Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin †

 , and
, and
Abstract
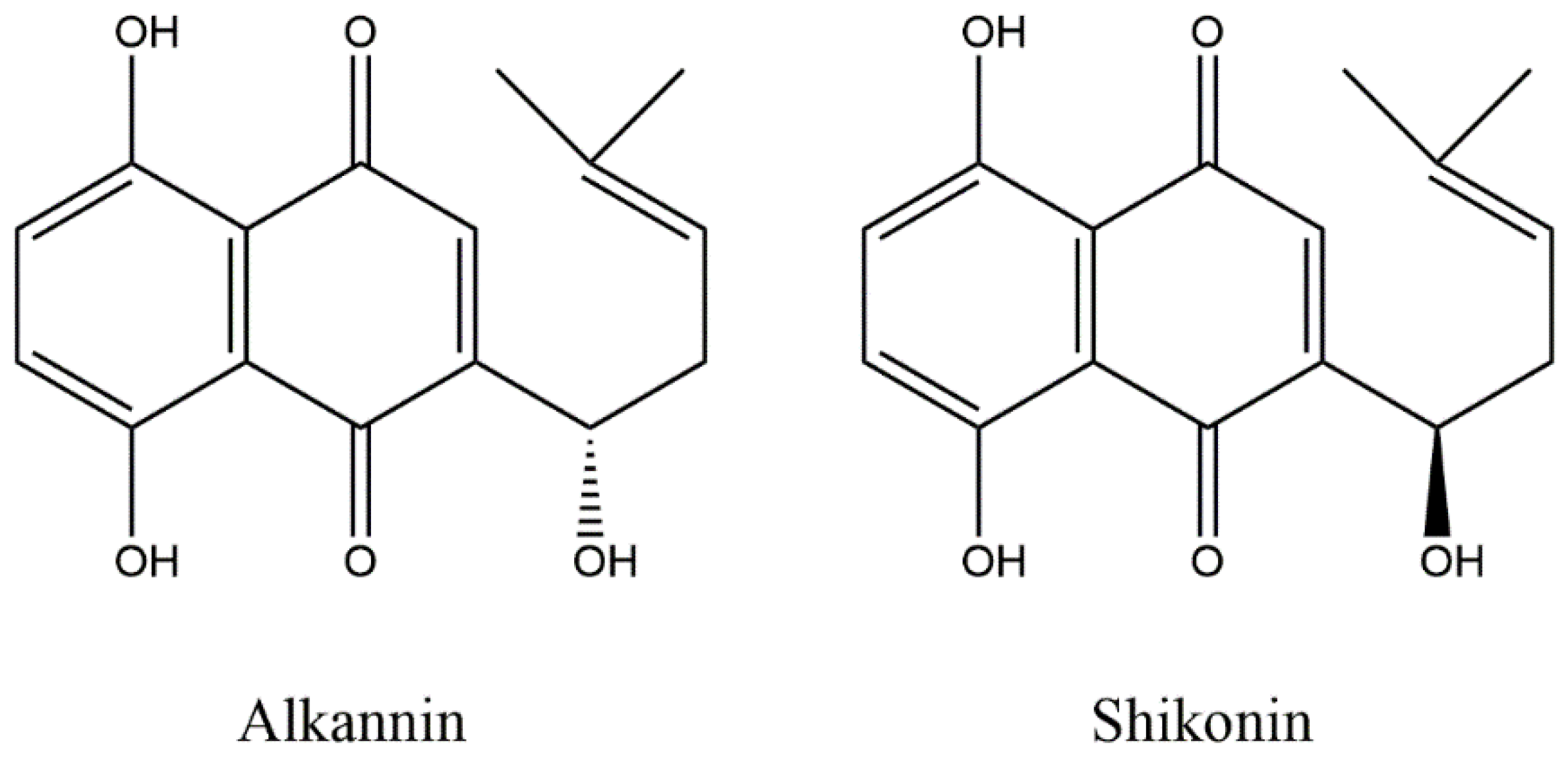
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Liposome Preparation
2.3. Characterization of Shikonin-Loaded Liposomes
2.3.1. Particle Size and ζ-Potential
2.3.2. Entrapment Efficiency
2.3.3. In Vitro Release
2.4. Stability
2.5. Statistical Analysis
3. Results and Discussion
3.1. Liposome Characterization
3.1.1. Particle Size Measurement
3.1.2. ζ-Potential
3.1.3. Entrapment Efficiency
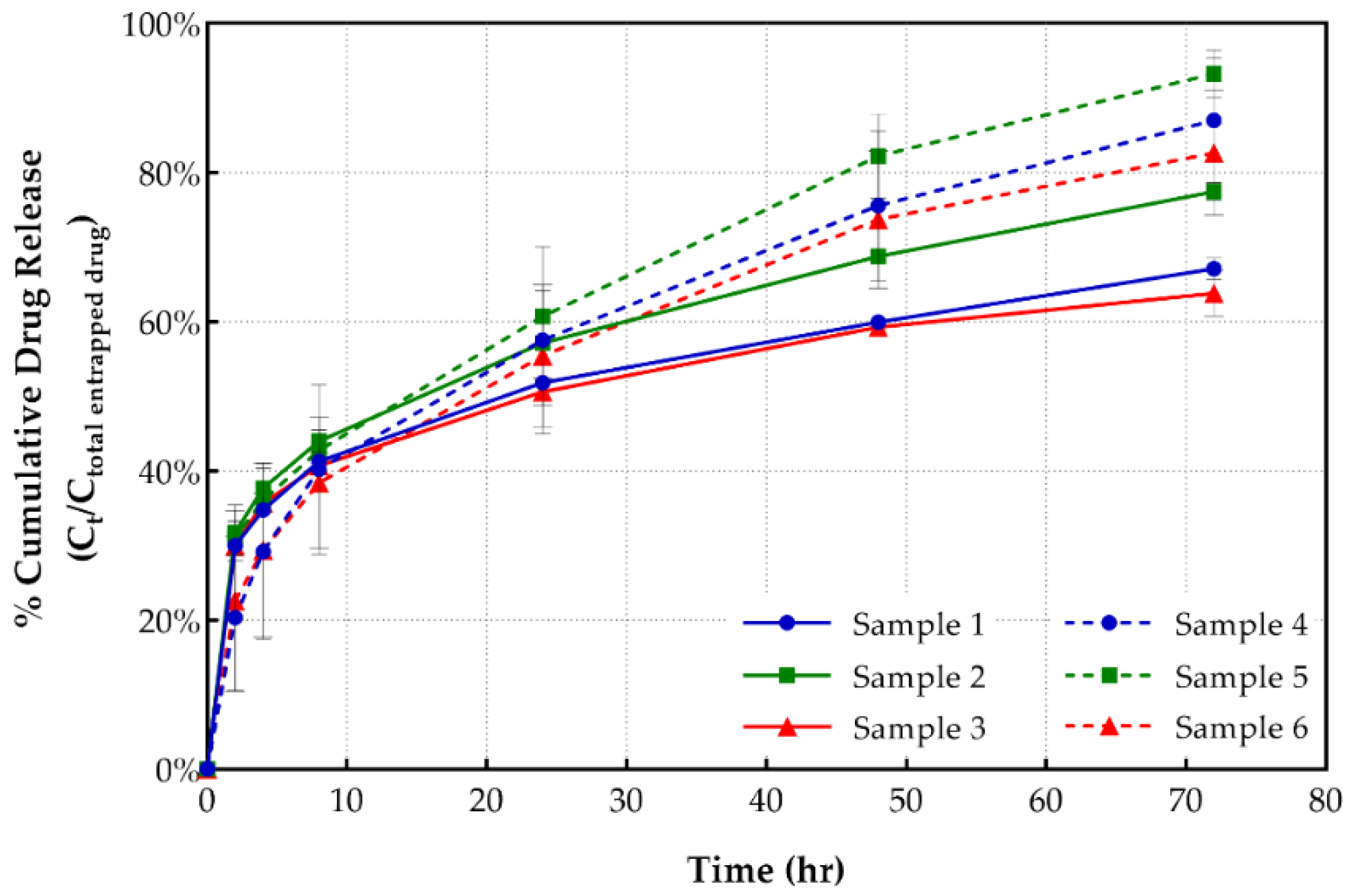
3.1.4. In Vitro Drug Release
3.2. Stability Study
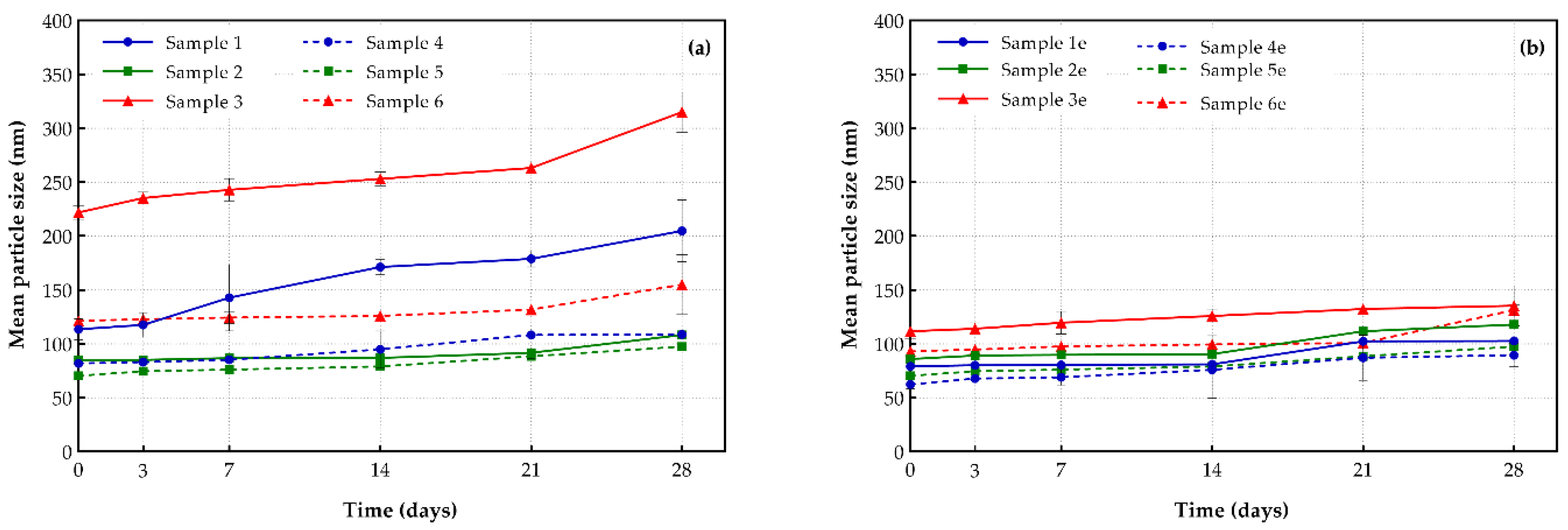
3.2.1. Particle Size Distribution
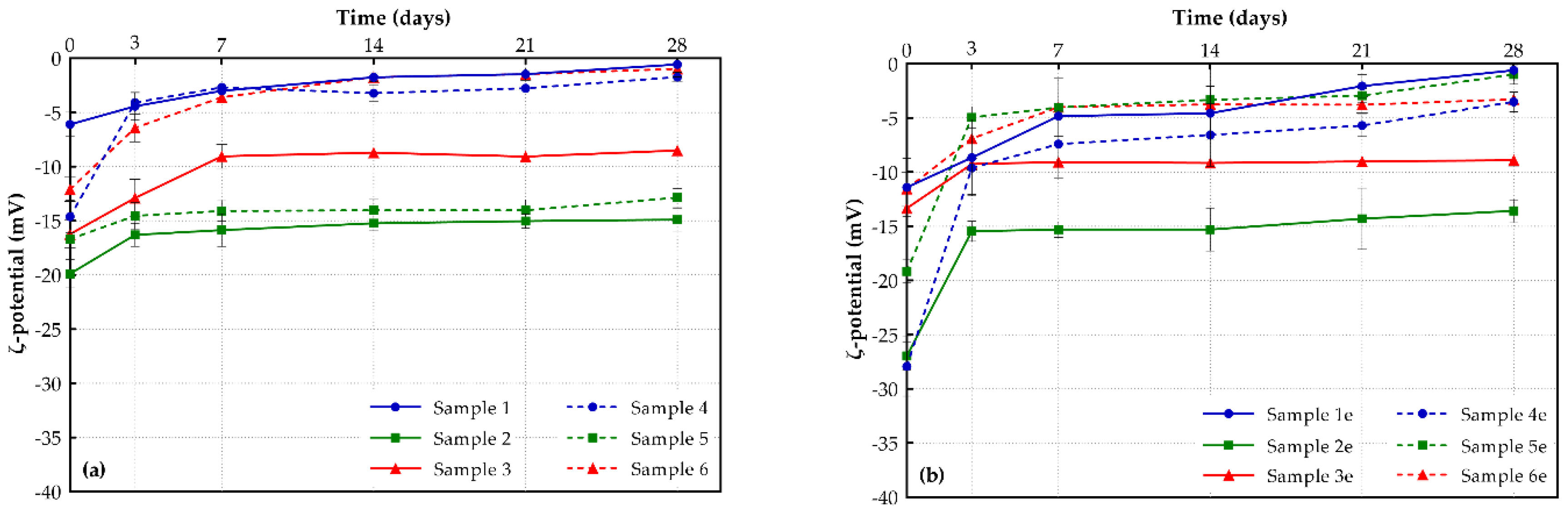
3.2.2. ζ-Potential
3.2.3. Drug Leakage
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ordoudi, S.A.; Tsermentseli, S.K.; Nenadis, N.; Assimopoulou, A.N.; Tsimidou, M.Z.; Papageorgiou, V.P. Structure-radical scavenging activity relationship of alkannin/shikonin derivatives. Food Chem. 2011, 124, 171–176. [Google Scholar] [CrossRef]
