Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications

1
Department of Chemical and Biological Engineering, Illinois Institute of Technology, 10 West 33rd Street, Chicago, IL 60616, USA
2
Departamento de Ingeniería Química, Universidad de Guadalajara, Blvd. Gral. Marcelino García Barragán # 1451, Guadalajara, Jalisco C.P. 44430, Mexico
*
Author to whom correspondence should be addressed.
Gels 2018, 4(3), 61; https://doi.org/10.3390/gels4030061
Submission received: 10 May 2018
/
Revised: 26 June 2018
/
Accepted: 27 June 2018
/
Published: 11 July 2018
(This article belongs to the Special Issue Hydrogels for Drug Delivery)
Abstract
:Hydrogels are materials specially suited for encapsulation of biological elements. Their large water content provides an environment compatible with most biological molecules. Their crosslinked nature also provides an ideal material for the protection of encapsulated biological elements against degradation and/or immune recognition. This makes them attractive not only for controlled drug delivery of proteins, but they can also be used to encapsulate cells that can have therapeutic applications. Thus, hydrogels can be used to create systems that will deliver required therapies in a controlled manner by either encapsulation of proteins or even cells that produce molecules that will be released from these systems. Here, an overview of hydrogel encapsulation strategies of biological elements ranging from molecules to cells is discussed, with special emphasis on therapeutic applications.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Polymeric hydrogels are materials consisting of crosslinked polymer chains with high affinity for water. Their crosslinked nature confers them with adequate mechanical properties for a variety of technological applications [1,2]. Hydrogels can be engineered to retain very large water contents while retaining their shape. In fact, it is not unusual to find hydrogels containing more than 99% water [2]. Their large water content was proposed, very early, to result in small interfacial tensions in aqueous environments that would make them highly biocompatible in a variety of biomedical applications [3]. In particular, hydrogels are especially useful for the encapsulation of biological agents because they provide the natural aqueous environment required for biomolecules to function in biological systems. Hydrogels are ideal not only for the encapsulation of proteins and other biomolecules, but also for the encapsulation of living cells. Inherent advantages of hydrogels for these applications are (1) the aqueous environment helps maintain the biological function of the encapsulated material; (2) the crosslinked nature of hydrogels provide a diffusion barrier capable of allowing the passage of molecules of a given size threshold while excluding larger molecules from interacting with the encapsulated elements; (3) the size threshold can be tailored according to the degree of crosslinking of the hydrogel; (4) hydrogels can be created in situ under mild reaction conditions (e.g., temperature and pH), which make them convenient for minimally invasive surgery; (5) hydrogel properties can be selected according to specific applications (e.g., stimuli responsive, degradable, highly or lightly crosslinked, etcetera); (6) hydrogels can be functionalized with biological molecules in order to provide the required biochemical cues for the encapsulated cells (e.g., extracellular matrix proteins or peptides or degradation sequences); and (7) they can provide adequate 3-D microstructure that can modulate the mechano-biochemical transduction signals of encapsulated cells.
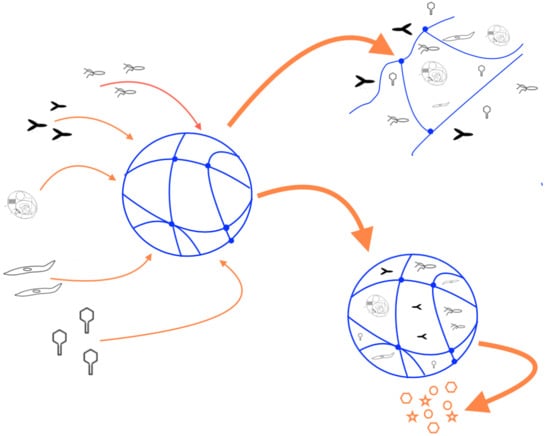
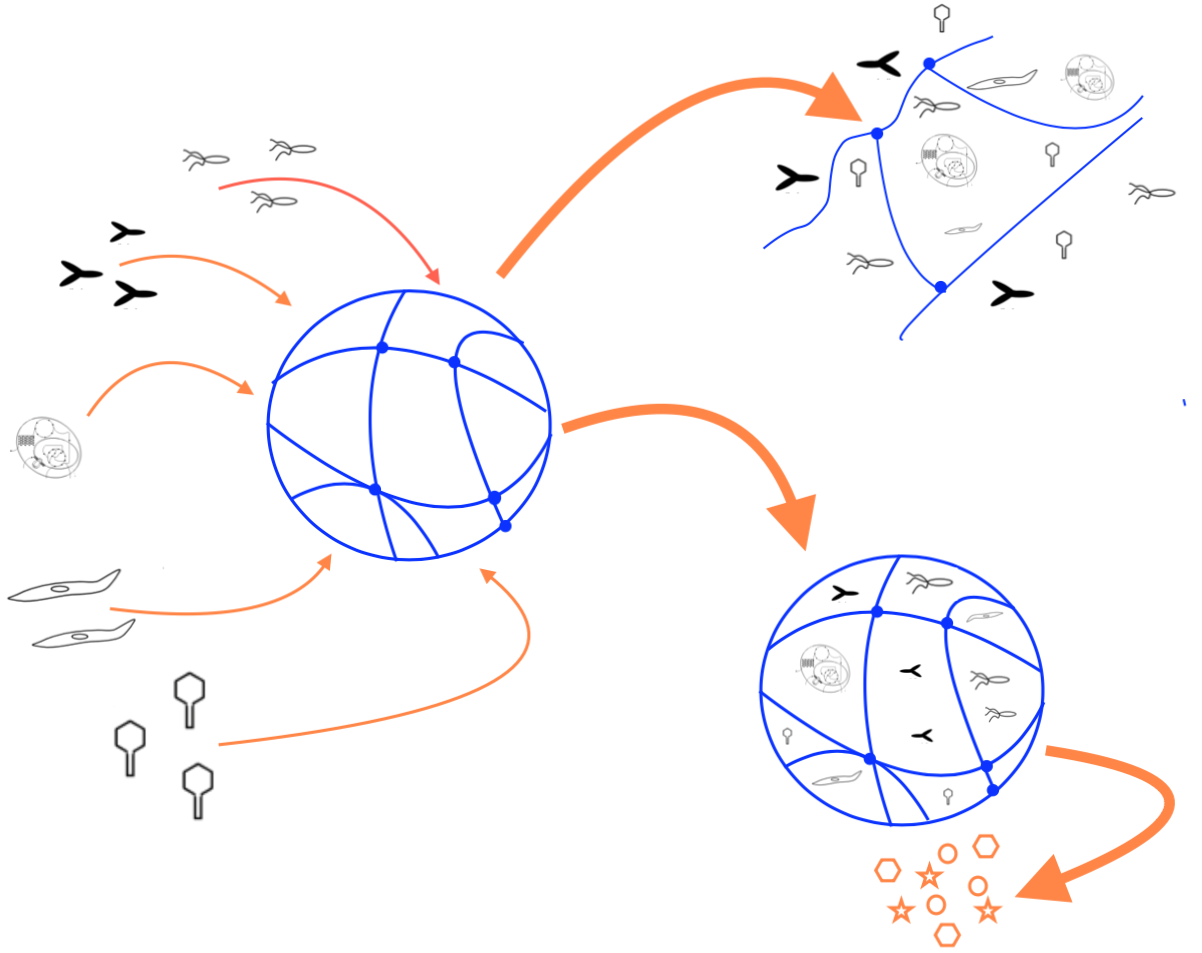
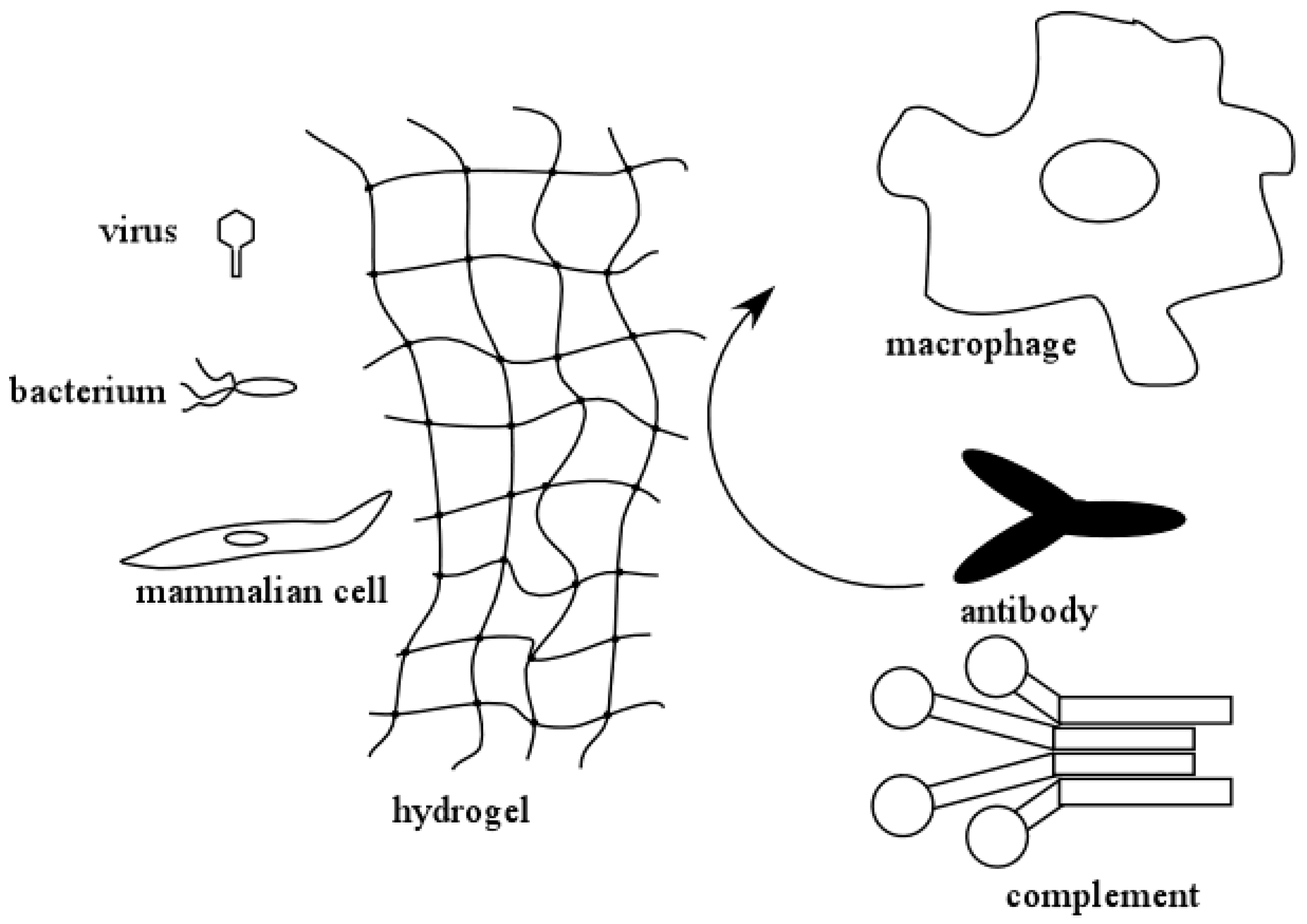
An important feature of hydrogels used to encapsulate molecules or cells is that the crosslinked hydrogel layer can protect the encapsulated biological agents from degradation and/or immune rejection. Figure 1 illustrates this concept. In an encapsulated system, small molecules and solutes can freely permeate and diffuse through the hydrogel layer. However, larger molecules, such as antibodies and components of the complement system, can be excluded from interacting with the encapsulated biological agents. This protects them from immune surveillance and clearance by the humoral (complement system and antibodies) and the cellular (macrophages, B- and T-cells, etc.) immune responses. As a result of this, hydrogels can be conveniently used in medical applications where the therapeutic molecules are being produced by encapsulated cells. The size exclusion capabilities of hydrogels also help protect the encapsulated biological agents against degradation by proteases. The latter is another convenient feature that makes hydrogels attractive for controlled released of biomolecules.
2. Hydrogel Preparation
Different preparation methods exist to create hydrogel materials and the polymeric chain can be designed to provide useful properties to these materials. Hydrogels can be formed as homopolymers, copolymers, or multiple polymers. The choice of the hydrogel precursor and/or composition is often made according to the required application. This can include requirements such as biocompatibility, mechanical and transport properties, degradation versus chemical stability, the ability to respond to changes in the environment, and the nature of crosslinks (e.g., covalent, ionic, hydrogen bonds, hydrophobic interactions, and others). The nature of the crosslink interaction in hydrogels is an important factor in their performance [4]. In the drug delivery of biomolecules, it is often desirable to release encapsulated biomolecules over time. This can be accomplished through degradable crosslinks, or by implementing crosslinks that slowly change over time. In other applications, like those involving transplanted cells, it is often desirable to have hydrogel systems that remain stable over time, in order to prevent immune recognition of the encapsulated cells.
The type of crosslinks in hydrogels can have an influence in the properties of hydrogels [4]. By far, the majority of hydrogels are crosslinked by means of covalent interactions. This is because of the ease by which multifunctional monomers can be added in the synthesis process, which often times involves free radical polymerization. However, once they are formed, covalent bonds will not break unless they form part of a degradable moiety. Also, the degradation of the moieties is irreversible, and this can limit the extent to which hydrogel properties can be controlled. In addition to covalent bonds, other types of intermolecular interactions can confer unique characteristics to hydrogels, and non-covalent crosslinks often times can provide hydrogels with unique stimuli responsive or mechanical properties [4]. For example, hydrogen bond crosslinks in hydrogels have been exploited in order to create hydrogels with large elongation and mechanical toughness [5]. In other instances, a combination of ionic and hydrogen bond interactions have been exploited to achieve self-healing properties as well as unique mechanical properties [4,5,6,7]. Crosslinks can also be based on van der Waals and hydrophobic interactions, in order to create hydrogel systems. Approaches used to make hydrogels based on hydrophobic crosslinks often involved amphiphilic ABA triblock copolymers. For example, cholesterol functionalized carbonates with a central hydrophilic block were shown to assemble into gels that could encapsulate drug-loaded polymeric micelles [8]. Poly(ethylene glycol) end capped at both ends with hydrophobic dipeptides, such as tyrosine, have been shown to assemble into hydrogels and have a gel transition close to body temperature [9]. Fluoralkyl-ended poly(ethylene glycol) polymers also assemble into hydrogels at body temperature, and they were exploited for the release of human growth hormone [10,11,12]. Ionic interactions have also been exploited in order to create crosslinks in hydrogels, and sometimes, the properties of hydrogels are conferred by a combination of different intermolecular interactions. In a recent publication, the combination of hydrogen bonds and hydrophobic interactions were exploited in order to create hydrogels with high toughness [13]. As such, the implementation of various types of intermolecular interactions in hydrogels can often be exploited to provide unique properties to these systems. Nonetheless, the flexibility for introducing crosslinks in hydrogels by means of different interactions, such as covalent bonds, hydrogen bonds, ionic and van der Waals interactions, allow encapsulation of biological agents in many different formulations according to the intended application, manufacturing method, and other characteristics of the encapsulated system.
Formation of crosslinks among polymeric chains that have large affinity for water is fundamental for the preparation of hydrogels. Thus, a large number of hydrogel preparation methods involve free radical polymerization. In free radical polymerization, an initiator is decomposed by a variety of mechanisms in such a way that it generates free radicals. These radicals react with the unsaturated double bonds or pi electrons of a monomer in such a manner that they are added to the molecule and transfer the free radical to the monomer. This monomer itself becomes a free radical capable of interacting with more monomers, thus creating a mechanism for chain-growth polymerization [2,14,15]. In order to form crosslinked networks, multifunctional monomers are needed for the formation of crosslinks. Hydrogels intended for biomedical applications can be formed using free radicals chemistry using water-soluble initiators through bulk polymerization [1,16,17,18] or interfacial polymerization [19,20,21,22,23,24,25,26,27,28,29,30]. Furthermore, the decomposition of the initiator can proceed by thermal decomposition, photolysis, or redox reactions [1,2,3,16,17,18]. Free radicals can also be formed using ionizing radiation, electrochemical decomposition, plasma-generated free radicals, and even sonication [2,3,14,16,31]. By far, the use of water-soluble initiators at moderate temperatures using a sensitizer, decomposition of potassium persulfate, or photolytically induced polymerizations, are the preferred methods in applications involving encapsulation of biological elements, due to the fact that it allows the reaction to proceed at mild conditions of temperature and pH.
Other polymerization methods have been used in the formation of hydrogels. Living free radical polymerization methods have been employed to make novel hydrogel materials or precursors. The two main methods are reversible addition fragmentation chain transfer (RAFT) [32,33,34,35] and atom transfer radical polymerization (ATRP) [36,37,38,39,40,41,42,43,44,45]. These methods have been particularly important and useful for the creation of novel hydrogel systems because they allow polymerization to proceed without chain termination by chain transfer. As a result of this, they have been widely used to create polymer brushes and interfaces functionalized with polymers [43,45,46,47,48,49], and to produce block copolymers capable of self-assembling into hydrogel systems [50,51,52,53,54,55,56,57,58,59,60,61]. Additional advantages of these systems are that the block copolymers can be chosen in such manner that they assemble into hydrogels in response to changes in their environment (e.g., pH or temperature).
Other methods exist for the formation of hydrogels, and the mechanisms can involve different types of intermolecular interactions, such as hydrogen bonding, ionic, acid–base, and van der Waals interactions, or a combination of two or more of these interactions [4,5,6,7,8,9,10,11,12,13]. In order to further advance the field, a more complete understanding of the complex interplay of molecular structures and intermolecular interactions will be necessary to further enhance the repertoire of “smart” materials for drug delivery.
