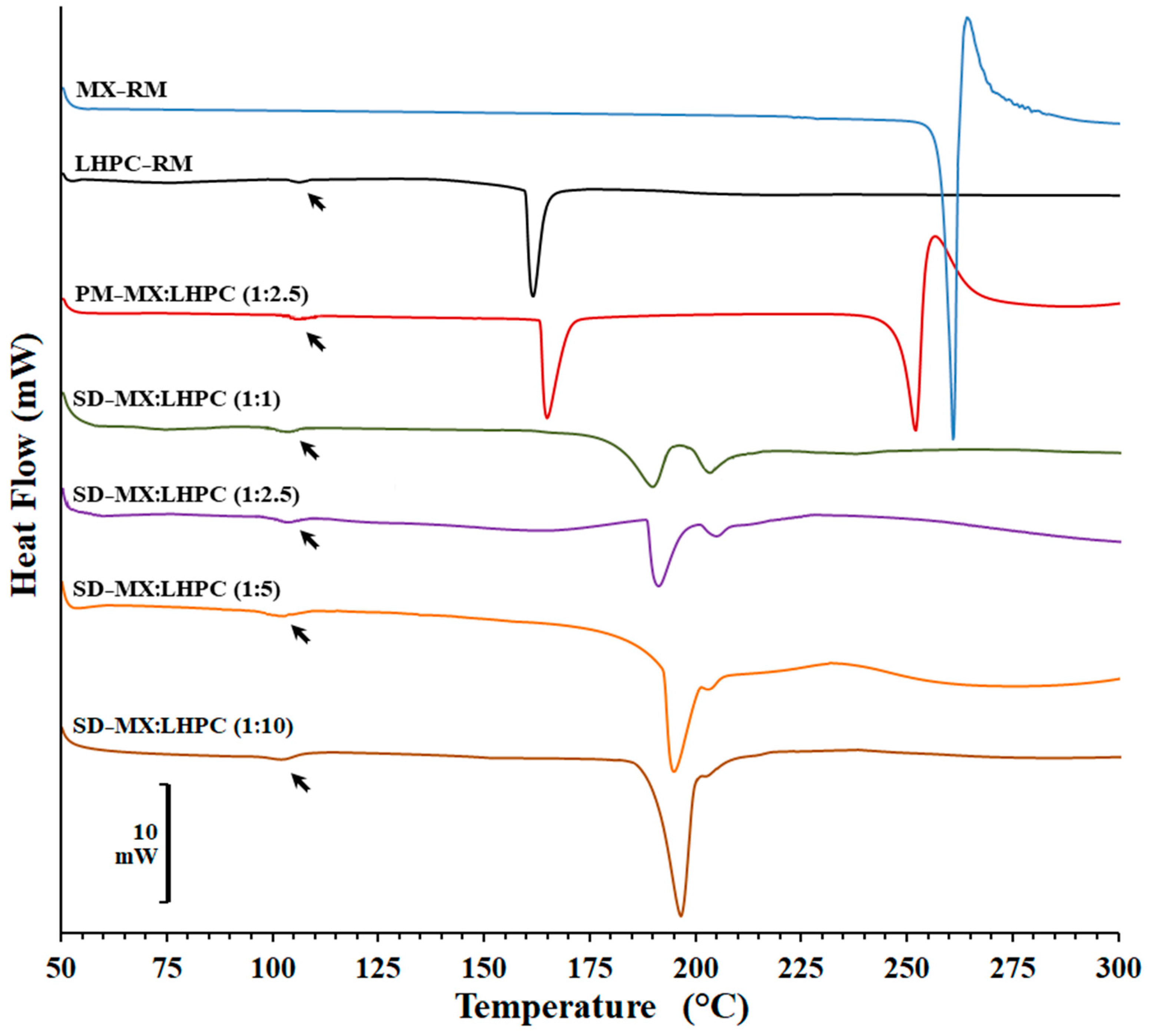
2.2. DSC Studies
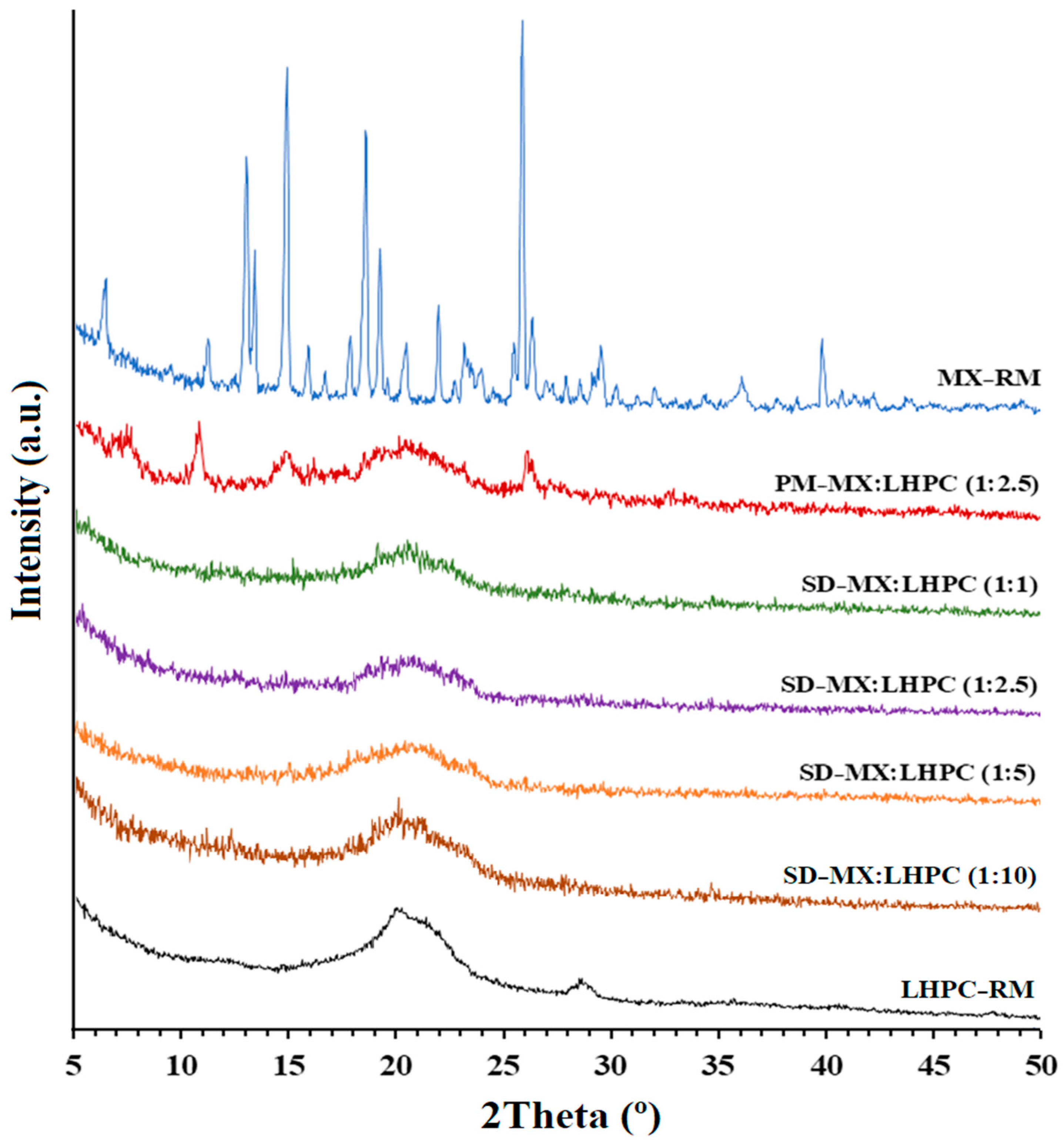
2.3. X-ray Powder Diffractometry (XRPD)
2.4. FTIR Spectroscopy Study
2.5. Characterization of HPG Hydrogels
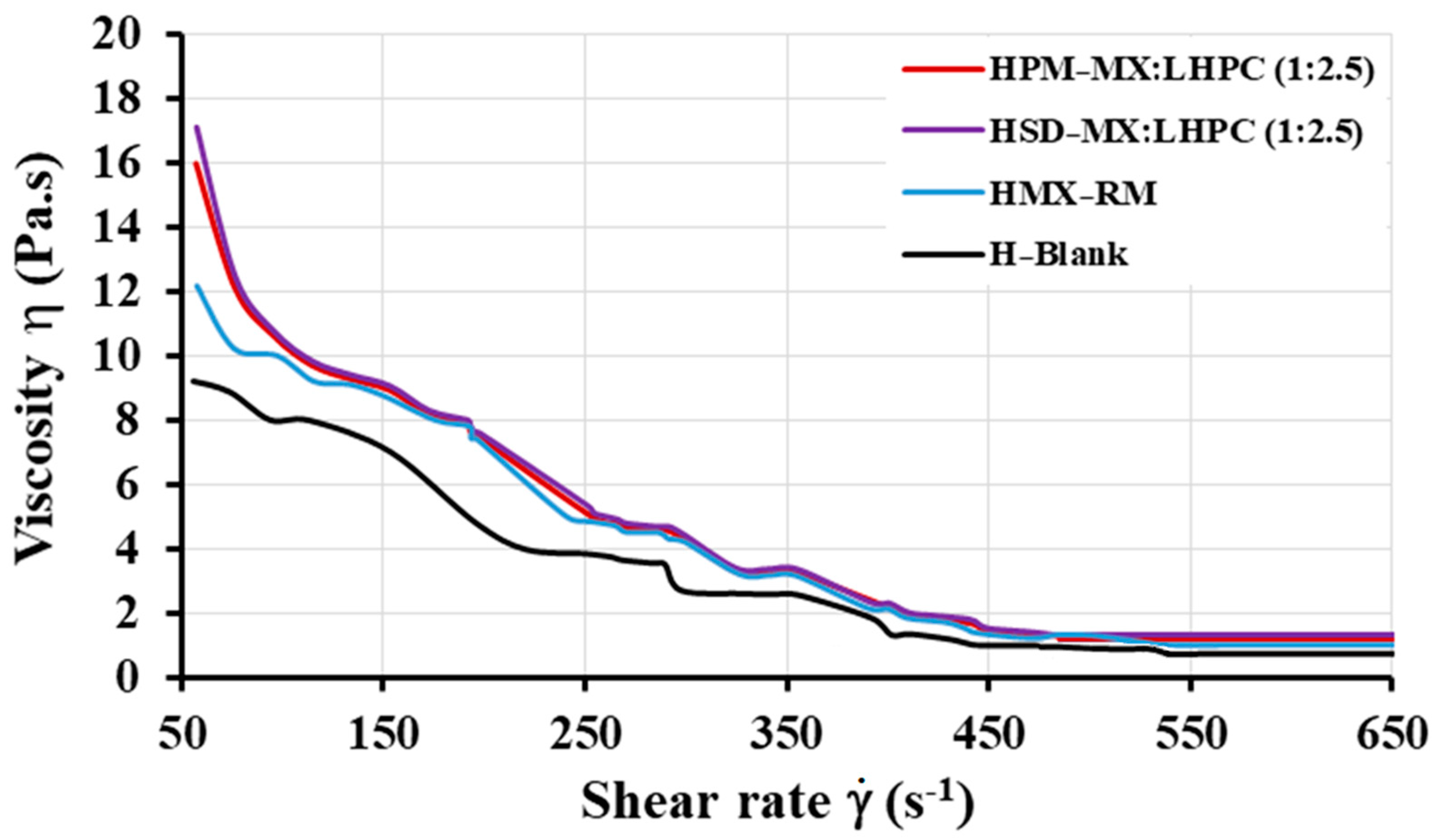
2.5.1. Viscosity Studies
2.5.2. Scanning Electron Microscopy (SEM)
2.5.3. Fourier Transform Infrared Spectroscopy for the Different Hydrogels
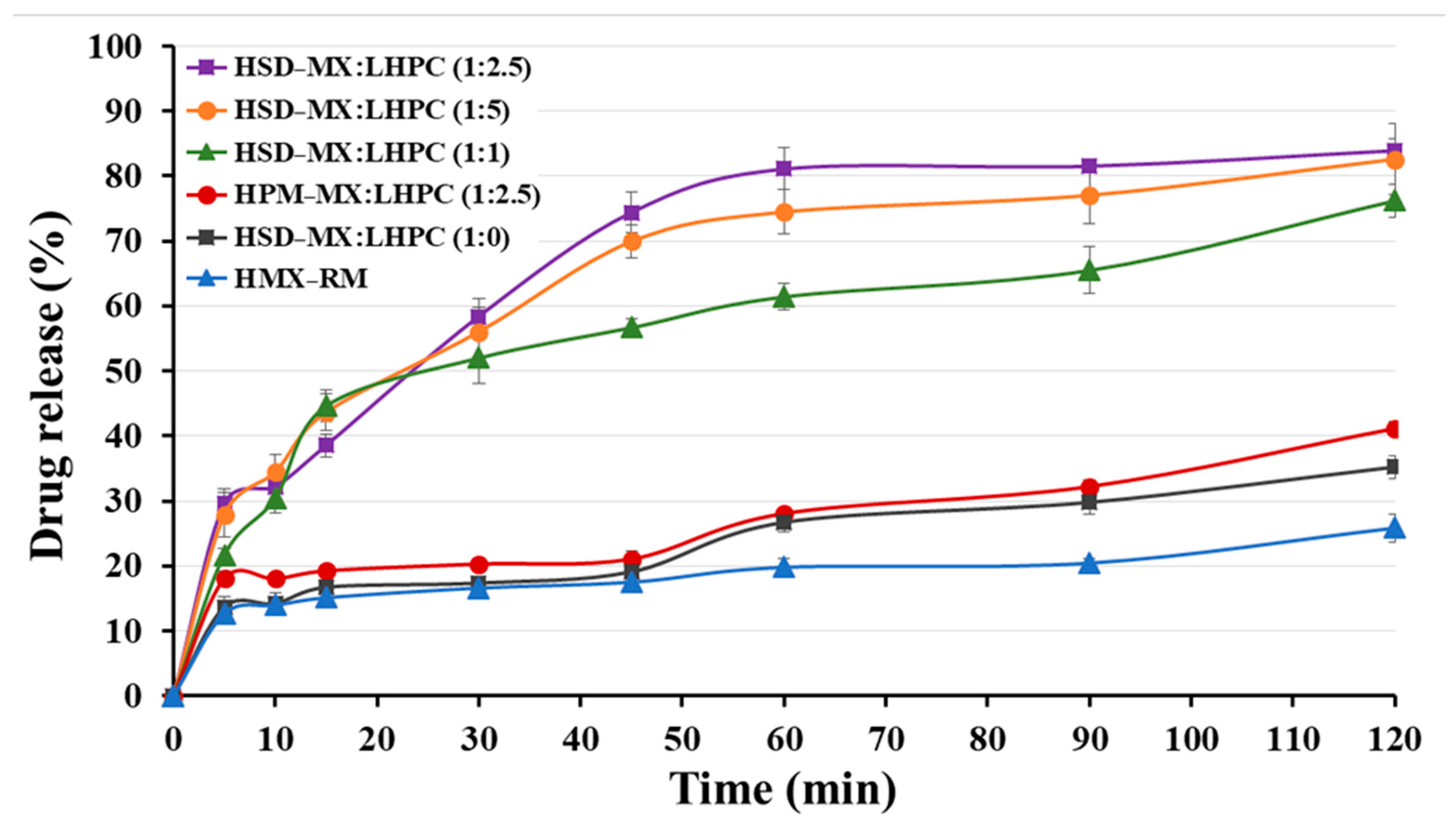
2.5.4. In Vitro Release Profile Study
2.5.5. Kinetic Studies
3. Conclusions
4. Materials and Methods
4.1. Substances and Reagents
4.2. Preparation of Solid Dispersions (SDs)
4.3. Preparation of Hydrogel Formulations
4.4. Solubility Study
4.5. Differential Scanning Calorimetry (DSC)
4.6. X-ray Powder Diffractometry (XRPD)
4.7. Fourier-Transform Infrared Spectroscopy (FTIR)
4.8. Viscosity Study
4.9. Scanning Electron Microscopy (SEM)
4.10. In Vitro Drug Release
4.11. Drug Release Kinetics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Amico, C.; Fontana, F.; El-Sayed, N.; Elbadri, K.; Correia, A.; Rahikkala, A.; Saarinen, J.; Shahbazi, M.A.; Santos, H.A. Double-Layered Polyvinylpyrrolidone–Poly (Methyl Vinyl Ether-Alt-Maleic Acid) based Microneedles to Deliver Meloxicam: An In Vitro, In Vivo and Short-Term Stability Evaluation Study. Adv. Therap. 2023, 6, 2300138. [Google Scholar] [CrossRef]
- Aljubailah, A.; Alqahtani, S.M.; Al-Garni, T.S.; Saeed, W.S.; Semlali, A.; Aouak, T. Naproxen-loaded Poly (2-hydroxyalkyl methacrylates): Preparation and drug release dynamics. Polymers 2022, 14, 450. [Google Scholar] [CrossRef]
- Albarahmieh, E.; Qi, S.; Craig, D.Q. Hot melt extruded transdermal films based on amorphous solid dispersions in Eudragit RS PO: The inclusion of hydrophilic additives to develop moisture-activated release systems. Int. J. Pharm. 2016, 514, 270–281. [Google Scholar] [CrossRef]