- Papageorgiou, V.P. Naturally occurring isohexenylnaphthazarin pigments: A new class of drugs. Planta Med. 1980, 38, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Ballis, A.C. Alkannins and shikonins: A new class of wound healing agents. Curr. Med. Chem. 2008, 15, 3248–3267. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Couladouros, E.A.; Hepworth, D.; Nicolaou, K.C. The chemistry and biology of alkannin, shikonin, and related naphthazarin natural products. Angew. Chem. Int. Ed. 1999, 38, 270–300. [Google Scholar] [CrossRef]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Samanidou, V.F.; Papadoyannis, I.N. Recent advances in chemistry, biology and biotechnology of alkannins and shikonins. Curr. Org. Chem. 2006, 10, 2123–2142. [Google Scholar] [CrossRef]
- Karapanagioti, E.G.; Assimopoulou, A.N. Naturally occurring wound healing agents: An evidence-based review. Curr. Med. Chem. 2016, 23, 3285–3321. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jia-Hua, C.; Qing-Qing, M.; Shao-Shun, L.; Wen, Z.; Sui, X. Advance in anti-tumor mechanisms of shikonin, alkannin and their derivatives. Mini-Rev. Med. Chem. 2018, 18, 164–172. [Google Scholar]
- Xie, Y.; Hou, X.L.; Wu, C.L. The research progress of cell apoptosis induced by shikonin and signal pathway of apoptosis. Chin. J. Pharm. Biotechnol. 2016, 23, 173–178. [Google Scholar]
- Zhao, Q.; Assimopoulou, A.N.; Klauck, S.M.; Damianakos, H.; Chinou, I.; Kretschmer, N.; Rios, J.L.; Papageorgiou, V.P.; Bauer, R.; Efferth, T. Inhibition of c-MYC with involvement of ERK/JNK/MAPK and AKT pathways as a novel mechanism for shikonin and its derivatives in killing leukemia cells. Oncotarget 2015, 6, 38934–38951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-Y.; Hu, Y.; Que, Z.-Y.; Wang, P.; Liu, Y.-H.; Wang, Z.-H.; Xue, Y.-X. Shikonin inhibits the migration and invasion of human glioblastoma cells by targeting phosphorylated β-catenin and phosphorylated PI3K/Akt: A potential mechanism for the anti-glioma efficacy of a traditional chinese herbal medicine. Int. J. Mol. Sci. 2015, 16, 23823–28848. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Zhang, B.; Yao, J.; Liu, Y.; Fang, J. Shikonin targets cytosolic thioredoxin reductase to induce ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Free Radic. Biol. Med. 2014, 70, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Wiench, B.; Eichhorn, T.; Paulsen, M.; Efferth, T. Shikonin directly targets mitochondria and causes mitochondrial dysfunction in cancer cells. Evid.-Based Complement. Altern. Med. 2012, 2012, 726025. [Google Scholar] [CrossRef] [PubMed]
- Spyrelli, E.D.; Kyriazou, A.V.; Virgiliou, C.; Nakas, A.; Deda, O.; Papageorgiou, V.P.; Assimopoulou, A.N.; Gika, H.G. Metabolic profiling study of shikonin’s cytotoxic activity in the Huh7 human hepatoma cell line. Mol. BioSyst. 2017, 13, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Shikonin and NRF2 Chemoprevention; University of Maryland Baltimore; National Cancer Institute: Baltimore, MD, USA, 2014. Available online: http://projectreporter.nih.gov/project_info_description.cfm?aid=8685189 (accessed on 25 May 2018).