3. Encapsulation within Hydrogels for Medical Applications
Hydrogels are useful materials in medical applications because of their biocompatibility and high water content [3,62]. Biological molecules can also be incorporated within these systems, in order to guide the desired biological responses [24,63,64,65,66,67,68,69,70,71,72,73,74,75,76]. Not surprisingly, it has been said that hydrogels were the first biomaterials that were rationally designed for medical applications [77]. The fact that the composition of the polymeric chains and the nature of the crosslinks can be manipulated independently, allow for wide variations in their properties according to specific applications. Hydrogel crosslinks can be engineered to exhibit properties which makes it possible for a hydrogel to degrade with time. In this regard, hydrogel crosslinks can be designed so they degrade by hydrolysis [78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95], or by enzymatic action [64,69,73,75,76,96,97,98,99,100,101,102,103,104,105]. An examination of the scientific literature shows how versatile hydrogels can be in many biomedical applications. The fact that hydrogels can be synthesized under mild conditions of pH and temperature in aqueous environments makes them highly suitable for minimally invasive applications [10,22,24,64,91,106,107,108,109,110,111,112,113,114,115]. It is possible for hydrogels to form, in situ, through photopolymerization of water-soluble monomers and crosslinkers, as hydrogel precursors under physiological conditions of pH and temperature [20,21,22,27,28,67,69,86,110,116,117,118,119,120,121], or by the self-assembly of macromers [6,8,9,10,12,21,50,77,122,123]. It is also possible to form lightly crosslinked hydrogels based on stimuli responsive polymers (e.g., thermoresponsive polymers) that incorporate biomolecules or even cells that can be delivered through minimally invasive procedures. When such materials reach body temperature, the thermoresponsive moieties change the properties of the material from a mostly viscous gel to a solid for localized drug delivery or cell therapies [24,50,73,77,93,97,112,124,125,126,127,128,129]. All these characteristics make hydrogels ideally suited for the delivery of biological agents, ranging from simple biomolecules to entire cells and cell clusters. Thus, they can easily be implemented not only for the delivery of encapsulated drugs, but also to deliver cells and/or when the molecule(s) to be delivered is (are) encapsulated within the hydrogel environment. However, during the formation of hydrogels in vivo, the equilibrium swelling of the material needs to be taken into account. Often times, the swelling of the hydrogel as synthesized may not correspond to its equilibrium swelling. In those situations, the volume occupied by the hydrogel upon formation can change with time. This could have important implications in situations where hydrogels are used in reconstructive surgery (e.g., hydrogel materials containing cells and molecules that are intended to fill a defect in tissue). In those situations, it is important to understand and predict the expected characteristics of the formed hydrogels, so that the formulation of hydrogels precursors can lead, as close as possible, to the formation of hydrogels in a state that is as close as possible to the equilibrium swelling. This can be particularly difficult in the case of stimuli-responsive hydrogels or when the crosslinks are intended to slowly degrade with time. In those situations, it is important to understand the potential effects that the varying volume and/or mesh size of the hydrogel can have in a particular application.
4. Proteins and Biomolecules
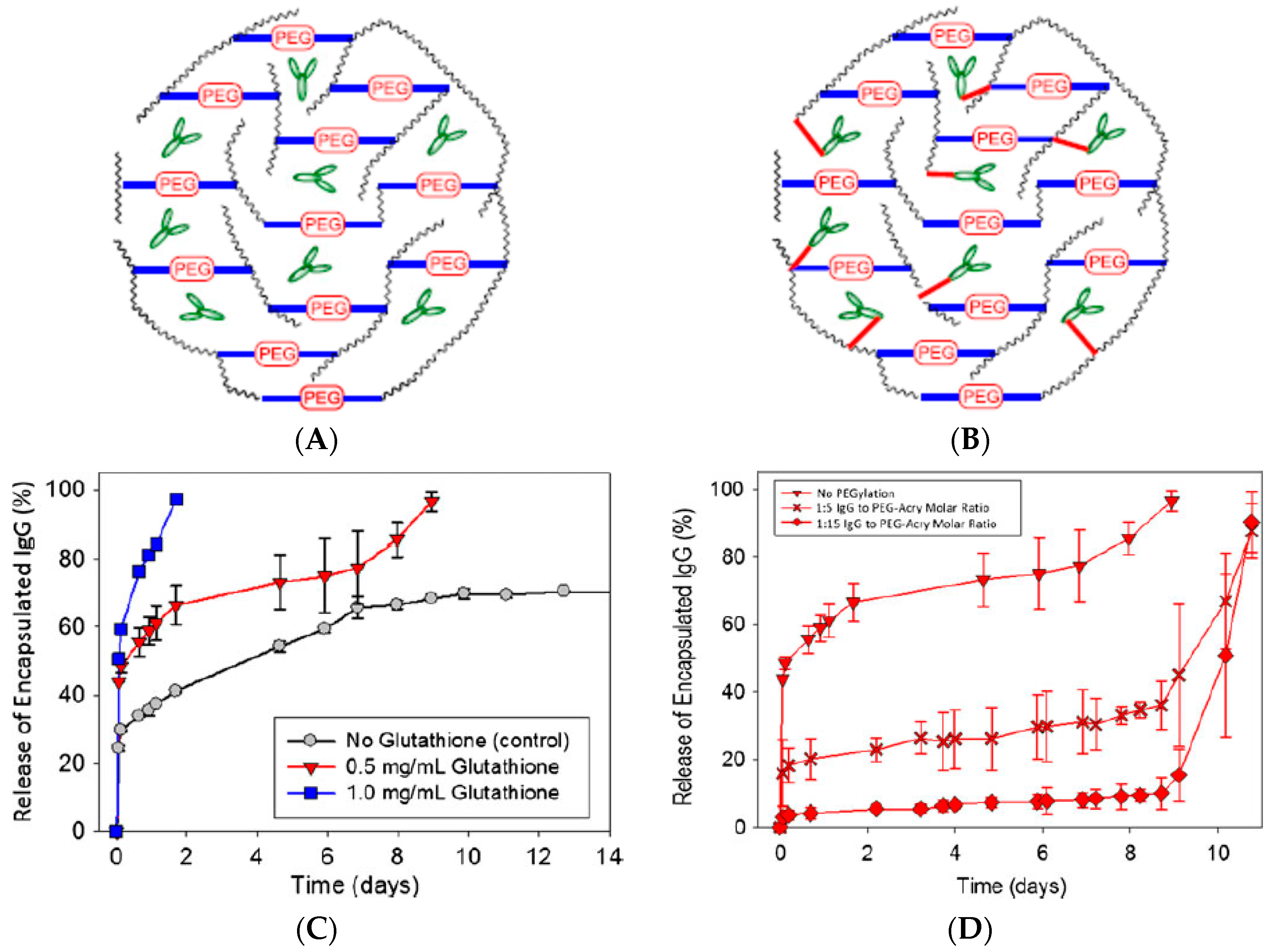
The fact that hydrogels are cytocompatible and preserve the complex structure and function of biomolecules makes them ideal materials for drug delivery of encapsulated molecules. Many examples abound in the literature on the use of hydrogels for the encapsulation and controlled release of proteins for therapeutic applications. Among the possible proteins to be released from hydrogels, the delivery of growth factors has been widely reported as a strategy to improve wound healing [64,76,130,131,132,133,134,135,136,137,138,139,140,141], release of bone morphogenetic protein 2 (BMP-2) has been used for bone regeneration [142,143,144,145,146,147,148,149,150,151,152], encapsulation of insulin has been explored for oral delivery and/or triggered release under various conditions [153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172], encapsulation of different antigens in hydrogels has been explored in order to enhance the immune response for vaccine boosting effects and other immunotherapies [173,174,175,176,177,178,179,180], and encapsulation of antibodies has been use in localized delivery applications [181,182,183,184]. These are just a few of the many applications that have been explored involving the encapsulation of biomolecules, but they serve to exemplify why hydrogels are such versatile materials for applications involving the encapsulation and release of biological macromolecules. Many times, a burst release is observed in these and other drug delivery systems. In the case of proteins and other macromolecules encapsulated within hydrogels, the burst release can be ascribed to proteins weakly bound to the hydrogel and/or proteins that are encapsulated just within the periphery of the hydrogel. In a recent paper on the encapsulation of antibodies specific for vascular endothelial growth factor (VEGF), we showed that the burst release could go from as little to 25% of the total amount encapsulated to almost 50%, depending on the composition of the hydrogel [129]. Thus, methods to decrease this burst release could involve changing the hydrogel composition. In the same paper, the approach to delay the burst release consisted in covalently linking the antibodies directly to the hydrogel using a bifunctional PEG [129]. These ideas are illustrated in Figure 2. It can be seen that the covalent linkage does not prevent the burst release, but it can delay it by almost 9 days. In this case, the reason was that the linkage did not degrade faster than the hydrogel. Thus, the release occurred once the hydrogel degraded to a significant extent. However, this illustrates that pulsed delivery could be achieved if the rates of degradation of the hydrogel crosslinkers and the molecule linking the antibodies to the hydrogel are carefully chosen.
Controlled Release of Encapsulated Molecules from Hydrogels
Understanding the release of encapsulated molecules from hydrogels is important in drug delivery applications. Depending on the characteristics of the system, release can occur by a combination of mechanisms that need to be identified in order to better design drug delivery systems based on these materials. One of the most widely used models to analyze drug release involves the use of the Higuchi equation [185] which, in relative amounts released from a drug delivery system, can be expressed as
where represents the relative or fractional drug release, is a kinetic constant that depends on the diffusion coefficient of the drug and dimensions of the drug delivery system, and t is the release time. This equation is based on a model of Fickian diffusion, assuming an infinite sink. In general, it can provide adequate estimates of released drugs during the initial stages of the process, as long as release proceeds by diffusion alone. A system that follows Fickian diffusion then would be expected to show a dependence of initial release that is linear with the square root of time. However, other factors can affect the release of the encapsulated drug in such ways that the release can proceed through the contribution of other mechanisms in addition to diffusion. In those cases, it is not uncommon to observe systems where the initial release does not follow a dependence with the square root of time. Ritger and Peppas proposed the following more general equation in order to model and elucidate potential mechanisms of release [186]:
Here, represents the relative or fractional drug release, is a kinetic constant, t is the release time, and n has been termed a “diffusional exponent” that provides insights as to the release mechanism from hydrogels. For a thin hydrogel, if n = 0.5 the equation becomes similar to Higuchi’s equation, and indicates Fickian diffusion as the release mechanism. When n approaches a value of 1, the mechanism can be assumed to be the so called Case II transport, which corresponds to zero order release (the rate of release is constant). This can occur because of concomitant swelling and/or dissolution of the drug delivery system during release. If 0.5 < n < 1, the release mechanism can be a complex process involving various mechanisms (e.g., diffusion, dissolution and/or swelling together).
The equations above are normally limited to the initial release stages, but are useful to identify drug release behaviors. For longer release times, the distribution of the drug within the hydrogel changes and decreases with time, leading to slowing of the release rates. An analysis of the release process can be complex when the properties of the hydrogel also change with time (e.g., because of swelling and/or degradation). In the case of diffusion-controlled release, the process has been modeled using Fick’s equation, in which case, the solution depends on boundary conditions and geometry of the system. For initial homogeneous distribution of the drug within the hydrogel and release into a very large system, analytical solutions have been obtained for the following geometries with drugs of a constant diffusion coefficient, D, within the hydrogel [187];
Thin film with negligible “edge” effects
L: thickness of the film.
Spherical hydrogel
R: sphere radius.
Cylindrical hydrogel device
qn: are the nth roots of the Bessel function of the first kind of zero order; R: cylinder radius; H: cylinder length.
Since models that predict release of drugs encapsulated within hydrogels are mainly based on the assumption of a diffusion mechanism, they require knowledge of diffusion coefficients within hydrogels. Since normally only the bulk diffusion coefficients are available, it is necessary to account for the effects that hydrogels can have in the diffusion coefficients of the encapsulated molecules. For example, if the size of the released molecules was much smaller than the mesh size of the hydrogels, the use of bulk diffusion coefficients could provide adequate predictions of the drug release process. However, in the case of release of macromolecules such as proteins from hydrogels, the size of these molecules can be comparable to the mesh or “pore size” of the hydrogel. In those cases, the diffusion coefficients could differ significantly from bulk diffusion coefficients. As a result of this, several models have been developed that attempt to model the effects that hydrogel chains have on the diffusion coefficients of macromolecules. Existing models are based on different mechanisms invoked to explain such effects, namely: free volume theory [188,189], hydrodynamic theory [189,190], or obstruction theory [189,191,192,193]. These theories have led to the development of different equations that attempt to describe how the hydrogel environment affects diffusion coefficients. Such models are able to represent the experimental data on diffusion of molecules within hydrogels to different extents. In other words, the models can predict diffusion coefficients in some polymer systems better than others, which indicate limitations in our knowledge of all the hydrogel characteristics that influence diffusion of molecules. Comparisons between models have been described elsewhere [189,192]. In what follows, only some representative models for the different theories are given.
In free volume theory, diffusion is described to occur when the solute “jumps” into voids formed in the solvent space by redistribution of the free volume within the liquid [189,192]. Solute diffusion depends on the jumping distance, thermal velocity of solute, and the probability that there is a hole free volume adjacent to the solute molecule. The hydrodynamic theory describes solute transport in terms of the Stokes–Einstein equation, whereby the solute is modeled as a hard sphere that is large compared to the space of solvent in which it moves, and the solvent is treated as a continuum [190]. In hydrodynamic theory models, the frictional drag on the solute is affected by the polymer chains, which enhance frictional drag by slowing down the solvent near the polymer chains. Thus, hydrodynamic theory models focus on modeling the friction drag coefficient [189,192]. Obstruction theory models assume that the polymer chains in the hydrogel cause an increase in the pathlength for diffusion. The polymer chains can be conceptualized as a sieve that allows the passage of molecules only if they can pass between the polymer chains.
The following are some equations derived from the different theories. It is not an extensive list of the different equations developed to predict diffusion coefficients within hydrogels, but they are given as illustrative examples. In these equations Dg is the diffusion coefficient of a molecule within the hydrogel, D0 is the bulk diffusion coefficient of the molecule, rS is the radius of the solute, rf is the radius of the polymer fiber, and is the volume fraction of the polymer in the gel.
Free volume theory model by Lustig and Peppas [194]
In this equation, k1 and k2 are treated as fitting parameters.
Hydrodynamic theory model by Cukier [195]
Here, kc is an undefined constant for a given polymer–solvent system that must be fitted to the data.
Hydrodynamic theory model by Philips et al. [196]
where k is the hydraulic permeability of the medium, which can be estimated for hydrogels using the correlation developed by Jackson and James [197]:
Obstruction model by Ogston et al. [198]
Obstruction model by Amsden [193]
An analysis of the effectiveness of these equations in predicting solute diffusion within hydrogels can be found elsewhere [189,192]. These equations also illustrate the complexities of modeling drug release from hydrogels, especially in situations where the swelling changes as a function of time, due to crosslink degradation, changes in activity coefficients of the systems with changing solute concentrations, and changes in swelling due to non-equilibrium or changing equilibrium conditions.
5. Viruses
Although viruses are normally thought as pathogenic biological entities causing a multitude of diseases, they can also be employed in some applications with potentially beneficial health effects. Two potential applications of these systems are as an alternative to antibiotics (bacteriophages) and as vectors for delivery of nucleic acids in genetic engineering applications. However, their use in therapeutic applications is often times limited because of immune clearance, inactivation by natural defenses or barriers in the body, and the need to direct them to specific sites in the body. In this regard, hydrogel materials can help circumvent some of these limitations. Similar to other applications, encapsulation of viruses within hydrogels offers protection against immune recognition because the hydrogel material surrounding the encapsulated viruses offers a physical barrier against the many elements of the immune system.
For some applications of phage therapy, oral administration is a convenient therapeutic method. However, in order for such a route of administration to be effective, two major requirements need to be addressed. One is avoiding the harsh acidic environment of the stomach, where the low pH can quickly inactivate the phages administered via oral ingestion. For this, it may be necessary to encapsulate the phages in a system that offers protection against acidic inactivation. The other is the need to release the encapsulated phages after passage through the stomach (e.g., in the small intestine). These requirements can be met with a hydrogel system that isolates encapsulated phages from the low pH environment and, subsequently, releases the phages from the hydrogel when the pH increases in the small intestine.