- Zhang, J.; Froelich, A.; Michniak-Kohn, B. Topical delivery of meloxicam using liposome and microemulsion formulation approaches. Pharmaceutics 2020, 12, 282. [Google Scholar] [CrossRef]
- Pironi, A.M.; Melero, A.; Eloy, J.O.; Guillot, A.J.; Santos, K.P.; Chorilli, M. Solid dipersions included in poloxamer hydrogels have favorable rheological properties for topical application and enhance the in vivo antiinflammatory effect of ursolic acid. J. Drug Deliv. Sci. Technol. 2022, 74, 103602. [Google Scholar] [CrossRef]
- Shehata, T.M.; Elnahas, H.M.; Elsewedy, H.S. Development, characterization and optimization of the anti-inflammatory influence of meloxicam loaded into a eucalyptus oil-based nanoemulgel. Gels 2022, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Marinko, N.; Zámostný, P. Meloxicam carrier systems having enhanced release and aqueous wettability prepared using micro-suspensions in different liquid media. AAPS Pharmscitech 2020, 21, 155. [Google Scholar] [CrossRef]
- Nair, A.R.; Lakshman, Y.D.; Anand, V.S.; Sree, K.N.; Bhat, K.; Dengale, S.J. Overview of extensively employed polymeric carriers in solid dispersion technology. AAPS PharmSciTech 2020, 21, 309. [Google Scholar] [CrossRef] [PubMed]
- Bakhrushina, E.O.; Dubova, A.I.; Nikonenko, M.S.; Grikh, V.V.; Shumkova, M.M.; Korochkina, T.V.; Krasnyuk-Jr, I.I.; Krasnyuk, I.I. Thermosensitive Intravitreal In Situ Implant of Cefuroxime Based on Poloxamer 407 and Hyaluronic Acid. Gels 2023, 9, 693. [Google Scholar] [CrossRef]
- Jadach, B.; Misek, M.; Ferlak, J. Comparison of Hydroxypropyl Methylcellulose and Alginate Gel Films with Meloxicam as Fast Orodispersible Drug Delivery. Gels 2023, 9, 687. [Google Scholar] [CrossRef]