- Ni, F.; Huang, X.; Chen, Z.; Qian, W.; Tong, X. Shikonin exerts antitumor activity in Burkitt’s lymphoma by inhibiting C-MYC and PI3K/AKT/mTOR pathway and acts synergistically with doxorubicin. Sci. Rep. 2018, 8, 3317. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Arima, N.; Yoshimitsu, M.; Honda, S.I.; Soeda, S. Liposomes and nanotechnology in drug development: Focus on oncotargets. Int. J. Nanomed. 2012, 7, 4943–4951. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Rhee, J.W.; Richie, J.P.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. New frontiers in nanotechnology for cancer treatment. Urol. Oncol. Semin. Orig. Investig. 2008, 26, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G.; Wills, E.J.; Swain, C.P.; Tavill, A.S. Drug-carrier potential of liposomes in cancer chemotherapy. Lancet 1974, 1, 1313–1316. [Google Scholar] [CrossRef]
- Gumulec, J.; Fojtu, M.; Raudenska, M.; Sztalmachova, M.; Skotakova, A.; Vlachova, J.; Skalickova, S.; Nejdl, L.; Kopel, P.; Knopfova, L.; et al. Modulation of induced cytotoxicity of doxorubicin by using apoferritin and liposomal cages. Int. J. Mol. Sci. 2014, 15, 22960–22977. [Google Scholar] [CrossRef] [PubMed]
- Heger, Z.; Polanska, H.; Merlos Rodrigo, M.A.; Guran, R.; Kulich, P.; Kopel, P.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Kizek, R.; et al. Prostate tumor attenuation in the nu/nu murine model due to anti-sarcosine antibodies in folate-targeted liposomes. Sci. Rep. 2016, 6, 33379. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M.C.; Lasic, D.D. Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171–199. [Google Scholar] [CrossRef]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 1999, 51, 691–744. [Google Scholar] [PubMed]
- Andresen, T.L.; Jensen, S.S.; Jørgensen, K. Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release. Prog. Lipid Res. 2005, 44, 68–97. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.H.; Alzghari, S.K.; Chee, W.; Sankari, S.S.; La-Beck, N.M. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J. Control. Release 2016, 232, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Tang, C.; Gui, H.; Wang, X.; Qi, J.; Wang, X.; Yang, Y. Preparation, cellular uptake and angiogenic suppression of shikonin-containing liposomes in vitro and in vivo. Biosci. Rep. 2013, 33, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Kontogiannopoulos, K.N.; Assimopoulou, A.N.; Dimas, K.; Papageorgiou, V.P. Shikonin–loaded liposomes as a new drug delivery system: Physicochemical characterization and in vitro cytotoxicity. Eur. J. Lipid Sci. Technol. 2011, 113, 1113–1123. [Google Scholar] [CrossRef]
- Kontogiannopoulos, K.N.; Tsermentseli, S.K.; Assimopoulou, A.N.; Papageorgiou, V.P. Sterically stabilized liposomes as a potent carrier for shikonin. J. Liposome Res. 2014, 24, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Han, H.K.; Shin, H.J.; Ha, D.H. Improved oral bioavailability of alendronate via the mucoadhesive liposomal delivery system. Eur. J. Pharm. Sci. 2012, 46, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Crosasso, P.; Ceruti, M.; Brusa, P.; Arpicco, S.; Dosio, F.; Cattel, L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J. Control. Release 2000, 63, 19–30. [Google Scholar] [CrossRef]
- Kelly, C.; Jefferies, C.; Cryan, S.A. Targeted liposomal drug delivery to monocytes andmacrophages. J. Drug Deliv. 2011, 2011, 727241. [Google Scholar] [CrossRef] [PubMed]
- Assimopoulou, A.N.; Ganzera, M.; Stuppner, H.; Papageorgiou, V.P. Simultaneous determination of monomeric and oligomeric alkannins and shikonins by high-performance liquid chromatography–diode array detection–mass spectrometry. Biomed. Chromatogr. 2008, 22, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Krasnici, S.; Werner, A.; Eichhorn, M.E.; Schmitt-Sody, M.; Pahernik, S.A.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Naujoks, K.; et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int. J. Cancer 2003, 105, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.C.; Lim, H.J.; Shim, J.; Kim, J.; Chang, I.S. Improved stability of liposome in oil/water emulsion by association of amphiphilic polymer with liposome and its effect on bioactive skin permeation. Colloids Surf. A Physicochem. Eng. Asp. 2007, 299, 160–168. [Google Scholar] [CrossRef]
- Gardikis, K.; Hatziantoniou, S.; Bucos, M.A.; Fessas, D.; Signorelli, M.; Felekis, T.; Zervou, M.; Screttas, C.G.; Steele, B.R.; Ionov, M.; et al. New drug delivery nanosystem combining liposomal and dendrimeric technology (liposomal locked-in dendrimers) for cancer therapy. J. Pharm. Sci. 2010, 99, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Bonacucina, G.; Cespi, M.; Misici-Falzi, M.; Palmieri, G.F. Colloidal soft matter as drug delivery system. J. Pharm. Sci. 2009, 98, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L. Guidance for Industry Liposome Drug Products. Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation; Center for Drug Evaluation and Research (CDER), U.S. Department of Health and Human Services, Food and Drug Administration: Washington, DC, USA, 2002.