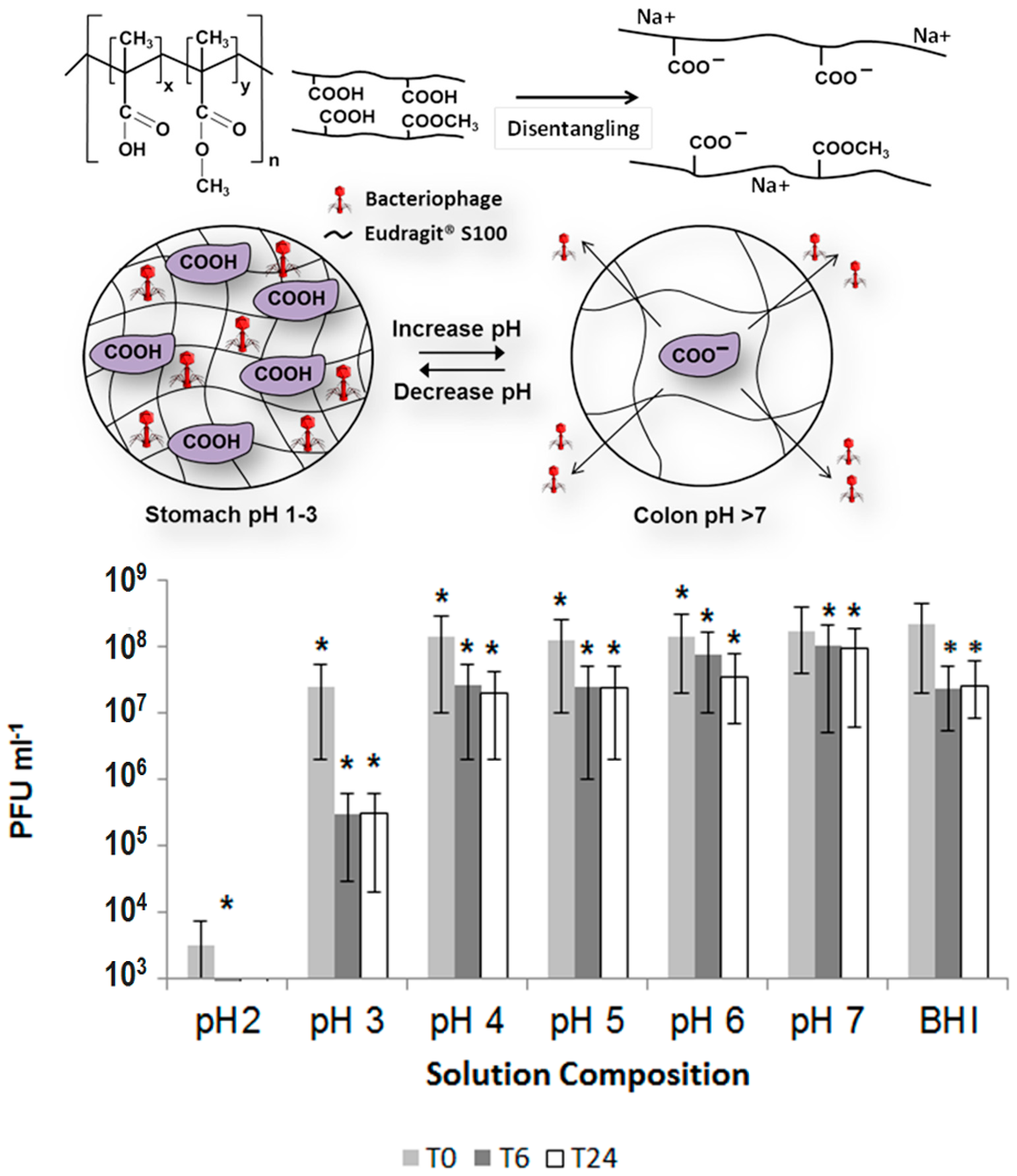
Oral administration of bacteriophages has been proposed as an alternative to the use of antibiotics for controlling the presence of enterohemorrhagic E. coli in cattle [199,200]. This is an important application, since enterohemorrhagic E. coli is the main cause of hemolytic uremic syndrome, and cattle are the main reservoir of the pathogenic forms of E. coli causing this disease [201]. This approach is also attractive as an alternative to the indiscriminate use of antibiotics, which can result in the emergence of antibiotic resistant bacteria. As mentioned, oral delivery of bacteriophages for controlling these bacteria presents the challenge of the need to bypass the harsh acidic conditions of the stomach, which could quickly deactivate the bacteriophages before they reach the intestine, where enterohemorrhagic E. coli reside. Also, it is necessary for any delivery system to respond to the conditions of the intestine, such that bacteriophages are quickly released in order to control the pathogenic bacteria. Phage CA933P is capable of infecting enterohemorrhagic strains of E. coli, such as O157:H7, O145:H25, O13:H8, and ONT:H12, as well as other Gram-negative bacteria. However, it is quite sensitive to pH values below 4, which precludes direct oral administration [202]. For this reason, phage CA993P was encapsulated within alginate and low methoxylated pectin materials, in order to see their suitability for oral delivery to the intestine [199,200]. Hydrogel microspheres containing the encapsulated phages were formed by ionic crosslinking of these polysaccharide materials in the presence of calcium ions. The microspheres were made using the corresponding polymer solutions or using the polymer solutions emulsified with oleic acid [199,200]. It was reported that emulsified low methoxylated pectin was more efficient as a delivery system than unemulsified pectin, alginate, or emulsified alginate microspheres. Exposure of free (non-encapsulated phages) to pH 2.5 alone resulted in the reduction of bacteriophage titer by 3.7 logarithmic orders after only 10 min. Furthermore, at pH 2.5 and in the presence of 0.5 mg/mL pepsin, the phage titer decreased to undetectable levels within only 10 min. On the other hand, when bacteriophage CA993P was encapsulated within emulsified low methoxylated pectin spheres, there was no significant decrease in phage titer at pH 2.5 and concentrations of pepsin up to 4.2 mg/mL after 3 h [199]. Release of phages proceeded almost to completion after 4 h in phosphate buffer solution (PBS) at pH 7.2 and 37 °C [199,200], which are conditions of pH and temperature similar to what would be expected in the intestine. These are two important factors for the intended application, namely, that the encapsulated phages are protected from the harsh environment of the stomach, and that they can be quickly released under the conditions of the intestine. The release was also found to follow the Korsmeyer–Peppas model, which includes swelling, diffusion, and dissolution of the polymeric matrix [200].
Encapsulation of bacteriophages offers significant promise to address some medical problems. Clostridium difficile often causes intestinal infections that are especially difficult to treat [203] and the treatment can come at significant costs [204]. Treatment of C. difficile is particularly difficult with antibiotics, and recurrent infections often times occur [205]. Thus, delivery of bacteriophages is a promising treatment for this condition. The C. difficile specific bacteriophage, myovirus CDKM9 was encapsulated within the commercial pH responsive polymer Eudragit® S100 using a microfluidics system as proof of concept of the potential for treatment of C. difficile [206]. The bacteriophages were encapsulated within monodisperse microspheres about 100 μm in diameter. The encapsulated phages remained stable and viable for 4 weeks at 4 °C. There was no significant release of phages under simulated gastric conditions at pH 2 for 3 h. However, the microsphere showed significant release of active phages for 24 h at pH 7, which offers the potential of this technology for the treatment of C. difficile. The concept of encapsulating C. difficile-specific phages for delivery to the intestine is shown in Figure 3. Here, the polymer contains carboxyl groups that remain protonated within the acidic environment of the stomach. This allows for protection of the encapsulated phages because the hydrogel remains collapsed and does not swell with the acidic fluids of the stomach. Once these particles reach the small intestine, the pH change causes the carboxylic groups to deprotonate, which causes the hydrogel to suddenly swell because of electrostatic repulsion and the more polar carboxylate groups, which causes the encapsulated bacteriophages to be released in the small intestine.
In another further medical application, encapsulation of bacteriophages has been explored in order to address the problem caused by the formation of biofilms of Proteus mirabilis on urinary catheters. Proteus mirabilis can cause significant complications in catheter-associated urinary tract infections because it produces bacterial urease enzymes that generate ammonia upon reaction with urea, thus increasing the pH of urine. This can result in local supersaturation and precipitation of struvite and apatite, causing their accumulation in the catheter walls, and eventually leading to obstruction of the catheter [207,208,209,210]. In order to address this potential problem, a coating for catheters was designed consisting of a hydrogel layer containing encapsulated bacteriophages on the surface of the catheter. This layer was covered with the pH-responsive polymer EUDRAGIT®S 100 [210]. This coating showed that, using an in vitro bladder system, catheter blockage was doubled under conditions of P. mirabilis infection. Also, under such conditions of catheter infection, a “burst release” of the encapsulated phages occurred in response to the rapid local pH [210]. Thus, this approach also shows the potential of phage encapsulation for medical applications.
Even for tissue engineering applications, the encapsulation of viruses can offer useful applications. Encapsulation of lentiviruses within degradable poly(ethylene glycol)-based hydrogels has been proposed as a method to enhance transgene expression [211]. By increasing the residence time within the hydrogels, it was shown that prolonged and localized gene expression of pLenti-CMV-GFP and pLenti-CMV-GLuc could be achieved [211].
In some applications, it is also necessary to study the long-term protection effects of encapsulated bacteriophages. The Salmonella enteritidis specific phage f3αSE, when encapsulated within alginate spheres crosslinked with Ca2+ ions, was shown to remain viable at low pH conditions and higher temperatures for extended periods of time [212]. In fact, alginate-encapsulated bacteriophages remained infectious for long periods of time. In a water flow system, the encapsulated phages could be kept at concentrations of 102 to 104 PFU/mL for 250 h, which makes them suitable for oral administration in water feeding systems for poultry.
Other concepts on the release of encapsulated bacteriophages have been explored. The antimicrobial bacteriophage K (ΦK) has been encapsulated within agarose gels upon which a layer of methacrylate modified hyaluronan hydrogel were photocrosslinked [213]. These types of materials would respond to infectious agents such as Staphylococcus aureus strains, which are known to release hyaluronidase as an important virulence factor secreted by some pathogenic bacteria. In the presence of hyaluronidase positive pathogenic bacteria, the degradation of the methacrylated hyaluronan hydrogel would result in the release of the encapsulated bacteriophage K, which would subsequently infect the pathogenic bacteria. In this manner, this system was designed as a hydrogel capable of responding to bacterial infection.
6. Bacteria
With the increasing understanding of the role of the microbiome in human health, it is becoming evident that there is a need to have methods to efficiently deliver probiotics to the intestine. Oral administration of probiotics, although widely used, cannot be effective without a method of protection against the low pH of the stomach, and the need for quick release at the pH of the intestine. Furthermore, any encapsulation method also needs to maintain the viability of encapsulated microorganisms during storage of the product until administration. Thus, the hydrogel system must offer the capability of quickly releasing the encapsulated bacteria in the intestine in order to allow colonization of the intestinal tract with beneficial bacteria [214,215,216,217].
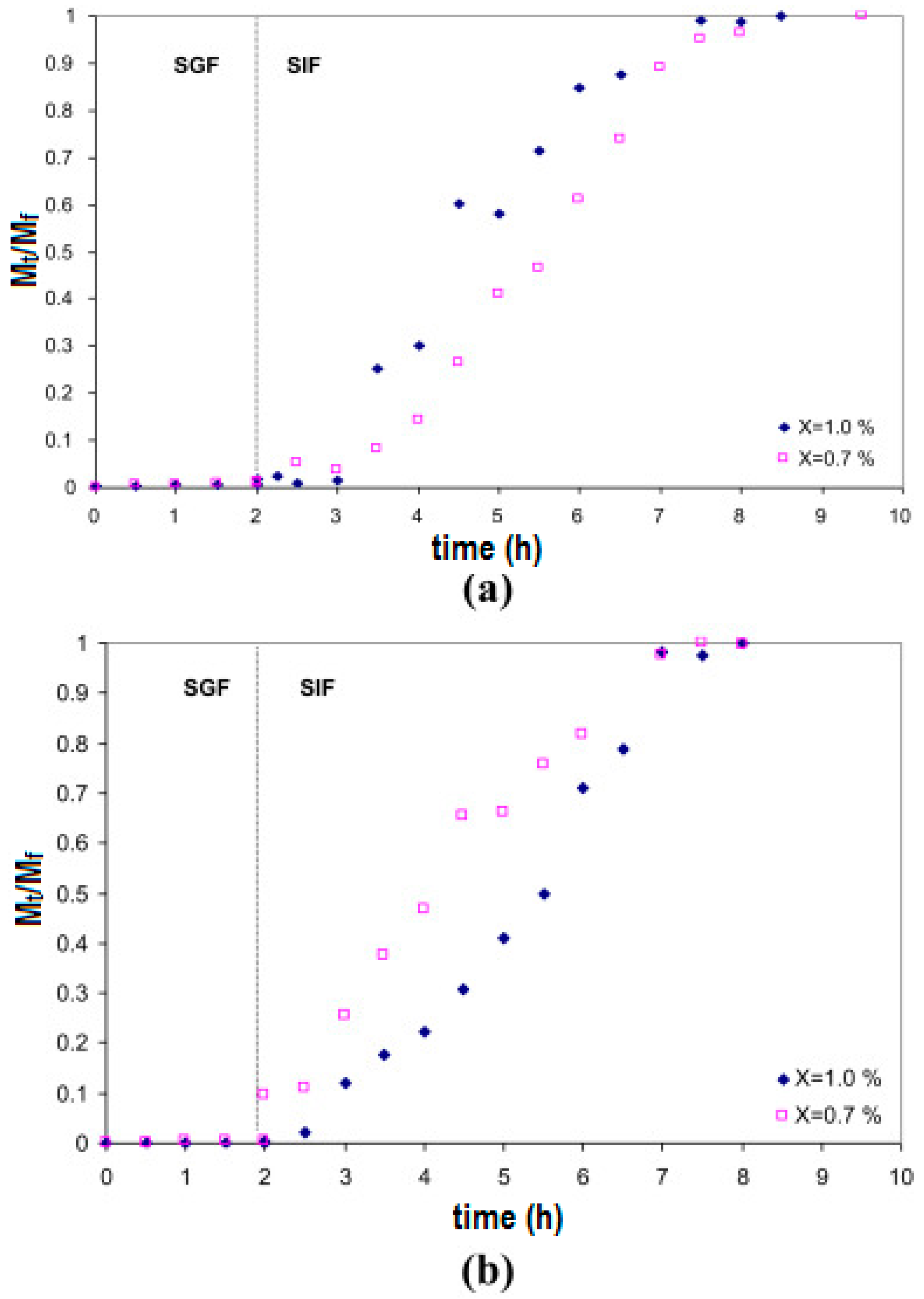
Work on the encapsulation of probiotics for oral delivery to the intestines is emerging in the literature. Different hydrogels have been explored and shown to be able to protect encapsulated bacteria from the low pH environment of the stomach, while allowing quick release at the pH conditions of the intestine. Xanthan–chitosan hydrogels exhibited negligible release of encapsulated Pediococcus acidilactici in water or simulated gastric fluid at pH 2.0 for two hours [218]. The pH of simulated gastric fluid seemed more critical than the presence of pepsin in the release of encapsulated bacteria. The encapsulated bacteria were completely released in simulated intestinal fluid in 5 h. Furthermore, the xanthan–chitosan capsules were stable in water for at least 3 days, and encapsulation within these hydrogels showed six-log retention of viability over non-encapsulated P. acidilactici for 1 h at pH 2.0, and four-log retention of viability at pH 2.0 after 4 h [218]. Figure 4 shows that the encapsulated bacteria remain within the hydrogel capsules under low pH conditions (simulated gastric fluid or SGF) for at least two hours, and that when the pH is changed to that found in the intestine (simulated intestinal fluid or SIF) swelling of the hydrogel capsules and release of the bacteria occur soon after, and continue for at least 5 h. This shows how hydrogels allow the formulation of release systems, where bacteria are protected from the harsh stomach environment for the typical duration of food passage through the digestive system.
Alginate/pectin was also explored for the encapsulation of Lactococcus lactis within beads of these hydrogel materials. L. lactis is of importance because it produces and releases nisin, a polycyclic antibacterial peptide. Nisin, in combination with miconazole, has been used in the treatment of C. difficile, a bacterial intestinal infection that is particularly difficult to treat [219]. Thus, the encapsulation of L. lactis could be considered as another potential application for the controlled release from hydrogels of molecules produced by encapsulated bacteria for therapeutic applications. A 75/25 alginate/pectin ratio was found to be optimum for providing the best mechanical properties of beads containing L. lactis, and also allowed for the best release of nisin during the storage period. Lactobacillus casei encapsulated within an alginate–pea protein isolate matrix and was found to be protected during freeze drying compared to free cells. Storage in the frozen state was achieved for 84 days, although the freeze-dried cells exhibited a weaker protection against the low pH of simulated gastric acid conditions, but showed similar release profiles in simulated intestinal fluid [220]. Bifidobacterium longum, an obligate anaerobe that establishes a protective gut microbiome in infants through adulthood, was encapsulated within alginate microgels that were subsequently coated with chitosan [221]. Interestingly, the chitosan layer did not seem to improve the viability of the encapsulated B. longum. In fact, storage and gastrointestinal stability decreased when compared to alginate beads without the chitosan coating, which further indicates the need to more fully understand the complexity of these systems. Another less elegant approach explored was the formation of compressed pellets of lyophilized probiotics (L. acidophilus) together with dry sodium alginate. In this case, the pressed cell pellet was carefully surrounded by dry alginate before further compression into pellets. Upon hydration, this system provided probiotics surrounded by the hydrated hydrogel, which also offer protection against acidic challenges and release under simulated intestinal fluid conditions [222].
7. Islet Cells
Hydrogels can help isolate cells against immune recognition. Semipermeable hydrogel membranes around cells protect them against immune recognition, while at the same time, they allow the passage of nutrients, oxygen, and small molecules. Because of this, hydrogels are ideal materials for transplantation of cells that offer therapeutic benefits. This is why hydrogel capsules have long been explored for the encapsulation of cells. Probably one of the first applications of encapsulated mammalian cells for therapeutic applications in humans was for the purpose of treating type I diabetes [223]. The first clinical trial of immunoprotected islet cells in humans was published in 1994 [224]. This involved alginate as the encapsulating material for islet allografts implanted in humans. Islet cells were isolated from human cadaver donors by collagenase digestion and gradient separation. Then they were cultured and coated by an alginate–polylysine–alginate membrane, and implanted in the peritoneal cavity of a 38 years old adult patient who had been insulin dependent for 30 years. The procedure was successful in that the patient was able to become insulin independent for 9 months following the procedure. This success followed the earlier work by the same group on islet transplantation in diabetic dogs, which showed they could become insulin independent following transplantation of alginate encapsulated islets for 172 days, and that the implanted islets remained viable for as long as 726 days [225]. Encapsulation of islets within alginate capsules is not without problems. Earlier attempts at using this technology for the treatment of insulin dependent diabetes failed because mannuronic acid monomers from alginate capsules were found to stimulate interleukins 1 and 6, as well as tumor necrosis factor, which resulted in the formation of fibrosis due to fibroblast proliferation around the implanted capsules [225]. Mechanical integrity was also found to be important, and directly influenced by the amount of mannuronic acid in the alginate [225]. For these reasons, alginate high in guluronic acid needed to be used. Also, after formation of alginate capsules around the islets, a layer of polylysine was formed in order to confer better mechanical stability. Since polycations can elicit fibrous capsule formation [226], another layer of alginate needed to be placed around the capsules, which resulted in the alginate–polylysine–alginate coating. This also illustrates the need to better understand the biocompatibility of hydrogel materials in applications where encapsulated cells need to be implanted [223,224,225].