- Elim, D.; Fitri, A.M.; Mahfud, M.A.; Afika, N.; Sultan, N.A.; Asri, R.M.; Permana, A.D. Hydrogel forming microneedle-mediated transdermal delivery of sildenafil citrate from polyethylene glycol reservoir: An ex vivo proof of concept study. Colloids Surf. B. Biointerfaces 2023, 222, 113018. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Giri, B.R.; Song, E.S.; Bae, J.; Lee, J.; Kim, D.W. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef]
- Torrado-Salmerón, C.; Guarnizo-Herrero, V.; Henriques, J.; Seiça, R.; Sena, C.M.; Torrado-Santiago, S. Multiparticulate Systems of Ezetimibe Micellar System and Atorvastatin Solid Dispersion Efficacy of Low-Dose Ezetimibe/Atorvastatin on High-Fat Diet-Induced Hyperlipidemia and Hepatic Steatosis in Diabetic Rats. Pharmaceutics 2021, 13, 421. [Google Scholar] [CrossRef]
- Mai, N.N.; Otsuka, Y.; Kawano, Y.; Hanawa, T. Preparation and characterization of solid dispersions composed of curcumin, hydroxypropyl cellulose and/or sodium dodecyl sulfate by grinding with vibrational ball milling. Pharmaceuticals 2020, 13, 383. [Google Scholar] [CrossRef]
- Alencar, L.J.; Medeiros, G.C.; Sobrinho, J.L.; Lee, P.I.; Soares, M.F. Amorphous solid dispersions in high-swelling, low-substituted hydroxypropyl cellulose for enhancing the delivery of poorly soluble drugs. Int. J. Pharm. 2023, 642, 123122. [Google Scholar] [CrossRef]
- Popescu, I.; Constantin, M.; Solcan, G.; Ichim, D.L.; Rata, D.M.; Horodincu, L.; Solcan, C. Composite Hydrogels with Embedded Silver Nanoparticles and Ibuprofen as Wound Dressing. Gels 2023, 9, 654. [Google Scholar] [CrossRef]
- Fatima, F.; Aleemuddin, M.; Ahmed, M.M.; Anwer, M.K.; Aldawsari, M.F.; Soliman, G.A.; Mahdi, W.A.; Jafar, M.; Hamad, A.M.; Alshehri, S. Design and Evaluation of Solid Lipid Nanoparticles loaded topical gels: Repurpose of fluoxetine in diabetic wound healing. Gels 2023, 9, 21. [Google Scholar] [CrossRef]