- Tenzel, R.A.; Aitcheson, D.F. Preparation of uniform-size liposomes and other lipid structures. WO1989011335A1, 30 November 1989. [Google Scholar]
- Dadashzadeh, S.; Mirahmadi, N.; Babaei, M.H.; Vali, A.M. Peritoneal retention of liposomes: Effects of lipid composition, PEG coating and liposome charge. J. Control. Release 2010, 148, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Sriwongsitanont, S.; Ueno, M. Effect of a PEG lipid (DSPE-PEG2000) and freeze-thawing process on phospholipid vesicle size and lamellarity. Colloid Polym. Sci. 2004, 282, 753–760. [Google Scholar] [CrossRef]
- Shenoy, V.S.; Gude, R.P.; Murthy, R.S.R. Investigations on paclitaxel loaded HSPC based conventional and PEGylated liposomes: In vitro release and cytotoxic studies. Asian J. Pharm. Sci. 2011, 6, 1–7. [Google Scholar]
- Muller, R.H. Colloidal Carriers for Controlled Drug Delivery and Targeting: Modification, Characterization, and in vivo Distribution; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Gentile, E.; Cilurzo, F.; Di Marzio, L.; Carafa, M.; Ventura, C.A.; Wolfram, J.; Paolino, D.; Celia, C. Liposomal chemotherapeutics. Future Oncol. 2013, 9, 1849–1859. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.R.; Weston, N.; Coombes, A.G.A.; Fitzgerald, M.; Perrie, Y. Liposome formulation of poorly water soluble drugs: Optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 2004, 285, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Cui, F.D.; Choi, M.K.; Cho, J.W.; Chung, S.J.; Shim, C.K.; Kim, D.D. Enhanced solubility and stability of PEGylated liposomal paclitaxel: In vitro and in vivo evaluation. Int. J. Pharm. 2007, 338, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Vali, A.M.; Toliyat, T.; Shafaghi, B.; Dadashzadeh, S. Preparation, optimization, and characterization of topotecan loaded PEGylated liposomes using factorial design. Drug Dev. Ind. Pharm. 2008, 34, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Mourelatou, E.A.; Libster, D.; Nir, I.; Hatziantoniou, S.; Aserin, A.; Garti, N.; Demetzos, C. Type and location of interaction between hyperbranched polymers and liposomes. Relevance to design of a potentially advanced drug delivery nanosystem (aDDnS). J. Phys. Chem. B 2011, 115, 3400–3408. [Google Scholar] [CrossRef] [PubMed]