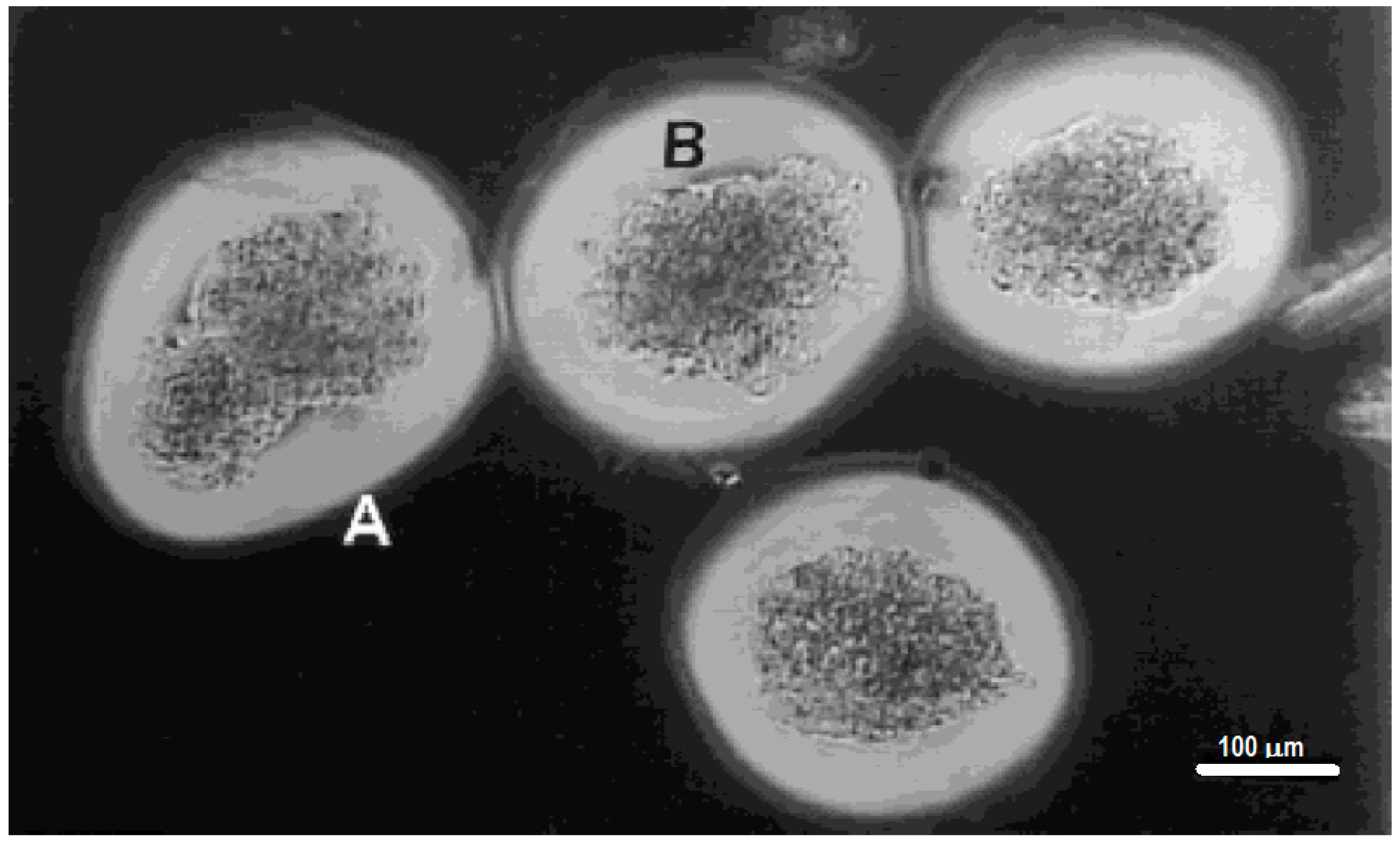
A slightly different approach for the encapsulation of islets within hydrogels involves the photopolymerization of poly(ethylene glycol) diacrylate (PEG-DA) around islets [116,117,227]. This system involves the interfacial photopolymerization of PEG-DA around eosin-Y stained islets. The photopolymerization reaction starts at the eosin-Y molecules [28] and produces a layer of crosslinked PEG-DA hydrogel that extends only about a 100 to 300 μm from the interface [28,30,228,229], and can be controlled by varying the polymerization conditions [20,228,229]. The advantages of this approach are various: (1) poly(ethylene glycol) has been found to exhibit excellent biocompatibility in most biomaterial applications; (2) the polymerization conditions maintain cell viability; (3) the thin layers of hydrogel formed around the islets permit efficient transport of oxygen and nutrients to the islets; (4) the crosslinked nature of the hydrogels presents an immunoisolation barrier against components of the immune system, such as antibodies and complement components; (5) the covalent nature of the crosslinks is more stable than divalent cation crosslinked alginate; and (6) the coating is non-ionic (unlike alginate). The last two factors contribute to the biggest advantages compared with alginate capsules in terms of biocompatibility and stability. Encapsulation of islets within photopolymerized PEG-DA was shown to be successful in xenograft transplants of porcine islets in the peritoneal cavity of Sprague-Dawley rats. The encapsulated islets were shown to be glucose responsive. Normoglycemia was maintained in diabetic induced rats for up to 110 days after implantation of 5000 to 8000 encapsulated islets. This shows promise on this approach for the treatment of insulin dependent diabetes [117]. Figure 5 shows the uniformity and conformal coating of PEG diacrylate hydrogels around porcine islet cells [20].
8. Tissue Engineering
Encapsulation of cells within hydrogels can serve many applications in medicine. The applications discussed above involved the delivery of cells or molecules produced by the cells and the hydrogel was, for the most part, an inert material. However, there are many applications in which the hydrogel provides more than an encapsulating material, a physical barrier against the immune system, or a simple vehicle for the delivery of cells. The hydrogel itself can constitute the environment that allows cells to differentiate and/or maintain certain phenotype. In this regard, the system that is delivered in the body consists of hydrogel and cells altogether. Thus, the hydrogel itself, along with cells and biochemical molecules, can constitute an active part of the system that is delivered to certain tissues for cell therapy purposes.
Work on cartilage tissue engineering often relies on the use of hydrogels to deliver chondrocytes as well as maintaining their phenotype. An advantage of hydrogels is that they can be created under mild conditions compatible with the viability of cells, such as chondrocytes, and often times, the hydrogel properties are designed in order to allow for minimal invasive surgery applications [87,91,107,109,112,230,231,232,233,234]. Many different types of hydrogels have been studied for the formation of cartilage using encapsulated chondrocytes. Alginate has been shown to induce redifferentiation of chondrocytes that were dedifferentiated by monolayer cell culture [235]. Alginate/hyaluronan composite hydrogels loaded with TGF-beta 1 have also been demonstrated to allow the differentiation of periodontal ligament stem cells and human bone marrow mesenchymal stem cells into a chondrogenic phenotype. Such differentiation of these cells into a chondrogenic phenotype was shown to be dependent on the modulus of elasticity of the hydrogel delivery system [236]. It is likely that the similarity of hydrogel mechanical properties and cartilage allow the cells to be differentiated into a chondrogenic phenotype.
Other hydrogels have also been explored for cartilage regeneration. Thermoresponsive polymers with low critical solution temperatures below 37 °C are promising materials because of their capability to be made into lightly crosslinked hydrogels that can be delivered through a needle into the body. When they reach body temperature, they solidify and remain in place, adopting the shape of the environment they were delivered to. Healy et al. has synthesized poly(N-isopropyl acrylamide) based hydrogels containing peptide-based crosslinks that are degraded specifically by metalloproteinases involved in the remodeling of the extracellular matrix during the wound healing reaction [23,112]. Such hydrogels were found to support bovine articular chondrocyte viability for at least 28 days in vitro. They could be injected through a small needle and quickly stabilized in situ by the thermoresponsive properties of the hydrogel materials. The chondrocytes encapsulated in such thermoresponsive hydrogels produced extracellular matrix with properties similar to natural cartilage. They stained for sulfated polysaccharides, and there was evidence that the hydrogel material allowed for the proliferation of chondrocytes within those hydrogels [112]. Thus, thermoresponsive hydrogels hold great promise for minimal invasive applications.
Photopolymerization reactions are fast and can be used to create hydrogels in situ under mild reaction conditions compatible with cell viability. Thus, they also have the potential to be implemented as minimally invasive surgical procedures for cell-based therapies. For this reason, photopolymerized poly(ethylene glycol) based hydrogels have been explored for tissue engineering of cartilage. Polyethyleneoxide dimethacrylate photopolymerized into thick, 2 to 8 mm thick disks of 9 mm diameter, was shown to successfully encapsulate and maintain the viability and differentiation of chondrocytes. The chondrocytes were shown to produce extracellular matrix components characteristic of cartilage tissue. The specimens showed homogeneity in the distribution of cells and extracellular matrix components throughout the sample. Furthermore, they were shown to produce glycosaminoglycan and collagen homogeneously throughout the hydrogels, which is promising as a technology for cartilage repair [237].
Hydrogels have been also implemented in hard tissue applications. Their versatility to be formed in situ under mild conditions, which are compatible with the integrity of biomolecules and cells, made them ideal materials for the treatment of bone defects and to accelerate healing in orthopedic applications [148]. Different hydrogel systems have been explored for bone healing applications. In a good number of these applications, the encapsulation and delivery of growth factors and molecules that induce the stimulation of osteoblasts is employed. The encapsulation and delivery of bone morphogenetic protein 2 (BMP-2) has been widely employed in bone healing/regeneration applications. This molecule has been shown to successfully allow for bone regeneration when incorporated in hydrogels [149]. BMP-2 has been used in PEG [75,151,152,238], alginate [142,144,147,149,236], polyphosphazenes [146], and elastin-like peptides [143]. Some systems combine more than one hydrogel component, and often incorporate a component of the extracellular matrix, such as hyaluronic acid [142], or the introduction of RGD peptide sequences that allow cell adhesion [75,143,148]. The encapsulation and delivery of BMP-2 has also been explored alone or together with other biomolecules, such as platelet-derived growth factor (PDGF) [143,144]. It is noteworthy to indicate the advantage to incorporate molecules such as BMP-2 with hydrogels in bone-healing applications. In a recent paper involving the encapsulation of human bone marrow cells within hydrogels containing alginate/hyaluronic acid and BMP-2, there was an enhanced osteogenic differentiation in vitro and bone regeneration in vivo, compared with the hydrogel alone or BMP-2 [142]. This is an indication that hydrogels play a synergistic role when combined with cells and signaling molecules.
The fact that hydrogels can be compatible with the viability of cells, can preserve the biological function of proteins and other biomolecules, and also can be injectable or polymerized in situ under mild conditions, make them important materials for stem cell-based therapies [70,100,137,142,143,144,152,236,238,239,240,241]. Research on stem cells indicates that their differentiation is dependent on a number of environmental signals, among which the rigidity of the matrix plays an important role [242]. The mechanical properties (hence the rigidity) of hydrogels are strongly dependent on their degree of crosslinking. For this reason, there have been a number of studies that indicate that the mechanical properties of hydrogels, which can be controlled through the degree of crosslinking, can have an effect in the differentiation of stem cells [243,244,245,246]. Many different applications can be found in the literature about the use of hydrogels in stem cells therapy. Injectable hydrogels carrying stem cells have been explored for cardiac tissue repair [247,248]. Injectable hydrogels based on PEG, or Tetronic® conjugated with fibrinogen, were shown to result in improvement of heart function after inducing infarcts in a rodent model through ligation of the left descending coronary artery. The results indicated that the hydrogels with the highest modulus provided greater recovery of heart function and neovascularization. This indicated that hydrogels, by themselves, had a therapeutic effect in treating myocardial damage after an infarct by preventing adverse remodeling of tissue after a heart attack [249]. Thus, it would be reasonable to expect that encapsulated stem cells would further contribute to recovery of damaged myocardium after an infarct. Injectable fibrin gels have been used to deliver bone marrow mononuclear cells to the cryoinjured infarcted myocardium of a rat animal model. Implantation of bone marrow mononuclear cells delivered in a fibrin matrix resulted in significantly higher neovascularization than cells implanted alone [250]. Other approaches have involved bone marrow-derived mononuclear cells in PEG–fibrin hydrogels with covalently bound hepatocyte growth factor, which resulted in cell prevalence increases of 15-fold in hearts receiving the matrix containing the hydrogel, cells, and growth factors, compared to control that received a saline injection only [251]. Other uses of hydrogels in conjunction with stem cells have included stem cells for neural tissue repair [243,252,253,254]. In those applications as well, the hydrogels extend the survival of the stem cells, and can modulate their differentiation. The differentiation is influenced by the mechanical properties of the hydrogel, as well as more complex factors, such as their porosity and whether signaling molecules are also encapsulated in the hydrogel systems.
9. Conclusions and Future Directions
Hydrogels are highly versatile materials for uses in many medical applications. There are many characteristics of these systems that make them attractive for biomedical applications. Their high water content is compatible with the biological activity of biomolecules and the viability of cells. Hydrogels offer many possibilities in the delivery of biological systems, such as molecules and cells, that would not be possible with other materials. Many examples can be found in the literature on their use for the encapsulation and delivery of biomolecules, viruses, bacteria, and mammalian cells for medical applications. The fact that hydrogels can be made under mild conditions of temperature and pH make them ideal not only for the encapsulation of the fragile living systems and biomolecules involved, but also permit their formation in situ for minimally invasive applications. Given that other molecules, such as adhesive [24,63,64,67,69,70,71,72,73,74,75,96,106,125,143,255,256,257,258,259] and/or degradation [64,73,76,97,98,99,106,109] sequences, and signaling molecules, such as growth factors [4,68,91,107,130,131,133,134,135,136,137,138,139,140,141,144,148,150,151,152,248,255,260,261,262,263,264,265], can be incorporated into these systems, offers the possibility not only to deliver cells, but also to modulate their function according to the required response. These are just some of the most salient characteristics of hydrogels that makes them useful in medical applications. However, these systems can be made with many other interesting characteristics. Their mechanical properties can be controlled or modulated according to the degree of crosslinking and chemical composition, which is important in many soft tissue applications, but also because the mechanical properties can influence the function and differentiation of cells [12,138,149,243,244,254,266,267]. Some hydrogels can be stimuli-responsive, such that they change properties in response to environmental cues, like temperature, pH, light, electrical stimuli, and others [77,93,268,269,270,271,272]. With the variety of properties that hydrogels can be prepared with, it is no surprise that they are widely used in many medical applications that combine many useful characteristics. It is likely that in the future, more examples with higher levels of sophistication will begin to emerge. For these, it will be necessary to develop a larger variety of stimuli-responsive hydrogels, degradable hydrogels, micro and nanostructured hydrogels, microfabricated hydrogel structures, and self-assembly methods to create supramolecular structures with these materials. Other potential properties would be gradient materials, where the gradient could be not only in terms of mechanical properties or crosslinking, but also the extent of biological function that could be incorporated into these materials to emulate the biological environment. A large number of hydrogel materials are currently made using free radical polymerization, and this creates materials that are difficult to degrade or cannot degrade in vivo. Although degradation sequences have been implemented in these materials by means of degradable crosslinkers, the main chains still have the properties of polymers made by free radical polymerization. The latter limits, to some extent, the current use of these materials in many applications. A further need still exists to make hydrogel materials that degrade completely, and new research may offer solutions to this limitation in the future.