- Guarnizo-Herrero, V.; Torrado-Salmerón, C.; Torres-Pabón, N.S.; Torrado, G.; Morales, J.; Torrado-Santiago, S. Study of different chitosan/sodium carboxymethyl cellulose proportions in the development of polyelectrolyte complexes for the sustained release of clarithromycin from matrix tablets. Polymers 2021, 13, 2813. [Google Scholar] [CrossRef] [PubMed]
- Parmar, P.K.; Wadhawan, J.; Bansal, A.K. Pharmaceutical nanocrystals: A promising approach for improved topical drug delivery. Drug Discov. Today 2021, 26, 2329–2349. [Google Scholar] [CrossRef]
- Wlodarski, K.; Sawicki, W.; Paluch, K.J.; Tajber, L.; Grembecka, M.; Hawelek, L.; Wojnarowska, Z.; Grzybowska, K.; Talik, E.; Paluch, M. The influence of amorphization methods on the apparent solubility and dissolution rate of tadalafil. Eur. J. Pharm. Sci. 2014, 62, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wei, C.; Qian, F.; Li, S. Surface wettability modulated by surfactant and its effects on the drug release and absorption of fenofibrate solid dispersions. AAPS PharmSciTech 2019, 20, 234. [Google Scholar] [CrossRef]
- Kim, D.S.; Choi, H.G.; Jin, S.G. Influence of Hydroxypropylmethylcellulose and Sodium Lauryl Sulfate on the Solubility and Dissolution of Sirolimus in Solvent-evaporated Solid Dispersions. Bull. Korean Chem. Soc. 2018, 39, 778–783. [Google Scholar] [CrossRef]
- Fathanah, A.; Setyawan, D.; Sari, R. Improving solubility and dissolution of meloxicam by solid dispersion using hydroxypropyl methylcellulose 2910 3 cps and nicotinamide. J. Basic Clin. Physiol. Pharmacol. 2019, 30, 20190249. [Google Scholar] [CrossRef]
- Tekade, A.R.; Yadav, J.N. A review on solid dispersion and carriers used therein for solubility enhancement of poorly water soluble drugs. Adv. Pharm. Bull. 2020, 10, 359–369. [Google Scholar] [CrossRef]