- Fatouros, D.G.; Antimisiaris, S.G. Effect of amphiphilic drugs on the stability and zeta-potential of their liposome formulations: A study with prednisolone, diazepam, and griseofulvin. J. Colloid Interface Sci. 2002, 251, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Dadashzadeh, S.; Vali, A.M.; Rezaie, M. The effect of PEG coating on in vitro cytotoxicity and in vivo disposition of topotecan loaded liposomes in rats. Int. J. Pharm. 2008, 353, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Huang, Y.Y. Encapsulating protein into preformed liposomes by ethanol-destabilized method. Artif. Cells Nanomed. Biotechnol. 2003, 31, 303–312. [Google Scholar] [CrossRef]
- Abra, R.M.; Mihalko, P.J.; Schreier, H. The effect of lipid composition upon the encapsulation and in vitro leakage of metaproterenol sulfate from 0.2 μm diameter, extruded, multilamellar liposomes. J. Control. Release 1990, 14, 71–78. [Google Scholar] [CrossRef]
- Colletier, J.P.; Chaize, B.; Winterhalter, M.; Fournier, D. Protein encapsulation in liposomes: Efficiency depends on interactions between protein and phospholipid bilayer. BMC Biotechnol. 2002, 2, 9. [Google Scholar] [CrossRef]
- Haeri, A.; Alinaghian, B.; Daeihamed, M.; Dadashzadeh, S. Preparation and characterization of stable nanoliposomal formulation of fluoxetine as a potential adjuvant therapy for drug-resistant tumors. Iran. J. Pharm. Res. 2014, 13, 3–14. [Google Scholar] [PubMed]
- Ho, E.A.; Osooly, M.; Strutt, D.; Masin, D.; Yang, Y.; Yan, H.; Bally, M. Characterization of long-circulating cationic nanoparticle formulations consisting of a two-stage PEGylation step for the delivery of siRNA in a breast cancer tumor model. J. Pharm. Sci. 2013, 102, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, X.; Guo, Y.; Zhang, Z. Lipid-enveloped hybrid nanoparticles for drug delivery. Nanoscale 2013, 5, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Begum, M.Y.; Abbulu, K.; Sudhakar, M. Preparation, characterization and in-vitro release study of flurbiprofen loaded stealth liposomes. Chem. Sci. Trans. 2012, 1, 201–209. [Google Scholar] [CrossRef][Green Version]