Acknowledgments
Support from the Department of Chemical and Biological Engineering at the Illinois Institute of Technology and the Universidad de Guadalajara are gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Ottenbrite, R.M.; Park, K.; Okano, T.; Peppas, N.A. Biomedical Applications of Hydrogels Handbook, 2010th ed.; Springer: New York, NY, USA, 2010; p. 432. [Google Scholar]
- Ratner, B.D.; Hoffman, A.S. Synthetic Hydrogels for Biomedical Applications. ACS Symp. Ser. 1976, 31, 1–36. [Google Scholar]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 13–36. [Google Scholar] [CrossRef]
- Song, G.; Zhang, L.; He, C.; Fang, D.-C.; Whitten, P.G.; Wang, H.; Jiang, L. Facile Fabrication of Tough Hydrogels Physically Cross-Linked by Strong Cooperative Hydrogen Bonding. Macromolecules 2013, 46, 7423–7435. [Google Scholar] [CrossRef]
- Zhang, G.; Lv, L.; Deng, Y.; Wang, C. Self-Healing Gelatin Hydrogels Cross-Linked by Combining Multiple Hydrogen Bonding and Ionic Coordination. Macromol. Rapid Commun. 2017, 38, 1700018. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Zhang, Y.; Li, Y.; Xua, B.; Liu, W. Hydrogen bonded and ionically crosslinked high strength hydrogels exhibiting Ca2+-triggered shape memory properties and volume shrinkage for cell detachment. J. Mater. Chem. B 2015, 3, 6347–6354. [Google Scholar] [CrossRef]
- Lee, A.L.Z.; Venkataraman, S.; Fox, C.H.; Coady, D.J.; Frank, C.W.; Hedrick, J.L.; Yang, Y.Y. Modular composite hydrogels from cholesterol-functionalized polycarbonates for antimicrobial applications. J. Mater. Chem. B 2015, 3, 6953–6963. [Google Scholar] [CrossRef]
- Hamley, I.W.; Cheng, G.; Castelletto, V. A Thermoresponsive Hydrogel Based on Telechelic PEG End-Capped with Hydrophobic Dipeptides. Macromol. Biosci. 2011, 11, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Tae, G.; Kornfield, J.A.; Hubbell, J.A. Sustained release of human growth hormone from in situ forming hydrogels using self-assembly of fluoroalkyl-ended poly(ethylene glycol). Biomaterials 2005, 26, 5259–5266. [Google Scholar] [CrossRef] [PubMed]
- Tae, G.; Kornfield, J.A.; Hubbell, J.A.; Johannsmann, D. Anomalous sorption in thin films of fluoroalkyl-ended poly(ethylene glycol)s. Langmuir 2002, 18, 8241–8245. [Google Scholar] [CrossRef]
- Tae, G.; Kornfield, J.A.; Hubbell, J.A.; Johannsmann, D.; Hogen-Esch, T.E. Hydrogels with controlled, surface erosion characteristics from self-assembly of fluoroalkyl-ended poly(ethylene glycol). Macromolecules 2001, 34, 6409–6419. [Google Scholar] [CrossRef]
- Chang, X.; Geng, Y.; Cao, H.; Zhou, J.; Tian, Y.; Shan, G.; Bao, Y.; Wu, Z.L.; Pan, P. Dual-Crosslink Physical Hydrogels with High Toughness Based on Synergistic Hydrogen Bonding and Hydrophobic Interactions. Macromol. Rapid Commun. 2018, 1700806. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Cowie, J.M.G.; Arrighi, V. Polymers: Chemistry and Physics of Modern Materials, 3rd ed.; CRC Press: Boca Raton, FA, USA, 2008. [Google Scholar]
- Hoffman, A.S. Hydrogels for Biomedical Applications. Adv. Drug. Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Maitra, J.; Shukla, V.K. Cross-linking in Hydrogels—A Review. Am. J. Polym. Sci. 2014, 4, 25–31. [Google Scholar]
- Cruise, G.M.; Scharp, D.S.; Hubbell, J.A. Characterization of permeability and network structure of interfacially photopolymerized poly(ethylene glycol) diacrylate hydrogels. Biomaterials 1998, 19, 1287–1294. [Google Scholar] [CrossRef]
- Cruise, G.M.; Hegre, O.D.; Scharp, D.S.; Hubbell, J.A. A sensitivity study of the key parameters in the interfacial photopolymerization of poly(ethylene glycol) diacrylate upon porcine islets. Biotechnol. Bioeng. 1998, 57, 655–665. [Google Scholar] [CrossRef]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Interfacial photopolymerization of poly(ethylene glycol)-based hydrogels upon alginate-poly(l-lysine) microcapsules for enhanced biocompatibility. Biomaterials 1993, 14, 1008–1016. [Google Scholar] [CrossRef]
- Hill-West, J.L.; Chowdhury, S.M.; Slepian, M.J.; Hubbell, J.A. Inhibition of thrombosis and intimal thickening by in situ photopolymerization of thin hydrogel barriers. Proc. Natl. Acad. Sci. USA 1994, 91, 5967–5971. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Shull, K.R.; Healy, K.E. Axisymmetric Adhesion Test To Examine the Interfacial Interactions between Biologically-Modified Networks and Models of the Extracellular Matrix. Langmuir 2003, 19, 1853–1860. [Google Scholar] [CrossRef]
- Healy, K.E.; Rezania, A.; Stile, R.A. Designing biomaterials to direct biological responses. Ann. N. Y. Acad. Sci. 1999, 875, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F. Sequential formation of hydrogel multilayers through surface initiated photopolymerization. Polym. Prepr. 2004, 45, 41–42. [Google Scholar]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F.A. Poly(ethylene glycol) diacrylate hydrogels on covalently attached eosin surface: Contact angle measurements of the surface. Polym. Prepr. 2003, 44, 206–207. [Google Scholar]
- Pathak, C.P.; Sawhney, A.S.; Hubbell, J.A. Rapid photopolymerization of immunoprotective gels in contact with cells and tissue. J. Am. Chem. Soc. 1992, 114, 8311–8312. [Google Scholar] [CrossRef]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F. Photopolymerization of Poly(Ethylene Glycol) Diacrylate on Eosin-Functionalized Surfaces. Langmuir 2004, 20, 8652–8658. [Google Scholar] [CrossRef] [PubMed]
- Papavasiliou, G.; Songprawat, P.; Perez-Luna, V.; Hammes, E.; Morris, M.; Chiu, Y.-C.; Brey, E. Three-Dimensional Patterning of Poly(Ethylene Glycol) Hydrogels Through Surface-Initiated Photopolymerization. Tissue Eng. Part C 2008, 14, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Kizilel, S.; Sawardecker, E.; Teymour, F.; Perez-Luna, V.H. Sequential formation of covalently bonded hydrogel multilayers through surface initiated photopolymerization. Biomaterials 2006, 27, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Odian, G. Principles of Polymerization, 4th ed.; Wiley-Interscience: New York, NY, USA, 2004. [Google Scholar]
- Moad, G.; Chiefari, J.; Chong, Y.K.; Krstina, J.; Mayadunne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living free radical polymerization with reversible addition—Fragmentation chain transfer (the life of RAFT). Polym. Int. 2000, 49, 993–1001. [Google Scholar] [CrossRef]
- Lowe, A.B.; Sumerlin, B.S.; Donovan, M.S.; McCormick, C.L. Facile Preparation of Transition Metal Nanoparticles Stabilized by Well-Defined (Co)polymers Synthesized via Aqueous Reversible Addition-Fragmentation Chain Transfer Polymerization. J. Am. Chem. Soc. 2002, 124, 11562–11563. [Google Scholar] [CrossRef] [PubMed]
- Yusa, S.; Shimada, Y.; Mitsukami, Y.; Yamamoto, T.; Morishima, Y. pH-Responsive Micellization of Amphiphilic Diblock Copolymers Synthesized via Reversible Addition-Fragmentation Chain Transfer Polymerization. Macromolecules 2003, 36, 4208–4215. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Rao, G.V.R.; Basame, S.B.; Keller, D.J.; Artyushkova, K.; Fulghum, J.E.; Lopez, G.P. Reversible Control of Free Energy and Topography of Nanostructured Surfaces. J. Am. Chem. Soc. 2004, 126, 8904–8905. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Rao, G.V.R.; Ista, L.K.; Wu, Y.; Andrzejewski, B.P.; Sklar, L.A.; Ward, T.L.; Lopez, G.P. Control of molecular transport through stimuli-responsive ordered mesoporous materials. Adv. Mater. 2003, 15, 1262–1266. [Google Scholar] [CrossRef]
- Ma, H.; Hyun, J.; Stiller, P.; Chilkoti, A. “Non-Fouling” Oligo(ethylene glycol)-Functionalized Polymer Brushes Synthesized by Surface-Initiated Atom Transfer Radical Polymerization. Adv. Mater. 2004, 16, 338–341. [Google Scholar] [CrossRef]
- Zhao, B.; Brittain, W.J. Synthesis of Tethered Polystyrene-block-Poly(methyl methacrylate) Monolayer on a Silicate Substrate by Sequential Carbocationic Polymerization and Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 1999, 121, 3557–3558. [Google Scholar] [CrossRef]
- Kim, J.-B.; Bruening, M.L.; Baker, G.L. Surface-Initiated Atom Transfer Radical Polymerization on Gold at Ambient Temperature. J. Am. Chem. Soc. 2000, 122, 7616–7617. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Miller, P.J.; Shukla, N.; Immaraporn, B.; Gelman, A.; Luokala, B.B.; Siclovan, T.M.; Kickelbick, G.; Vallant, T.; Hoffmann, H.; et al. Polymers at Interfaces: Using Atom Transfer Radical Polymerization in the Controlled Growth of Homopolymers and Block Copolymers from Silicon Surfaces in the Absence of Untethered Sacrificial Initiator. Macromolecules 1999, 32, 8716–8724. [Google Scholar] [CrossRef]
- Li, X.; Husson, S.M. Adsorption of dansylated amino acids on molecularly imprinted surfaces: A surface plasmon resonance study. Biosens. Bioelectron. 2006, 22, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, Y.; Wang, X.; Liu, S. Fabrication of Hybrid Silica Nanoparticles Densely Grafted with Thermoresponsive Poly(N-isopropylacrylamide) Brushes of Controlled Thickness via Surface-Initiated Atom Transfer Radical Polymerization. Chem. Mater. 2008, 20, 101–109. [Google Scholar] [CrossRef]
- Chakraborty, S.; Bishnoi, S.W.; Pérez-Luna, V.H. Gold Nanoparticles with Poly(N-isopropyl acrylamide) Formed via Surface Initiated Atom Transfer Free Radical Polymerization Exhibit Unusually Slow Aggregation Kinetics. J. Phys. Chem. C 2010, 114, 5947–5955. [Google Scholar] [CrossRef]
- Lou, X.; Wang, C.; He, L. Core-Shell Au Nanoparticle Formation with DNA-Polymer Hybrid Coatings Using Aqueous ATRP. Biomacromolecules 2007, 8, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; He, L. Surface passivation using oligo(ethylene glycol) in ATRP-assisted DNA detection. Sens. Actuators B 2008, 129, 225–230. [Google Scholar] [CrossRef]
- Couet, J.; Biesalski, M. Surface-Initiated ATRP of N-Isopropylacrylamide from Initiator-Modified Self-Assembled Peptide Nanotubes. Macromolecules 2006, 39, 7258–7268. [Google Scholar] [CrossRef]
- Raula, J.; Shan, J.; Nuopponen, M.; Niskanen, A.; Jiang, H.; Kauppinen, E.I.; Tenhu, H. Synthesis of Gold Nanoparticles Grafted with a Thermoresponsive Polymer by Surface-Induced Reversible-Addition-Fragmentation Chain-Transfer Polymerization. Langmuir 2003, 19, 3499–3504. [Google Scholar] [CrossRef]
- Dimitrova, I.; Trzebickab, B.; Müllerc, A.H.E.; Dworakb, A.; Tsvetanov, C.B. Thermosensitive water-soluble copolymers with doubly responsive reversibly interacting entities. Prog. Polym. Sci. 2007, 32, 1275–1343. [Google Scholar] [CrossRef]
- He, M.; Jiang, H.Y.; Wang, R.; Xie, Y.; Zhao, W.F.; Zhao, C.S. A versatile approach towards multi-functional surfaces via covalently attaching hydrogel thin layers. J. Colloid Interface Sci. 2016, 484, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Rwei, S.P.; Anh, T.H.N.; Chiang, W.Y.; Way, T.F.; Hsu, Y.J. Synthesis and Drug Delivery Application of Thermo- and pH-Sensitive Hydrogels: Poly(β-CD-co-N-Isopropylacrylamide-co-IAM). Materials 2016, 9, 1003. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.L.; Liu, X.Y.; Xu, X.L.; Chen, S.; Zhu, X.Y.; Du, Y.C.; Li, F. Temperature and pH dual-responsive polyhedral oligomeric silsesquioxane/poly[2-(dimethyl amino)-ethyl methacrylate]-b-poly(N-isopropylacrylamide) hybrid materials synthesized via RAFT polymerization and thiol-ene reaction: Potential candidates as drug delivery systems. Mater. Chem. Phys. 2016, 179, 65–71. [Google Scholar]
- Li, S.; Zhao, Z.X.; Wu, W.; Ding, C.M.; Li, J.S. Dual pH-responsive micelles with both charge-conversional property and hydrophobic-hydrophilic transition for effective cellular uptake and intracellular drug release. Polym. Chem. 2016, 7, 2202–2208. [Google Scholar] [CrossRef]
- Zhu, H.Z.; You, L.Q.; Wei, H.L.; Wang, G.F.; Chu, H.J.; Zhu, J.; He, J. Preparation and Characterization of pH-Sensitive Hydrogel Microspheres Based on Atom Transfer Radical Polymerization. Polym. Eng. Sci. 2015, 55, 2775–2782. [Google Scholar] [CrossRef]
- Wang, H.Y.; Qin, A.W.; Li, X.; Zhao, X.Z.; Liu, D.P.; He, C.J. Biocompatible Amphiphilic Conetwork Based on Crosslinked Star Copolymers: A Potential Drug Carrier. J. Polym. Sci. Part A 2015, 53, 2537–2545. [Google Scholar] [CrossRef]
- Yang, B.G.; Wang, C.Y.; Zhang, Y.B.; Ye, L.; Qian, Y.F.; Shu, Y.; Wang, J.M.; Li, J.J.; Yao, F.L. A thermoresponsive poly(N-vinylcaprolactam-co-sulfobetaine methacrylate) zwitterionic hydrogel exhibiting switchable anti-biofouling and cytocompatibility. Polym. Chem. 2015, 6, 3431–3442. [Google Scholar] [CrossRef]
- Ghaemy, M.; Ziaei, S.; Alizadeh, R. Synthesis of pH-sensitive amphiphilic pentablock copolymers via combination of ring-opening and atom transfer radical polymerization for drug delivery. Eur. Polym. J. 2014, 58, 103–114. [Google Scholar] [CrossRef]
- Forbes, D.C.; Creixell, M.; Frizzell, H.; Peppas, N.A. Polycationic nanoparticles synthesized using ARGET ATRP for drug delivery. Eur. J. Pharm. Biopharm. 2013, 84, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Siegwart, D.J.; Lee, H.I.; Sherwood, G.; Peteanu, L.; Hollinger, J.O.; Kataoka, K.; Matyjaszewski, K. Biodegradable nanogels prepared by atom transfer radical polymerization as potential drug delivery carriers: Synthesis, biodegradation, in vitro release, and bioconjugation. J. Am. Chem. Soc. 2007, 129, 5939–5945. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Liu, Y.Y.; Fan, X.D.; Huang, Y. Synthesis of thermo- and pH sensitive polymer beads containing beta-cyclodextrin and poly(N,N-dimethylaminoethyl metracrylate and their controlled drug release behavior. Acta Polym. Sin. 2005, 3, 357–362. [Google Scholar]
- Hoffman, A.S.; Cohn, D.; Hanson, S.R.; Harker, L.A.; Horbett, T.A.; Ratner, B.D.; Reynolds, L.O. Application of radiation-grafted hydrogels as blood-contacting biomaterials. Radiat. Phys. Chem. 1983, 22, 267–283. [Google Scholar] [CrossRef]
- Fittkau, M.H.; Zilla, P.; Bezuidenhout, D.; Lutolf, M.P.; Human, P.; Hubbell, J.A.; Davies, N. The selective modulation of endothelial cell mobility on RGD peptide containing surfaces by YIGSR peptides. Biomaterials 2005, 26, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Seliktar, D.; Zisch, A.H.; Lutolf, M.P.; Wrana, J.L.; Hubbell, J.A. MMP-2 sensitive, VEGF-bearing bioactive hydrogels for promotion of vascular healing. J. Biomed. Mater. Res. Part A 2004, 68, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Raeber, G.P.; Zisch, A.H.; Tirelli, N.; Hubbell, J.A. Cell-responsive synthetic hydrogels. Adv. Mater. 2003, 15, 888–892. [Google Scholar] [CrossRef]
- Lutolf, M.P.; Hubbell, J.A. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules 2003, 4, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.D.; Tirelli, N.; Hubbell, J.A. Photopolymerized hyaluronic acid-based hydrogels and interpenetrating networks. Biomaterials 2003, 24, 893–900. [Google Scholar] [CrossRef]
- Pratt Alison, B.; Weber Franz, E.; Schmoekel Hugo, G.; Muller, R.; Hubbell Jeffrey, A. Synthetic extracellular matrices for in situ tissue engineering. Biotechnol. Bioeng. 2004, 86, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halstenberg, S.; Panitch, A.; Rizzi, S.; Hall, H.; Hubbell Jeffrey, A. Biologically engineered protein-graft-poly(ethylene glycol) hydrogels: A cell adhesive and plasmin-degradable biosynthetic material for tissue repair. Biomacromolecules 2002, 3, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Nuttelman, C.R.; Tripodi, M.C.; Anseth, K.S. Synthetic hydrogel niches that promote human mesenchymal stem cell viability. Matrix Biol. 2005, 24, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Benoit, D.S.W.; Anseth, K.S. The effect on osteoblast function of colocalized RGD and PHSRN epitopes on PEG surfaces. Biomaterials 2005, 26, 5209–5220. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Anseth, K.S. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials 2002, 23, 4315–4323. [Google Scholar] [CrossRef]
- Kim, S.; Chung Eugene, H.; Gilbert, M.; Healy Kevin, E. Synthetic MMP-13 degradable ECMs based on poly(N-isopropylacrylamide-co-acrylic acid) semi-interpenetrating polymer networks. I. Degradation and cell migration. J. Biomed. Mater. Res. A 2005, 75, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Itle, L.J.; Koh, W.-G.; Pishko, M.V. Hepatocyte Viability and Protein Expression within Hydrogel Microstructures. Biotechnol. Prog. 2005, 21, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, S.C.; Ehrbar, M.; Halstenberg, S.; Raeber, G.P.; Schmoekel, H.G.; Hagenmueller, H.; Mueller, R.; Weber, F.E.; Hubbell, J.A. Recombinant Protein-co-PEG Networks as Cell-Adhesive and Proteolytically Degradable Hydrogel Matrixes. Part II: Biofunctional Characteristics. Biomacromolecules 2006, 7, 3019–3029. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, S.C.; Hubbell, J.A. Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrixes. Part I: Development and physicochemical characteristics. Biomacromolecules 2005, 6, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Kopecek, J.; Yang, J. Hydrogels as smart biomaterials. Polym. Int. 2007, 56, 1078–1098. [Google Scholar] [CrossRef]