- Cheney, M.L.; Weyna, D.R.; Shan, N.; Hanna, M.; Wojtas, L.; Zaworotko, M.J. Coformer selection in pharmaceutical cocrystal development: A case study of a meloxicam aspirin cocrystal that exhibits enhanced solubility and pharmacokinetics. J. Pharm. Sci. 2011, 100, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Emam, M.F.; El-Ashmawy, A.A.; Mursi, N.M.; Emara, L.H. Optimization of Meloxicam Solid Dispersion Formulations for Dissolution Enhancement and Storage Stability Using 33 Full Factorial Design Based on Response Surface Methodology. AAPS PharmSciTech 2022, 23, 248. [Google Scholar] [CrossRef] [PubMed]
- Torre-Iglesias, P.M.; García-Rodriguez, J.J.; Torrado, G.; Torrado, S.; Torrado-Santiago, S.; Bolás-Fernández, F. Enhanced bioavailability and anthelmintic efficacy of mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose. Drug Des. Devel. Ther. 2014, 8, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Tascon-Otero, E.; Torre-Iglesias, P.; Garcia-Rodriguez, J.J.; Peña, M.; Alvarez-Alvarez, C. Enhancement of the Dissolution Rate of Indomethacin by Solid Dispersions in Low-substituted Hydroxypropyl Cellulose. Ind. J. Pharm. Sci. 2019, 81, 824–833. [Google Scholar] [CrossRef]
- López-Manzanara, C.; Torres-Pabón, N.S.; Laguna, A.; Torrado, G.; Torre-Iglesias, P.M.; Torrado-Santiago, S.; Torrado-Salmerón, C. Development of Chitosan/Sodium Carboxymethylcellulose Complexes to Improve the Simvastatin Release Rate: Polymer/Polymer and Drug/Polymer Interactions’ Effects on Kinetic Models. Polymers 2023, 15, 4184. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Ruíz, E.; Álvarez-Álvarez, C.; Peña, M.Á.; Torrado-Salmerón, C.; Dahma, Z.; Torre-Iglesias, P.M. Multiparticulate systems of meloxicam for colonic administration in cancer or autoimmune diseases. Pharmaceutics 2022, 14, 1504. [Google Scholar] [CrossRef] [PubMed]
- Freitas, J.T.; Viana, O.M.; Bonfilio, R.; Doriguetto, A.C.; Araújo, M.B. Analysis of polymorphic contamination in meloxicam raw materials and its effects on the physicochemical quality of drug product. Eur. J. Pharm. Sci. 2017, 109, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Sarode, A.L.; Malekar, S.A.; Cote, C.; Worthen, D.R. Hydroxypropyl cellulose stabilizes amorphous solid dispersions of the poorly water soluble drug felodipine. Carbohydr. Polym. 2014, 112, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, H.K.; Rohera, B.D. Application of thermodynamic phase diagrams and Gibbs free energy of mixing for screening of polymers for their use in amorphous solid dispersion formulation of a non-glass-forming drug. J. Pharm. Sci. 2021, 110, 2703–2717. [Google Scholar] [CrossRef] [PubMed]
- Yegireddy, M.; Nadoor, P.; Rao, S.; Hanumanthu, P.B.; Rajashekaraiah, R.; Ramachandrappa, S.C.; Halemani, G.M.; Mannem, S.; Prasad, T.N.; Ubaradka, S. Chitosan encapsulated meloxicam nanoparticles for sustained drug delivery applications: Preparation, characterization, and pharmacokinetics in Wistar rats. Molecules 2022, 27, 7312. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, H.; Liu, R.; Long, L.; Xu, J.; Chen, M.; Qiu, H. Preparation of a fast water-based UV cured polyurethane-acrylate wood coating and the effect of coating amount on the surface properties of oak (Quercus alba L.). Polymers 2019, 11, 1414. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Meng, Z.; Li, Y.; Liu, J.; Xu, Y.; Wang, Y.; Li, X. Preparation and evaluation of mosapride citrate dual-release dry suspension. AAPS PharmSciTech 2019, 20, 155. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, N.; Rincón, M.; Silva-Abreu, M.; Sosa, L.; Pesantez-Narvaez, J.; Calpena, A.C.; Rodríguez-Lagunas, M.J.; Mallandrich, M. Semi-Solid Dosage Forms Containing Pranoprofen-Loaded NLC as Topical Therapy for Local Inflammation: In Vitro, Ex Vivo and In Vivo Evaluation. Gels 2023, 9, 448. [Google Scholar] [CrossRef]
- Malkin, A.; Kulichikhin, V.; Ilyin, S. A modern look on yield stress fluids. Rheol. Acta 2017, 56, 177–188. [Google Scholar] [CrossRef]