- Zhao, X.; Harris, J.M. Novel degradable poly(ethylene glycol) esters for drug delivery. ACS Symp. Ser. 1997, 680, 458–472. [Google Scholar]
- Zhao, X.; Harris, J.M. Novel degradable poly(ethylene glycol) hydrogels for controlled release of protein. J. Pharm. Sci. 1998, 87, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- West, J.L.; An, Y.; Hubbell, J.A. Photopolymerized intravascular hydrogels for protein delivery. In Proceedings of the International Symposium on Controlled Release of Bioactive Materials, Kyoto, Japan, 7–10 July 1996; Controlled Release Society: Kyoto, Japan, 1996; pp. 833–834. [Google Scholar]
- Sawhney, A.S.; Pathak, C.P.; van Rensburg, J.J.; Dunn, R.C.; Hubbell, J.A. Optimization of photopolymerized bioerodible hydrogel properties for adhesion prevention. J. Biomed. Mater. Res. 1994, 28, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, J.A.; Pathak, C.P.; Sawhney, A.S.; Desai, N.P.; Hill, J.L. Photopolymerizable Biodegradable Hydrogels as Tissue Contacting Material and Controlled-Release Carriers. U.S. Patent WO1993017669, 16 Septermber 1993. [Google Scholar]
- Reddy, S.K.; Anseth, K.S.; Bowman, C.N. Modeling of network degradation in mixed step-chain growth polymerizations. Polymer 2005, 46, 4212–4222. [Google Scholar] [CrossRef]
- Rice, M.A.; Anseth, K.S. Encapsulating chondrocytes in copolymer gels: Bimodal degradation kinetics influence cell phenotype and extracellular matrix development. J. Biomed. Mater. Res. Part A 2004, 70, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Bender, R.J.; Durand, K.L.; Anseth, K.S. Encapsulating chondrocytes in degrading PEG hydrogels with high modulus: Engineering gel structural changes to facilitate cartilaginous tissue production. Biotechnol. Bioeng. 2004, 86, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Durand, K.L.; Anseth, K.S. Manipulations in hydrogel chemistry control photoencapsulated chondrocyte behavior and their extracellular matrix production. J. Biomed. Mater. Res. Part A 2003, 67, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Martens, P.J.; Bryant, S.J.; Anseth, K.S. Tailoring the Degradation of Hydrogels Formed from Multivinyl Poly(ethylene glycol) and Poly(vinyl alcohol) Macromers for Cartilage Tissue Engineering. Biomacromolecules 2003, 4, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Metters, A.T.; Bowman, C.N.; Anseth, K.S. Verification of scaling laws for degrading PLA-b-PEG-b-PLA hydrogels. AIChE J. 2001, 47, 1432–1437. [Google Scholar] [CrossRef]
- Mason, M.N.; Metters, A.T.; Bowman, C.N.; Anseth, K.S. Predicting Controlled-Release Behavior of Degradable PLA-b-PEG-b-PLA Hydrogels. Macromolecules 2001, 34, 4630–4635. [Google Scholar] [CrossRef]
- Lu, S.; Anseth, K.S. Release Behavior of High Molecular Weight Solutes from Poly(ethylene glycol)-Based Degradable Networks. Macromolecules 2000, 33, 2509–2515. [Google Scholar] [CrossRef]
- Anseth, K.S.; Metters, A.T.; Bryant, S.J.; Martens, P.J.; Elisseeff, J.H.; Bowman, C.N. In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control. Release 2002, 78, 199–209. [Google Scholar] [CrossRef]
- Suggs, L.J.; Krishnan, R.S.; Garcia, C.A.; Peter, S.J.; Anderson, J.M.; Mikos, A.G. In vitro and in vivo degradation of poly(propylene fumarate-co-ethylene glycol) hydrogels. J. Biomed. Mater. Res. 1998, 42, 312–320. [Google Scholar] [CrossRef]
- Elvira, C.; Abraham, G.A.; Gallardo, A.; San Roman, J. Smart biodegradable hydrogels with applications in drug delivery and tissue engineering. In Biodegradable Systems in Tissue Engineering and Regenerative Medicine, 1st ed.; Reis, R.L., Román, J.S., Eds.; CRC Press: Boca Raton, FA, USA, 2005; pp. 493–508. [Google Scholar]
- Metters, A.T.; Anseth, K.S.; Bowman, C.N. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer 2000, 41, 3993–4004. [Google Scholar] [CrossRef]
- Murthy, N.; Thng, Y.X.; Schuck, S.; Xu, M.C.; Fréchet, J.M.J. A Novel Strategy for Encapsulation and Release of Proteins: Hydrogels and Microgels with Acid-Labile Acetal Cross-Linkers. J. Am. Chem. Soc. 2002, 124, 12398–12399. [Google Scholar] [CrossRef] [PubMed]
- Huebsch, N.; Gilbert, M.; Healy, K.E. Analysis of sterilization protocols for peptide-modified hydrogels. J. Biomed. Mater. Res. Part B 2005, 74, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Healy, K.E. Synthesis and Characterization of Injectable Poly(N-isopropylacrylamide-co-acrylic acid) Hydrogels with Proteolytically Degradable Cross-Links. Biomacromolecules 2003, 4, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Raeber, G.P.; Lutolf, M.P.; Hubbell, J.A. Molecularly engineered PEG hydrogels: A novel model system for proteolytically mediated cell migration. Biophys. J. 2005, 89, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Lauer-Fields, J.L.; Schmoekel, H.G.; Metters, A.T.; Weber, F.E.; Fields, G.B.; Hubbell, J.A. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. USA 2003, 100, 5413–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeloew, C.; Segura, T.; Hubbell, J.A.; Frey, P. The effect of enzymatically degradable poly(ethylene glycol) hydrogels on smooth muscle cell phenotype. Biomaterials 2007, 29, 314–326. [Google Scholar] [CrossRef] [PubMed]
- West, J.L.; Hubbell, J.A. Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules 1999, 32, 241–244. [Google Scholar] [CrossRef]
- Goetsch, K.P.; Bracher, M.; Bezuidenhout, D.; Zilla, P.; Davies, N.H. Regulation of tissue ingrowth into proteolytically degradable hydrogels. Acta Biomater. 2015, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Miller, J.S.; Moon, J.J.; West, J.L. Proteolytically degradable hydrogels with a fluorogenic substrate for studies of cellular proteolytic activity and migration. Biotechnol. Prog. 2005, 21, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Dikovsky, D.; Bianco-Peled, H.; Seliktar, D. Proteolytically Degradable Photo-Polymerized Hydrogels Made From PEG–Fibrinogen Adducts. Adv. Eng. Mater. 2010, 12, B200–B209. [Google Scholar] [CrossRef]
- Wade, R.J.; Bassin, E.J.; Rodell, C.B.; Burdick, J.A. Protease-degradable electrospun fibrous hydrogels. Nat. Commun. 2015, 6, 6639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutolf, M.P.; Weber, F.E.; Schmoekel, H.G.; Schense, J.C.; Kohler, T.; Mueller, R.; Hubbell, J.A. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat. Biotechnol. 2003, 21, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Elisseeff, J.; McIntosh, W.; Fu, K.; Blunk, T.; Langer, R. Controlled-release of IGF-I and TGF-b1 in a photopolymerizing hydrogel for cartilage tissue engineering. J. Orthop. Res. 2001, 19, 1098–1104. [Google Scholar] [CrossRef]
- Goessl, A.; Tirelli, N.; Hubbell, J.A. A hydrogel system for stimulus-responsive, oxygen-sensitive in situ gelation. J. Biomater. Sci. Polym. Ed. 2004, 15, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Lutolf, M.P.; Hubbell, J.A.; Hunziker, E.B.; Wong, M. Bovine Primary Chondrocyte Culture in Synthetic Matrix Metalloproteinase-Sensitive Poly(ethylene glycol)-Based Hydrogels as a Scaffold for Cartilage Repair. Tissue Eng. 2004, 10, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Hill-West, J.L.; Chowdhury, S.M.; Sawhney, A.S.; Pathak, C.P.; Dunn, R.C.; Hubbell, J.A. Prevention of postoperative adhesions in the rat by in situ photopolymerization of bioresorbable hydrogel barriers. Obstet. Gynecol. 1994, 83, 59–64. [Google Scholar] [PubMed]
- Suggs, L.J.; Mikos, A.G. Development of poly(propylene fumarate-co-ethylene glycol) as an injectable carrier for endothelial cells. Cell Transplant. 1999, 8, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Burghardt, W.R.; Healy, K.E. Synthesis and Characterization of Injectable Poly(N-isopropylacrylamide)-Based Hydrogels That Support Tissue Formation in Vitro. Macromolecules 1999, 32, 7370–7379. [Google Scholar] [CrossRef]
- Ruel-Gariepy, E.; Leroux, J.C. In situ-forming hydrogels—Review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, I.M.; Yacoub, M.H. Hydrogel scaffolds for tissue engineering: Progress and challenges. Glob. Cardiol. Sci. Pract. 2013, 2013, 316–342. [Google Scholar] [CrossRef] [PubMed]
- Nih, L.R.; Carmichael, S.T.; Segura, T. Hydrogels for brain repair after stroke: An emerging treatment option. Curr. Opin. Biotechnol. 2016, 40, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Modification of islet of Langerhans surfaces with immunoprotective poly(ethylene glycol) coatings via interfacial photopolymerization. Biotechnol. Bioeng. 1994, 44, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Cruise, G.M.; Hegre, O.D.; Lamberti, F.V.; Hager, S.R.; Hill, R.; Scharp, D.S.; Hubbell, J.A. In vitro and in vivo performance of porcine islets encapsulated in interfacially photopolymerized poly(ethylene glycol) diacrylate membranes. Cell Transplant. 1999, 8, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Pathak, C.P.; Sawhney, A.S.; Hubbell, J.A. In situ photopolymerization and gelation of water-soluble monomers: A new approach for local administration of peptide drugs. Polym. Prepr. 1992, 33, 65–66. [Google Scholar]
- Hubbell, J.A.; Pathak, C.P.; Sawhney, A.S. In vivo photopolymerization of PEG-based biodegradable hydrogels for the control of wound healing. Polym. Prepr. 1993, 34, 846–847. [Google Scholar]
- Rice, M.A.; Martens, P.J.; Bryant, S.J.; Mahoney, M.J.; Bowman, C.N.; Anseth, K.S. Photopolymerization of synthetic hydrogel niches for 3D cell culture and tissue regeneration. Polym. Prepr. 2004, 45, 11–12. [Google Scholar]
- Sawhney, A.S.; Pathak, C.P.; Hubbell, J.A. Bioerodible Hydrogels Based on Photopolymerized Poly(Ethylene Glycol)-co-Poly(Alpha-Hydroxy Acid) Diacrylate Macromers. Macromolecules 1993, 26, 581–587. [Google Scholar] [CrossRef]
- Cellesi, F.; Weber, W.; Fussenegger, M.; Hubbell, J.A.; Tirelli, N. Towards a fully synthetic substitute of alginate: Optimization of a thermal gelation/chemical cross-linking scheme (“tandem” gelation) for the production of beads and liquid-core capsules. Biotechnol. Bioeng. 2004, 88, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Cellesi, F.; Tirelli, N.; Hubbell, J.A. Towards a fully-synthetic substitute of alginate: Development of a new process using thermal gelation and chemical cross-linking. Biomaterials 2004, 25, 5115–5124. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Healy, K.E. Poly(N-isopropylacrylamide)-Based Semi-interpenetrating Polymer Networks for Tissue Engineering Applications. 1. Effects of Linear Poly(acrylic acid) Chains on Phase Behavior. Biomacromolecules 2002, 3, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Healy, K.E. Thermo-Responsive Peptide-Modified Hydrogels for Tissue Regeneration. Biomacromolecules 2001, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Stile, R.A.; Chung, E.; Burghardt, W.R.; Healy, K.E. Poly(N-isopropylacrylamide)-based semi-interpenetrating polymer networks for tissue engineering applications. Effects of linear poly(acrylic acid) chains on rheology. J. Biomater. Sci. Polym. Ed. 2004, 15, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Turturro, S.B.; Guthrie, M.J.; Appel, A.A.; Drapala, P.W.; Brey, E.M.; Pérez-Luna, V.H.; Mieler, W.F.; Kang-Mieler, J.J. The effects of cross-linked thermo-responsive PNIPAAm-based hydrogel injection on retinal function. Biomaterials 2011, 32, 3620–3626. [Google Scholar] [CrossRef] [PubMed]
- Drapala, P.W.; Brey, E.M.; Mieler, W.F.; Venerus, D.C.; Derwent, J.J.K.; Pérez-Luna, V.H. Role of Thermo-responsiveness and Poly(ethylene glycol) Diacrylate Cross-link Density on Protein Release from Poly(N-isopropylacrylamide) Hydrogels. J. Biomater. Sci. Polym. Ed. 2011, 22, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Drapala, P.W.; Jiang, B.; Chiu, Y.-C.; Mieler, W.F.; Brey, E.M.; Kang-Mieler, J.J.; Pérez-Luna, V.H. The Effect of Glutathione as Chain Transfer Agent in PNIPAAm-Based Thermo-responsive Hydrogels for Controlled Release of Proteins. Pharm. Res. 2014, 31, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Jain, E.; Sheth, S.; Dunn, A.; Zustiak, S.P.; Sell, S.A. Sustained release of multicomponent platelet-rich plasma proteins from hydrolytically degradable PEG hydrogels. J. Biomed. Mater. Res. A 2017, 105, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Mostafalu, P.; Kiaee, G.; Giatsidis, G.; Khalilpour, A.; Nabavinia, M.; Dokmeci, M.R.; Sonkusale, S.; Orgill, D.P.; Tamayol, A.; Khademhosseini, A. A Textile Dressing for Temporal and Dosage Controlled Drug Delivery. Adv. Funct. Mater. 2017, 27, 1702399. [Google Scholar] [CrossRef]
- Kasiewicz, L.N.; Whitehead, K.A. Recent advances in biomaterials for the treatment of diabetic foot ulcers. Biomater. Sci. 2017, 5, 1962–1975. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, Y.; Lan, Y.; Zuo, Q.; Li, C.; Zhang, Y.; Guo, R.; Xue, W. Acceleration of skin regeneration in full-thickness burns by incorporation of bFGF-loaded alginate microspheres into a CMCS-PVA hydrogel. J. Tissue Eng. Regener. Med. 2017, 11, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Chen, D.; Shen, C.; Shen, J.; Zhao, H.; He, Y. Platelet-Rich Plasma-Loaded Poly(d,l-lactide)-Poly(ethylene glycol)-Poly(d,l-lactide) Hydrogel Dressing Promotes Full-Thickness Skin Wound Healing in a Rodent Model. Int. J. Mol. Sci. 2016, 17, 1001. [Google Scholar] [CrossRef] [PubMed]
- Hajimiri, M.; Shahverdi, S.; Esfandiari, M.A.; Larijani, B.; Atyabi, F.; Rajabiani, A.; Dehpour, A.R.; Amini, M.; Dinarvand, R. Preparation of hydrogel embedded polymer-growth factor conjugated nanoparticles as a diabetic wound dressing. Drug Dev. Ind. Pharm. 2016, 42, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Takei, T.; Nakahara, H.; Tanaka, S.; Nishimata, H.; Yoshida, M.; Kawakami, K. Effect of chitosan-gluconic acid conjugate/poly(vinyl alcohol) cryogels as wound dressing on partial-thickness wounds in diabetic rats. J. Mater. Sci. Mater. Med. 2013, 24, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Moura, L.I.F.; Dias, A.M.A.; Carvalho, E.; de Sousa, H.C. Recent advances on the development of wound dressings for diabetic foot ulcer treatment—A review. Acta Biomater. 2013, 9, 7093–7114. [Google Scholar] [CrossRef] [PubMed]
- Fabiilli, M.L.; Wilson, C.G.; Padilla, F.; Martin-Saavedra, F.M.; Fowlkes, J.B.; Franceschi, R.T. Acoustic droplet-hydrogel composites for spatial and temporal control of growth factor delivery and scaffold stiffness. Acta Biomater. 2013, 9, 7399–7409. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.P.; Morgado, P.I.; Miguel, S.P.; Coutinho, P.; Correia, I.J. Dextran-based hydrogel containing chitosan microparticles loaded with growth factors to be used in wound healing. Mater. Sci. Eng. C 2013, 33, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.; Morck, D.; Schultz, C. Treatment of corneal defects with delayed re-epithelization with a medical device/drug delivery system for epidermal growth factor. Clin. Exp. Ophthalmol. 2012, 40, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Mishra, D.; Das, T.; Maiti, T.K. Wound pH-Responsive Sustained Release of Therapeutics from a Poly(NIPAAm-co-AAc) Hydrogel. J. Biomater. Sci. Polym. Ed. 2012, 23, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.W.; Byun, J.H.; Oh, S.H.; Kim, T.H.; Park, J.S.; Rho, G.J.; Lee, J.H. Multivalent ion-based in situ gelling polysaccharide hydrogel as an injectable bone graft. Carbohydr. Polym. 2018, 180, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Coletta, D.J.; Ibanez-Fonseca, A.; Missana, L.R.; Jammal, M.V.; Vitelli, E.J.; Aimone, M.; Zabalza, F.; Issa, J.P.M.; Alonso, M.; Rodriguez-Cabello, J.C.; et al. Bone Regeneration Mediated by a Bioactive and Biodegradable Extracellular Matrix-Like Hydrogel Based on Elastin-Like Recombinamers. Tissue Eng. Part A 2017, 23, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.A.; Jordan, J.; Roy, A.; Gottardi, R.; Fedorchak, M.V.; Kumta, P.N.; Little, S.R. Programmed Platelet-Derived Growth Factor-BB and Bone Morphogenetic Protein-2 Delivery from a Hybrid Calcium Phosphate/Alginate Scaffold. Tissue Eng. Part A 2017, 23, 1382–1393. [Google Scholar] [CrossRef] [PubMed]
- Santovena, A.; Monzon, C.; Delgado, A.; Evora, C.; Llabres, M.; Farina, J.B. Development of a standard method for in vitro evaluation of Triamcinolone and BMP-2 diffusion mechanism from thermosensitive and biocompatible composite hyaluronic acid-pluronic hydrogels. J. Drug Deliv. Sci. Technol. 2017, 42, 284–291. [Google Scholar] [CrossRef]