- Laureanti, E.J.; Paiva, T.S.; Matos, L.M.; Jorge, R.M. Microencapsulation of bioactive compound extracts using maltodextrin and gum arabic by spray and freeze-drying techniques. Int. J. Biol. Macromol. 2023, 253, 126969. [Google Scholar] [CrossRef]
- Oliveira, M.X.; Canafístula, F.V.; Ferreira, C.R.; Fernandes, L.V.; Araújo, A.R.; Ribeiro, F.O.; Souza, J.M.; Lima, I.C.; Assreuy, A.M.; Silva, D.A.; et al. Hydrogels dressings based on guar gum and chitosan: Inherent action against resistant bacteria and fast wound closure. Int. J. Biol. Macromol. 2023, 253, 127281. [Google Scholar] [CrossRef]
- Nowak, K.M.; Bodek, K.H.; Szterk, A.; Rudnicka, K.; Szymborski, T.; Kosieradzki, M.; Fiedor, P. Preclinical assessment of the potential of a 3D chitosan drug delivery system with sodium meloxicam for treating complications following tooth extraction. Int. J. Biol. Macromol. 2019, 133, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.N.; Soliman, G.A.; Rehman, N.U.; Anwer, M.K. Crisaborole Loaded Nanoemulsion Based Chitosan Gel: Formulation, Physicochemical Characterization and Wound Healing Studies. Gels 2022, 8, 318. [Google Scholar] [CrossRef] [PubMed]
- Ponnammal, P.; Kanaujia, P.; Yani, Y.; Ng, W.K.; Tan, R.B. Orally disintegrating tablets containing melt extruded amorphous solid dispersion of tacrolimus for dissolution enhancement. Pharmaceutics 2018, 10, 35. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Kinetic Models | K | r2 |
|---|---|---|---|
| H − MX − RM | Zero-order | 0.1073 | 0.9952 |
| First-order | −0.0013 | 0.9981 | |
| Higuchi model | 0.0099 | 0.9935 | |
| H − PM − MX:LHPC (1:2.5) | Zero-order | 0.2048 | 0.9975 |
| First-order | −0.0029 | 0.9934 | |
| Higuchi model | 2.0806 | 0.9820 | |
| H − SD − MX:LHPC (1:0) | Zero-order | 0.1919 | 0.9983 |
| First-order | −0.0025 | 0.9973 | |
| Higuchi model | 2.5637 | 0.9950 | |