- Seo, B.B.; Koh, J.T.; Song, S.C. Tuning physical properties and BMP-2 release rates of injectable hydrogel systems for an optimal bone regeneration effect. Biomaterials 2017, 122, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Wang, J.L.; Wu, J.J.; Zhang, J.; Wan, Y.; Wu, H. Injectable hydrogels embedded with alginate microspheres for controlled delivery of bone morphogenetic protein-2. Biomed. Mater. 2016, 11, 025010. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.M.R.; Black, C.R.M.; Dawson, J.I.; Oreffo, R.O.C. A review of hydrogel use in fracture healing and bone regeneration. J. Tissue Eng. Regener. Med. 2016, 10, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, L.; Priddy, L.B.; Esancy, C.; Li, M.T.A.; Stevens, H.Y.; Jiang, X.; Tran, L.; Rowe, D.W.; Guldberg, R.E. Hydrogel-based Delivery of rhBMP-2 Improves Healing of Large Bone Defects Compared With Autograft. Clin. Orthop. Relat. Res. 2015, 473, 2885–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelbach, R.J.; Fransen, P.; Pulido, D.; D’Este, M.; Duttenhoefer, F.; Sauerbier, S.; Freiman, T.; Niemeyer, P.; Albericio, F.; Alini, M.; et al. Injectable Hyaluronan Hydrogels with Peptide-Binding Dendrimers Modulate the Controlled Release of BMP-2 and TGF-beta 1. Macromol. Biosci. 2015, 15, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Brockmeyer, P.; Kramer, K.; Krohn, S.; Kauffmann, P.; Mauth, C.; Dard, M.; Schliephake, H.; Gruber, R.M. Influence of synthetic polyethylene glycol hydrogels on new bone formation during mandibular augmentation procedures in Goettingen minipigs. J. Mater. Sci. Mater. Med. 2015, 26, 194. [Google Scholar] [CrossRef] [PubMed]
- Metzger, S.; Lienemann, P.S.; Ghayor, C.; Weber, W.; Martin, I.; Weber, F.E.; Ehrbar, M. Modular Poly(ethylene glycol) Matrices for the Controlled 3D-Localized Osteogenic Differentiation of Mesenchymal Stem Cells. Adv. Healthc. Mater. 2015, 4, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Song, C.C.; Du, F.S.; Li, Z.C. Supersensitive Oxidation-Responsive Biodegradable PEG Hydrogels for Glucose-Triggered Insulin Delivery. ACS Appl. Mater. Interfaces 2017, 9, 25905–25914. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.L.; Wei, W.; Li, J.J.; Zuo, G.C.; Pan, X.H.; Su, T.; Zhang, J.F.; Dong, W. Salecan-Based pH-Sensitive Hydrogels for Insulin Delivery. Mol. Pharm. 2017, 14, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, G.H.; Yu, W.J.; Liu, D.P.; Chen, H.; Liu, Y.K.; Huang, Q.; Tong, Z.Z.; Yao, J.M.; Kong, X.D. A composite hydrogel system containing glucose-responsive nanocarriers for oral delivery of insulin. Mater. Sci. Eng. C 2016, 69, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, X.Y.; Zhang, Y.; Shang, Q. Fabrication and evaluation of a novel polymeric hydrogel of carboxymethyl chitosan-g-polyacrylic acid (CMC-g-PAA) for oral insulin delivery. RSC Adv. 2016, 6, 52858–52867. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Mukherjee, D.; Mishra, R.; Kundu, P.P. Development of pH sensitive polyurethane-alginate nanoparticles for safe and efficient oral insulin delivery in animal models. RSC Adv. 2016, 6, 41835–41846. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Chakraborty, S.; Bhattacharya, S.; Mishra, R.; Kundu, P.P. pH-sensitive chitosan/alginate core-shell nanoparticles for efficient and safe oral insulin delivery. Int. J. Biol. Macromol. 2015, 72, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Cao, Y.; Song, X.F.; Zhang, Z.; Zhuang, X.L.; He, C.L.; Chen, X.S. Biodegradable, pH-Responsive Carboxymethyl Cellulose/Poly(Acrylic Acid) Hydrogels for Oral Insulin Delivery. Macromol. Biosci. 2014, 14, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, A.; Al-Remawi, M.; Maghrabi, I.; Hamaidi, M.; Jaber, N. Development of insulin loaded mesoporous silica injectable particles layered by chitosan as a controlled release delivery system. Int. J. Pharm. 2014, 461, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Dang, T.T.; Ma, M.L.; Tang, B.C.; Cheng, H.; Jiang, S.; Dong, Y.Z.; Zhang, Y.L.; Anderson, D.G. Glucose-Responsive Microgels Integrated with Enzyme Nanocapsules for Closed-Loop Insulin Delivery. ACS Nano 2013, 7, 6758–6766. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Sarkar, K.; Soam, S.; Kundu, P.P. Formulation of pH-responsive carboxymethyl chitosan and alginate beads for the oral delivery of insulin. J. Appl. Polym. Sci. 2013, 129, 835–845. [Google Scholar] [CrossRef]
- Liu, G.; Ma, R.J.; Ren, J.; Li, Z.; Zhang, H.X.; Zhang, Z.K.; An, Y.L.; Shi, L.Q. A glucose-responsive complex polymeric micelle enabling repeated on-off release and insulin protection. Soft Matter 2013, 9, 1636–1644. [Google Scholar] [CrossRef]
- Deat-Laine, E.; Hoffart, V.; Cardot, J.M.; Subirade, M.; Beyssac, E. Development and in vitro characterization of insulin loaded whey protein and alginate microparticles. Int. J. Pharm. 2012, 439, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Mundargi, R.C.; Rangaswamy, V.; Aminabhavi, T.M. pH-Sensitive oral insulin delivery systems using Eudragit microspheres. Drug Dev. Ind. Pharm. 2011, 37, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ma, G.H.; Wang, L.Y.; Wu, J.; Su, Z.G. Hollow quaternized chitosan microspheres increase the therapeutic effect of orally administered insulin. Acta Biomater. 2010, 6, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Builders, P.F.; Kunle, O.O.; Okpaku, L.C.; Builders, M.O.; Attama, A.A.; Adikwu, M.U. Preparation and evaluation of mucinated sodium alginate microparticles for oral delivery of insulin. Eur. J. Pharm. Biopharm. 2008, 70, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, A.; Pruthi, V. Oral insulin delivery by polymeric nanospheres. J. Biomed. Nanotechnol. 2007, 3, 68–74. [Google Scholar] [CrossRef]
- Sajeesh, S.; Sharma, C.P. Interpolymer complex microparticles based on polymethacrylic acid-chitosan for oral insulin delivery. J. Appl. Polym. Sci. 2006, 99, 506–512. [Google Scholar]
- Sajeesh, S.; Sharma, C.P. Poly methacrylic acid-alginate semi-IPN microparticles for oral delivery of insulin: A preliminary investigation. J. Biomater. Appl. 2004, 19, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.K.K.; Sharma, C.P. Oral insulin—A perspective. J. Biomater. Appl. 2003, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Victor, S.P.; Sharma, C.P. Stimuli sensitive polymethacrylic acid microparticles (PMAA)—Oral insulin delivery. J. Biomater. Appl. 2002, 17, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Wu, Y.D.; Ye, H.B.; Yu, S.J.; He, C.L.; Chen, X.S. Interleukin-15 and cisplatin co-encapsulated thermosensitive polypeptide hydrogels for combined immuno-chemotherapy. J. Control. Release 2017, 255, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Bobbala, S.; Tamboli, V.; McDowell, A.; Mitra, A.K.; Hook, S. Novel Injectable Pentablock Copolymer Based Thermoresponsive Hydrogels for Sustained Release Vaccines. AAPS J. 2016, 18, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Walke, S.; Srivastava, G.; Nikalje, M.; Doshi, J.; Kumar, R.; Ravetkar, S.; Doshi, P. Fabrication of chitosan microspheres using vanillin/TPP dual crosslinkers for protein antigens encapsulation. Carbohydr. Polym. 2015, 128, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.H.; Li, S.; Howard, O.M.Z.; Dunlap, M.; Trivett, A.; Schneider, J.P.; Oppenheim, J.J. Enhanced immunostimulatory effects of DNA-encapsulated peptide hydrogels. Biomaterials 2015, 53, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Hariyadi, D.M.; Ma, Y.; Wang, Y.; Bostrom, T.; Malouf, J.; Turner, M.S.; Bhandari, B.; Coombes, A.G.A. The potential for production of freeze-dried oral vaccines using alginate hydrogel microspheres as protein carriers. J. Drug Deliv. Sci. Technol. 2014, 24, 178–184. [Google Scholar] [CrossRef]
- Yun, Y.H.; Goetz, D.J.; Yellen, P.; Chen, W.L. Hyaluronan microspheres for sustained gene delivery and site-specific targeting. Biomaterials 2004, 25, 147–157. [Google Scholar] [CrossRef]
- Murthy, N.; Xu, M.C.; Schuck, S.; Kunisawa, J.; Shastri, N.; Frechet, J.M.J. A macromolecular delivery vehicle for protein-based vaccines: Acid-degradable protein-loaded microgels. Proc. Natl. Acad. Sci. USA 2003, 100, 4995–5000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowersock, T.L.; HogenEsch, H.; Torregrosa, S.; Borie, D.; Wang, B.; Park, H.; Park, K. Induction of pulmonary immunity in cattle by oral administration of ovalbumin in alginate microspheres. Immunol. Lett. 1998, 60, 37–43. [Google Scholar] [CrossRef]
- Fletcher, N.A.; Babcock, L.R.; Murray, E.A.; Krebs, M.D. Controlled delivery of antibodies from injectable hydrogels. Mater. Sci. Eng. C 2016, 59, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Blasi, L.; Argentiere, S.; Morello, G.; Palama, I.; Barbarella, G.; Cingolani, R.; Gigli, G. Uptake and distribution of labeled antibodies into pH-sensitive microgels. Acta Biomater. 2010, 6, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Lira, J.C.; Camacho-Frias, E.; Pena-Alvarez, A.; Vera-Avila, L.E. Preparation and characterization of a sol-gel immunosorbent doped with 2,4-D antibodies. Chem. Mater. 2003, 15, 154–161. [Google Scholar] [CrossRef]
- Markowitz, M.A.; Turner, D.C.; Martin, B.D.; Gaber, B.P. Diffusion and transfer of antibody proteins from a sugar-based hydrogel. Appl. Biochem. Biotechnol. 1997, 68, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in form of slabs, sphere, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Zarzycki, R.; Modrzejewska, Z.; Nawrotek, K. Drug release from hydrogel matrices. Ecol. Chem. Eng. S 2010, 17, 117–136. [Google Scholar]
- Cohen, M.H.; Turnbull, D. Molecular transport in liquids and glasses. J. Chem. Phys. 1959, 31, 1164–1169. [Google Scholar] [CrossRef]
- Masaro, L.; Zhu, X.X. Physical models of diffusion for polymer solutions, gels and solids. Prog. Polym. Sci. 1999, 24, 731–775. [Google Scholar] [CrossRef]
- Bird, R.B.; Stewart, W.E.; Lightfoot, E.N. Transport Phenomena; John Wiley and Sons: Toronto, ON, Canada, 1960. [Google Scholar]
- Mackie, J.S.; Meares, P. The diffusion of electrolytes in a cation-exchange resin membrane I. Theoretical. Proc. R. Soc. A 1955, 232, 498–509. [Google Scholar] [CrossRef]
- Amsden, B. Solute Diffusion within Hydrogels. Mechanisms and Models. Macromolecules 1998, 31, 8382–8395. [Google Scholar] [CrossRef]
- Amsden, B. An Obstruction-Scaling Model for Diffusion in Homogenous Hydrogels. Macromolecules 1999, 32, 874–879. [Google Scholar] [CrossRef]
- Lustig, S.R.; Peppas, N.A. Solute Diffusion in Swollen Membranes. IX. Scaling Laws for Solute Diffusion in Gels. J. Appl. Polym. Sci. 1988, 36, 735–747. [Google Scholar] [CrossRef]
- Cukier, R.I. Diffusion of Brownian spheres in semidilute polymer solutions. Macromolecules 1984, 17, 252–255. [Google Scholar] [CrossRef]
- Phillips, R.J.; Deen, W.M.; Brady, J.F. Hindered transport of spherical macromolecules in fibrous membranes and gels. AIChE J. 1989, 35, 1761–1769. [Google Scholar] [CrossRef]
- Jackson, G.W.; James, D.F. The permeability of fibrous porous media. Can. J. Chem. Eng. 1986, 64, 364–374. [Google Scholar] [CrossRef]
- Ogston, A.G. The spaces in a uniform random suspension of fibres. Trans. Faraday Soc. 1958, 54, 1754–1757. [Google Scholar] [CrossRef]
- Dini, C.; Islan, G.A.; Urraza, P.J.D.; Castro, G.R. Novel Biopolymer Matrices for Microencapsulation of Phages: Enhanced Protection Against Acidity and Protease Activity. Macromol. Biosci. 2012, 12, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Dini, C.; Islan, G.A.; Castro, G.R. Characterization and Stability Analysis of Biopolymeric Matrices Designed for Phage-Controlled Release. Appl. Biochem. Biotechnol. 2014, 174, 2031–2047. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Taylor, C.M. What’s new in haemolytic uraemic syndrome? Eur. J. Pediatr. 2008, 167, 965. [Google Scholar] [CrossRef] [PubMed]
- Dini, C.; Urraza, P.J.D. Isolation and selection of coliphages as potential biocontrol agents of enterohemorrhagic and Shiga toxin-producing E. coli (EHEC and STEC) in cattle. J. Appl. Microbiol. 2010, 109, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.E.; Lamont, J.T. Clostridium difficile infection: A worldwide disease. Gut Liver 2014, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bouza, E.S. Consequences of Clostridium difficile infection: Understanding the healthcare burden. Clin. Microbiol. Infect. 2012, 18, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Polyzos, K.; Patouni, K.; Rafailidis, P.I.; Samonis, G.; Falagas, M.E. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: A systematic review of the evidence. Int. J. Antimicrob. Agents 2012, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vinner, G.K.; Vladisavljević, G.T.; Clokie, M.R.J.; Malik, D.J. Microencapsulation of Clostridium difficile specific bacteriophages using microfluidic glass capillary devices for colon delivery using pH triggered release. PLoS ONE 2017, 12, e0186239. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.M.; Stickler, D.J.; Mobley, H.L.T.; Shirtliff, M.E. Complicated catheter-associated urinary tract infections due to Escherichia coli and Proteus mirabilis. Clin. Microbiol. Rev. 2008, 21, 26–59. [Google Scholar] [CrossRef] [PubMed]
- Stickler, D.J.; Morgan, S.D. Modulation of crystalline Proteus mirabilis biofilm development on urinary catheters. J. Med. Microbiol. 2006, 55, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, C.; Mobley, H. Merging mythology and morphology: The multifaceted lifestyle of Proteus mirabilis. Nat. Rev. Microbiol. 2012, 10, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Milo, S.; Hathaway, H.; Nzakizwanayo, J.; Alves, D.R.; Esteban, P.P.; Jonesbe, B.V.; Jenkins, A.T.A. Prevention of encrustation and blockage of urinary catheters by Proteus mirabilis via pH-triggered release of bacteriophage. J. Mater. Chem. B 2017, 5, 5403–5411. [Google Scholar] [CrossRef]
- Skoumal, M.; Seidlits, S.; Shin, S.; Shea, L. Localized Lentivirus Delivery Via Peptide Interactions. Biotechnol. Bioeng. 2016, 113, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.J.; Retamales, J.; Palza, H.; Bastías, R. Encapsulation of specific Salmonella Enteritidis phage f3αSE on alginate-spheres as a method for protection and dosification. Electron. J. Biotechnol. 2018, 31, 57–60. [Google Scholar] [CrossRef]
- Bean, J.E.; Alves, D.R.; Laabei, M.; Esteban, P.P.; Thet, N.T.; Enright, M.C.; Jenkins, A.T.A. Triggered Release of Bacteriophage K from Agarose/Hyaluronan Hydrogel Matrixes by Staphylococcus aureus Virulence Factors. Chem. Mater. 2014, 26, 7201–7208. [Google Scholar] [CrossRef] [Green Version]
- Fuller, R. Probiotics in man and animals. J. Appl. Bacteriol. 1989, 66, 365–378. [Google Scholar] [PubMed]
- Hood, S.K.; Zottola, E.A. Effect of low pH on the ability of Lactobacillus acidophilus to survive and adhere to human intestinal-cells. J. Food Sci. 1988, 53, 1514–1516. [Google Scholar] [CrossRef]
- Hughes, D.B.; Hoover, D.G. Bifidobacteria: Their potential for use in American dairy products. Food Technol. 1991, 45, 74–83. [Google Scholar]
- Shah, N.P.; Lankaputhra, W.E.V. Improving viability of Lactobacillus acidophilus and Bifidobacterium spp. in yogurt. Int. Dairy J. 1997, 7, 349–356. [Google Scholar] [CrossRef]
- Argin, S.; Kofinas, P.; Lo, Y.M. The cell release kinetics and the swelling behavior of physically crosslinked xanthanechitosan hydrogels in simulated gastrointestinal conditions. Food Hydrocoll. 2014, 40, 138–144. [Google Scholar] [CrossRef]
- Le Lay, C.; Dridi, L.; Bergeron, M.G.; Ouellette, M.; Fliss, I.L. Nisin is an effective inhibitor of Clostridium difficile vegetative cells and spore germination. J. Med. Microbiol. 2016, 65, 169–175. [Google Scholar] [PubMed]
- Xu, M.; Gagné-Bourque, F.; Dumont, M.-J.; Jabaji, S. Encapsulation of Lactobacillus casei ATCC 393 cells and evaluation of their survival after freeze-drying, storage and under gastrointestinal conditions. J. Food Eng. 2016, 168, 52–59. [Google Scholar] [CrossRef]
- Yeung, T.W.; Üçok, E.F.; Tiani, K.A.; McClements, D.J.; Sela, D.A. Microencapsulation in Alginate and Chitosan Microgels to Enhance Viability of Bifidobacterium longum for Oral Delivery. Front. Microbiol. 2016, 7, 494. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Zhang, Z. Bioencapsulation by compression coating of probiotic bacteria for their protection in an acidic medium. Process Biochem. 2005, 40, 3346–3351. [Google Scholar] [CrossRef]
- Strand, B.L.; Coron, A.E.; Skjak-Braek, G. Current and Future Perspectives on Alginate Encapsulated Pancreatic Islet. STEM CELLS Transl. Med. 2017, 6, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon-Shiong, P.; Heintz, R.E.; Merideth, N.; Yao, Q.X.; Yao, Z.; Zheng, T.; Murphy, M.; Moloney, M.K.; Schmehl, M.; Harris, M.; et al. Insulin independence in a type 1 diabetic patient after encapsulated islet transplantation. Lancet 1994, 343, 950–951. [Google Scholar] [CrossRef]
- Soon-Shiong, P.; Feldman, E.; Nelson, R.; Heintz, R.; Yao, Q.; Yao, Z.; Zheng, T.; Merideth, N.; Skjak-Braek, G.; Espevik, T. Long-term reversal of diabetes by the injection of immunoprotected islets. Proc. Natl. Acad. Sci. USA 1993, 90, 5843–5847. [Google Scholar] [CrossRef] [PubMed]