| H − SD − MX:LHPC (1:1) | Zero-order | 0.2891 | 0.9814 |
| First-order | −0.0080 | 0.9992 | |
| Higuchi model | 6.0958 | 0.9873 | |
| H − SD − MX:LHPC (1:2.5) | Zero-order | 1.1700 | 0.9945 |
| First-order | −0.0160 | 0.9852 | |
| Higuchi model | 6.7841 | 0.9877 | |
| H − SD − MX:LHPC (1:5) | Zero-order | 0.486 | 0.9788 |
| First-order | −0.00125 | 0.9964 | |
| Higuchi model | 6.4260 | 0.9968 |
| Formulations | Korsmeyer–Peppas Kinetic Model | ||
|---|---|---|---|
| n | Kd (min−n) | r2 | |
| HMX − RM | 0.9862 | 0.1611 | 0.9964 |
| HPM − MX:LHPC (1:2.5) | 0.9798 | 0.2669 | 0.9947 |
| HSD − MX:LHPC (1:0) | 0.2964 | 0.2964 | 0.9816 |
| HSD − MX:LHPC (1:1) | 0.4462 | 0.4462 | 0.9375 |
| HSD − MX:LHPC (1:2.5) | 0.3820 | 0.3820 | 0.8982 |
| HSD − MX:LHPC (1:5) | 0.3993 | 0.3993 | 0.9880 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahma, Z.; Torrado-Salmerón, C.; Álvarez-Álvarez, C.; Guarnizo-Herrero, V.; Martínez-Alonso, B.; Torrado, G.; Torrado-Santiago, S.; de la Torre-Iglesias, P.M. Topical Meloxicam Hydroxypropyl Guar Hydrogels Based on Low-Substituted Hydroxypropyl Cellulose Solid Dispersions. Gels 2024, 10, 207. https://doi.org/10.3390/gels10030207
Dahma Z, Torrado-Salmerón C, Álvarez-Álvarez C, Guarnizo-Herrero V, Martínez-Alonso B, Torrado G, Torrado-Santiago S, de la Torre-Iglesias PM. Topical Meloxicam Hydroxypropyl Guar Hydrogels Based on Low-Substituted Hydroxypropyl Cellulose Solid Dispersions. Gels. 2024; 10(3):207. https://doi.org/10.3390/gels10030207
Chicago/Turabian StyleDahma, Zaid, Carlos Torrado-Salmerón, Covadonga Álvarez-Álvarez, Víctor Guarnizo-Herrero, Borja Martínez-Alonso, Guillermo Torrado, Santiago Torrado-Santiago, and Paloma Marina de la Torre-Iglesias. 2024. "Topical Meloxicam Hydroxypropyl Guar Hydrogels Based on Low-Substituted Hydroxypropyl Cellulose Solid Dispersions" Gels 10, no. 3: 207. https://doi.org/10.3390/gels10030207