- Strand, B.L.; Ryan, T.L.; In’t Veld, P.; Kulseng, B.; Rokstad, A.M.; Skjak-Brek, G.; Espevik, T. Poly-L-Lysine induces fibrosis on alginate microcapsules via the induction of cytokines. Cell Transplant. 2001, 10, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.S.; Cruise, G.M.; Hager, S.R.; Lamberti, F.V.; Yu, X.; Garufis, C.L.; Yu, Y.; Mundwiler, K.E.; Cole, J.F.; Hubbell, J.A.; et al. Immunoisolation of adult porcine islets for the treatment of diabetes mellitus. The use of photopolymerizable polyethylene glycol in the conformal coating of mass-isolated porcine islets. Ann. N. Y. Acad. Sci. 1997, 831, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Kızılel, S. Theoretical and Experimental Investigation for Interfacial Photopolymerization of Poly(ethylene glycol) Diacrylate. Ph.D. Thesis, Illinois Institute of Technology, Chicago, IL, USA, 2004. [Google Scholar]
- Kizilel, S.; Perez-Luna, V.H.; Teymour, F. Mathematical model for surface-initiated photopolymerization of poly(ethylene glycol) diacrylate. Macromol. Theory Simul. 2006, 15, 686–700. [Google Scholar] [CrossRef]
- Betre, H.; Setton, L.A.; Meyer, D.E.; Chilkoti, A. Characterization of a genetically engineered elastin-like polypeptide for cartilaginous tissue repair. Biomacromolecules 2002, 3, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Anseth, K.S.; Lee, D.A.; Bader, D.L. Crosslinking density influences the morphology of chondrocytes photoencapsulated in PEG hydrogels during the application of compressive strain. J. Orthop. Res. 2004, 22, 1143–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, S.J.; Anseth, K.S. Controlling the spatial distribution of ECM components in degradable PEG hydrogels for tissue engineering cartilage. J. Biomed. Mater. Res. Part A 2003, 64, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Anseth, K.S. Hydrogel properties influence ECM production by chondrocytes photoencapsulated in poly(ethylene glycol) hydrogels. J. Biomed. Mater. Res. 2002, 59, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Nuttelman, C.R.; Anseth, K.S. The effects of crosslinking density on cartilage formation in photocrosslinkable hydrogels. Biomed. Sci. Instrum. 1999, 35, 309–314. [Google Scholar] [PubMed]
- Bidarra, S.J.; Barrias, C.C.; Granja, P.L. Injectable alginate hydrogels for cell delivery in tissue engineering. Acta Biomater. 2014, 10, 1646–1662. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Diniz, I.M.; Chen, C.; Aghaloo, T.; Wu, B.M.; Shi, S.; Moshaverinia, A. Alginate/hyaluronic acid hydrogel delivery system characteristics regulate the differentiation of periodontal ligament stem cells toward chondrogenic lineage. J. Mater. Sci. Mater. Med. 2017, 28, 162. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Anseth, K.S. The effects of scaffold thickness on tissue engineered cartilage in photocrosslinked poly(ethylene oxide) hydrogels. Biomaterials 2001, 22, 619–626. [Google Scholar] [CrossRef]
- Nuttelman, C.R.; Benoit, D.S.W.; Tripodi, M.C.; Anseth, K.S. The effect of ethylene glycol methacrylate phosphate in PEG hydrogels on mineralization and viability of encapsulated hMSCs. Biomaterials 2006, 27, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Nuttelman, C.R.; Tripodi, M.C.; Anseth, K.S. Dexamethasone-functionalized gels induce osteogenic differentiation of encapsulated hMSCs. J. Biomed. Mater. Res. Part A 2006, 76, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Nuttelman, C.R.; Tripodi, M.C.; Anseth, K.S. In vitro osteogenic differentiation of human mesenchymal stem cells photoencapsulated in PEG hydrogels. J. Biomed. Mater. Res. Part A 2004, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Betre, H.; Ong, S.R.; Guilak, F.; Chilkoti, A.; Fermor, B.; Setton, L.A. Chondrocytic differentiation of human adipose-derived adult stem cells in elastin-like polypeptide. Biomaterials 2005, 27, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, L.; Sun, M.; Zhang, Y.; Chen, L.; Rong, Y.; Li, Y. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res. Ther. 2015, 6, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Arhaa, M.; Choudhary, S.; Ashton, R.S.; Bhatia, S.R.; Schaffer, D.V.; Kane, R.S. The influence of hydrogel modulus on the proliferation and differentiation of encapsulated neural stem cells. Biomaterials 2009, 30, 4695–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldshmid, R.; Seliktar, D. Hydrogel Modulus Affects Proliferation Rate and Pluripotency of Human Mesenchymal Stem Cells Grown in Three-Dimensional Culture. ACS Biomater. Sci. Eng. 2017, 7, 3433–3446. [Google Scholar] [CrossRef]
- Alakpa, E.V.; Jayawarna, V.; Lampel, A.; Burgess, K.V.; West, C.C.; Bakker, S.C.J.; Roy, S.; Javid, N.; Fleming, S.; Lamprou, D.A.; et al. Tunable Supramolecular Hydrogels for Selection of Lineage-Guiding Metabolites in Stem Cell Cultures. Chem 2016, 1, 298–319. [Google Scholar] [CrossRef] [Green Version]
- Lanniel, M.; Huq, E.; Allen, S.; Buttery, L.; Williams, P.M.; Alexander, M.R. Substrate induced differentiation of human mesenchymal stem cells on hydrogels with modified surface chemistry and controlled modulus. Soft Matter 2011, 7, 6501–6514. [Google Scholar] [CrossRef]
- Hasan, A.; Khattab, A.; Islam, M.A.; Hweij, K.A.; Zeitouny, J.; Waters, R.; Sayegh, M.; Hossain, M.M.; Paul, A. Injectable Hydrogels for Cardiac Tissue Repair after Myocardial Infarction. Adv. Sci. 2015, 2, 1500122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Marchant, R.E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, M.; Vaibavi, S.R.; Rufaihah, A.J.; Nithya, V.; Wang, J.; Shachaf, Y.; Kofidis, T.; Seliktar, D. The effect of matrix stiffness of injectable hydrogels on the preservation of cardiac function after a heart attack. Biomaterials 2014, 35, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Kim, I.K.; Cho, S.W.; Cho, M.C.; Hwang, K.K.; Piao, H.; Piao, S.; Lim, S.H.; Hong, Y.S.; Choi, C.Y.; et al. Implantation of bone marrow mononuclear cells using injectable fibrin matrix enhances neovascularization in infarcted myocardium. Biomaterials 2005, 26, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hu, Q.; Braunlin, E.A.; Suggs, L.J.; Zhang, J. Enhancing efficacy of stem cell transplantation to the heart with a PEGylated fibrin biomatrix. Tissue Eng. Part A 2008, 14, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zheng, J.; Wang, H.; Becker, M.L.; Leipzig, N.D. Neural stem cell encapsulation and differentiation in strain promoted crosslinked polyethylene glycol-based hydrogels. J. Biomater. Appl. 2018, 32, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wijekoon, A.; Leipzig, N.D. 3D Differentiation of Neural Stem Cells in Macroporous Photopolymerizable Hydrogel Scaffolds. PLoS ONE 2012, 7, e48824. [Google Scholar] [CrossRef] [PubMed]
- Aurand, E.R.; Wagner, J.L.; Shandas, R.; Bjugstad, K.B. Hydrogel formulation determines cell fate of fetal and adult neural progenitor cells. Stem Cell Res. 2014, 12, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Zisch, A.H.; Lutolf, M.P.; Ehrbar, M.; Raeber, G.P.; Rizzi, S.C.; Davies, N.; Schmoekel, H.; Bezuidenhout, D.; Djonov, V.; Zilla, P.; et al. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB J. 2003, 17, 2260–2262. [Google Scholar] [CrossRef] [PubMed]
- Masters, K.S.; Shah, D.N.; Walker, G.; Leinwand, L.A.; Anseth, K.S. Designing scaffolds for valvular interstitial cells: Cell adhesion and function on naturally derived materials. J. Biomed. Mater. Res. Part A 2004, 71, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-G.; Itle, L.J.; Pishko, M.V. Molding of hydrogel microstructures to create multiphenotype cell microarrays. Anal. Chem. 2003, 75, 5783–5789. [Google Scholar] [CrossRef] [PubMed]
- Zguris, J.C.; Itle, L.J.; Koh, W.-G.; Pishko, M.V. A Novel Single-Step Fabrication Technique to Create Heterogeneous Poly(ethylene glycol) Hydrogel Microstructures Containing Multiple Phenotypes of Mammalian Cells. Langmuir 2005, 21, 4168–4174. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-G.; Pishko, M. Photoreaction Injection Molding of Biomaterial Microstructures. Langmuir 2003, 19, 10310–10316. [Google Scholar] [CrossRef]
- Zisch, A.H.; Lutolf, M.P.; Hubbell, J.A. Biopolymeric delivery matrices for angiogenic growth factors. Cardiovasc. Pathol. 2003, 12, 295–310. [Google Scholar] [CrossRef]
- Burdick, J.A.; Mason, M.N.; Hinman, A.D.; Thorne, K.; Anseth, K.S. Delivery of osteoinductive growth factors from degradable PEG hydrogels influences osteoblast differentiation and mineralization. J. Control. Release 2002, 83, 53–63. [Google Scholar] [CrossRef]
- Zisch, A.H.; Zeisberger, S.M.; Ehrbar, M.; Djonov, V.; Weber, C.C.; Ziemiecki, A.; Pasquale, E.B.; Hubbell, J.A. Engineered fibrin matrixes for functional display of cell membrane-bound growth factor-like activities: Study of angiogenic signaling by ephrin-B2. Biomaterials 2004, 25, 3245–3257. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.K.A.; Richard, C.; Bessodes, M.; Scherman, D.; Merten, O.W. Growth Factor Delivery Approaches in Hydrogels. Biomacromolecules 2009, 10, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Ward, M.; Liang, E.; Young, M.J.; Langer, R. Stimulation of neurite outgrowth by neurotrophins delivered from degradable hydrogels. Biomaterials 2006, 27, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kang, Y.M.; Kang, K.N.; Kim, D.Y.; Kim, H.J.; Min, B.H.; Kim, J.H.; Kim, M.S. Wound Dressings for Wound Healing and Drug Delivery. Tissue Eng. Regener. Med. 2011, 8, 1–7. [Google Scholar]
- Itle, L.J.; Koh, W.-G.; Pishko, M.V. Multi-phenotypic cellular arrays for biosensing. BioMEMS Biomed. Nanotechnol. 2006, 3, 79–93. [Google Scholar]
- Pishko, M.V. Cell based bioassays in microfluidic systems. Polym. Prepr. 2006, 47, 1076–1077. [Google Scholar]
- Hoffman, A.S.; Stayton, P.S.; Bulmus, V.; Chen, G.; Chen, J.; Cheung, C.; Chilkoti, A.; Ding, Z.; Dong, L.; Fong, R.; et al. Founder’s Award, Society for Biomaterials. Sixth World Biomaterials Congress 2000, Kamuela, HI, May 15–20, 2000. Really smart bioconjugates of smart polymers and receptor proteins. J. Biomed. Mater. Res. 2000, 52, 577–586. [Google Scholar] [CrossRef]
- Itle, L.J.; Zguris, J.C.; Pishko, M.V. Cell-based bioassays in microfluidic systems. Proc. SPIE Int. Soc. Opt. Eng. 2004, 5588, 9–18. [Google Scholar]
- Kulkarni, R.V.; Biswanath, S. Electrically responsive smart hydrogels in drug delivery: A review. J. Appl. Biomater. Biomech. 2007, 5, 125–139. [Google Scholar] [PubMed]
- Velasco, D.; Elvira, C.; San Roman, J. New stimuli-responsive polymers derived from morpholine and pyrrolidine. J. Mater. Sci. Mater. Med. 2008, 19, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Prabaharan, M.; Mano, J.F. Stimuli-responsive hydrogels based on polysaccharides incorporated with thermo-responsive polymers as novel biomaterials. Macromol. Biosci. 2006, 6, 991–1008. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hydrogels can present barriers that prevent recognition of encapsulated viruses, bacteria, or cells by the immune system. The hydrogel can also provide an internal environment that allows the encapsulated cells to survive changes in environment.
Figure 1.
Hydrogels can present barriers that prevent recognition of encapsulated viruses, bacteria, or cells by the immune system. The hydrogel can also provide an internal environment that allows the encapsulated cells to survive changes in environment.

Figure 2.
IgG molecules entrapped within a degradable hydrogel. (A) The IgG molecules are encapsulated by means of physical entrapment. (B) Entrapped IgG molecules are covalently linked to the hydrogel. (C) Release of encapsulated IgG molecules (physically entrapped) versus time. Burst release (of IgG in this case) occurs when biomolecules are encapsulated within hydrogels (A). (D) Release of encapsulated IgG molecules that were covalently linked to the hydrogel. The burst release was delayed when the antibodies were linked to the hydrogel by means of covalent bonds using bifunctional PEG molecules (B). Adapted and reproduced with permission from [129].
Figure 2.
IgG molecules entrapped within a degradable hydrogel. (A) The IgG molecules are encapsulated by means of physical entrapment. (B) Entrapped IgG molecules are covalently linked to the hydrogel. (C) Release of encapsulated IgG molecules (physically entrapped) versus time. Burst release (of IgG in this case) occurs when biomolecules are encapsulated within hydrogels (A). (D) Release of encapsulated IgG molecules that were covalently linked to the hydrogel. The burst release was delayed when the antibodies were linked to the hydrogel by means of covalent bonds using bifunctional PEG molecules (B). Adapted and reproduced with permission from [129].

Figure 3.
(Top) Concept of encapsulation of bacteriophages within an ionic hydrogel. At low pH, the carboxylate groups remain protonated, and the hydrogel does not swell with the acidic fluids of the stomach. Once the particles reach the small intestine, the change in pH causes the carboxyl groups to become deprotonated (hence changed and more polar), which causes the hydrogel to swell and release the encapsulated bacteriophages. (Bottom) Number of plaque forming units from a system with encapsulated bacteriophages after 0 h (T0), 6 h (T6), and 24 h (T24) of residence within the hydrogel particles at the given pH values. (* indicates significantly different phage titres using a 2 sample t-test at each condition compared with phage at T0 for each composition (p < 0.05).). (Adapted and reproduced with permission from [206].)
Figure 3.
(Top) Concept of encapsulation of bacteriophages within an ionic hydrogel. At low pH, the carboxylate groups remain protonated, and the hydrogel does not swell with the acidic fluids of the stomach. Once the particles reach the small intestine, the change in pH causes the carboxyl groups to become deprotonated (hence changed and more polar), which causes the hydrogel to swell and release the encapsulated bacteriophages. (Bottom) Number of plaque forming units from a system with encapsulated bacteriophages after 0 h (T0), 6 h (T6), and 24 h (T24) of residence within the hydrogel particles at the given pH values. (* indicates significantly different phage titres using a 2 sample t-test at each condition compared with phage at T0 for each composition (p < 0.05).). (Adapted and reproduced with permission from [206].)

Figure 4.
Fractional release of P. acidilactici from xanthan–chitosan capsules prepared under four different conditions. The release pH conditions are pH = 2.0 for SGF and pH = 6.8 for SIF. (a) Chitosan: 0.7% (w/v), pH = 6.2; Xanthan: 0.7% (w/v) and 1.0% (w/v); (b) Chitosan: 0.7% (w/v), pH = 4.5; Xanthan: 0.7% (w/v) and 1.0% (w/v). (Reproduced and adapted with permission from [218].)
Figure 4.
Fractional release of P. acidilactici from xanthan–chitosan capsules prepared under four different conditions. The release pH conditions are pH = 2.0 for SGF and pH = 6.8 for SIF. (a) Chitosan: 0.7% (w/v), pH = 6.2; Xanthan: 0.7% (w/v) and 1.0% (w/v); (b) Chitosan: 0.7% (w/v), pH = 4.5; Xanthan: 0.7% (w/v) and 1.0% (w/v). (Reproduced and adapted with permission from [218].)

Figure 5.
Porcine islet cells encapsulated with a PEG diacrylate-based hydrogel using interfacial photopolymerization. The thin, outermost zone (labeled A) was presumed to be less crosslinked, and the filamentous nature of this outer zone may be due to the incomplete crosslinking of the hydrogel at the termination of laser illumination. Because the photoinitiator was present only at the islet surface, the thicker inner zone, closer to the islets (labeled B) and thus closer to the eosin Y photoinitiator, was presumably more highly crosslinking and thus had a more dense appearance. (Reproduced and adapted with permission from [20].)
Figure 5.
Porcine islet cells encapsulated with a PEG diacrylate-based hydrogel using interfacial photopolymerization. The thin, outermost zone (labeled A) was presumed to be less crosslinked, and the filamentous nature of this outer zone may be due to the incomplete crosslinking of the hydrogel at the termination of laser illumination. Because the photoinitiator was present only at the islet surface, the thicker inner zone, closer to the islets (labeled B) and thus closer to the eosin Y photoinitiator, was presumably more highly crosslinking and thus had a more dense appearance. (Reproduced and adapted with permission from [20].)

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pérez-Luna, V.H.; González-Reynoso, O. Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications. Gels 2018, 4, 61. https://doi.org/10.3390/gels4030061
AMA Style
Pérez-Luna VH, González-Reynoso O. Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications. Gels. 2018; 4(3):61. https://doi.org/10.3390/gels4030061
Chicago/Turabian StylePérez-Luna, Víctor H., and Orfil González-Reynoso. 2018. "Encapsulation of Biological Agents in Hydrogels for Therapeutic Applications" Gels 4, no. 3: 61. https://doi.org/10.3390/gels4030061
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.