- Pippa, N.; Psarommati, F.; Pispas, S.; Demetzos, C. The shape/morphology balance: A study of stealth liposomes via fractal analysis and drug encapsulation. Pharm. Res. 2013, 30, 2385–2395. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Type of Lipids | Lipid (mg) a | DSPE-mPEG2000 (mg) b | Cholesterol (mg) c | Shikonin (mg) d |
|---|---|---|---|---|---|
| 1 | DOPC | 120 | - | 13.2 | 1.47 |
| 1e | DOPC | 30 | - | 3.27 | - |
| 2 | DOPC/DSPG | 120 | - | 13.07 | 1.46 |
| 2e | DOPC/DSPG | 30 | - | 3.27 | - |
| 3 | DSPC/DSPG | 120 | - | 13.01 | 1.09 |
| 3e | DSPC/DSPG | 30 | - | 3.25 | - |
| 4 | DOPC | 120 | 33.12 | 13.2 | 1.47 |
| 4e | DOPC | 30 | 8.28 | 3.27 | - |
| 5 | DOPC/DSPG | 120 | 33.06 | 13.07 | 1.46 |
| 5e | DOPC/DSPG | 30 | 8.26 | 3.27 | - |
| 6 | DSPC/DSPG | 120 | 32.91 | 13.01 | 1.09 |
| 6e | DSPC/DSPG | 30 | 8.23 | 3.25 | - |
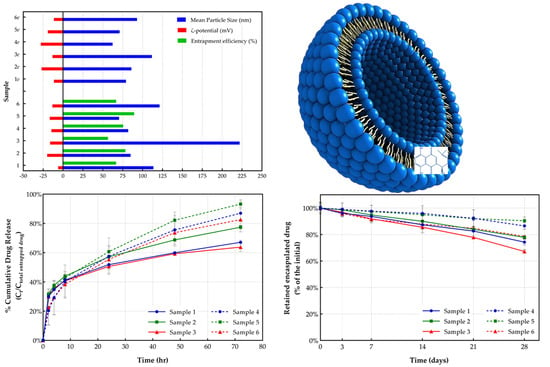
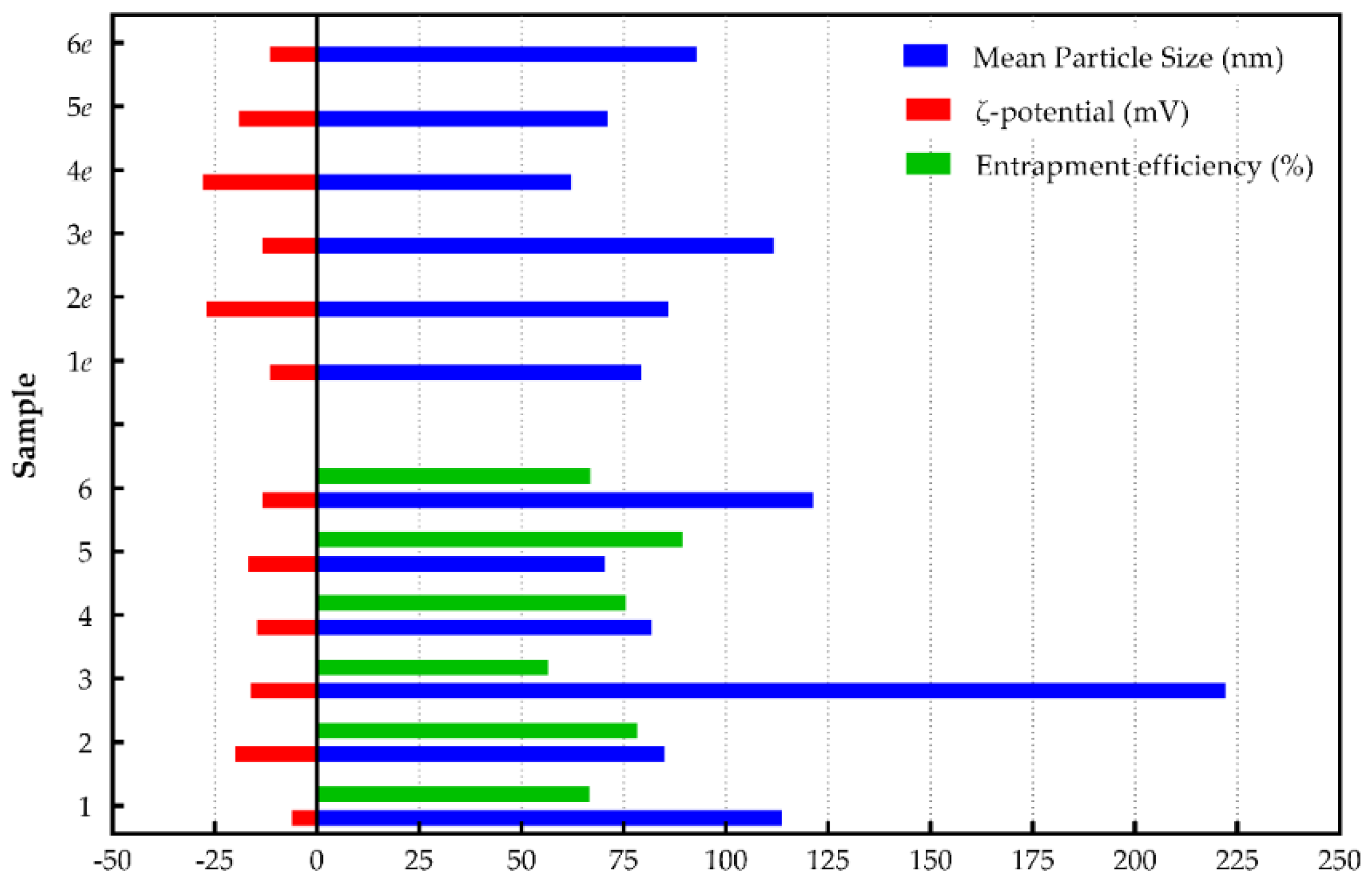
| Sample | Mean Particle Size (nm) | Polydispersity Index (PDI) | ζ-Potential (mV) | Entrapment Efficiency (%) |
|---|---|---|---|---|
| 1 | 113.62 ± 9.72 | 0.29 ± 0.02 | −6.10 ± 1.11 | 66.67 ± 0.04 |
| 1e | 79.15 ± 1.34 | 0.31 ± 0.01 | −11.41 ± 2.68 | --- |
| 2 | 84.90 ± 0.10 | 0.25 ± 0.03 | −19.88 ± 4.31 | 78.42 ± 0.01 |
| 2e | 85.95 ± 1.20 | 0.29 ± 0.07 | −26.97 ± 1.30 | --- |
| 3 | 222.0 ± 6.56 | 0.33 ± 0.05 | −16.23 ± 9.29 | 56.50 ± 0.03 |
| 3e | 111.67 ± 6.56 | 0.25 ± 0.02 | −13.33 ± 2.01 | --- |
| 4 | 81.92 ± 0.11 | 0.18 ± 0.02 | −14.62 ± 2.47 | 75.64 ± 0.04 |
| 4e | 62.33 ± 4.22 | 0.19 ± 0.01 | −27.93 ± 7.74 | --- |
| 5 | 70.42 ± 1.25 | 0.17 ± 0.02 | −16.68 ± 3.61 | 89.40 ± 0.02 |
| 5e | 71.15 ± 0.21 | 0.20 ± 0.01 | −19.17 ± 1.07 | --- |
| 6 | 121.33 ± 3.77 | 0.29 ± 0.01 | −13.38 ± 0.49 | 66.85 ± 0.01 |
| 6e | 93.00 ± 1.00 | 0.23 ± 0.03 | −11.54 ± 1.54 | --- |
| Sample | Total Drug Release (72 h) (%) | Drug Release at 8 h (%) | t50% (h) |
|---|---|---|---|
| 1 | 67.07 ± 0.01 | 41.24 ± 0.03 | 3.86 |
| 2 | 77.41 ± 0.01 | 43.95 ± 0.01 | 7.04 |
| 3 | 63.80 ± 0.03 | 40.67 ± 0.02 | 3.57 |
| 4 | 87.0 ± 0.08 | 40.22 ± 0.11 | 18.26 |
| 5 | 93.25 ± 0.03 | 42.76 ± 0.00 | 18.45 |
| 6 | 82.48 ± 0.08 | 38.36 ± 0.09 | 17.93 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsermentseli, S.K.; Kontogiannopoulos, K.N.; Papageorgiou, V.P.; Assimopoulou, A.N. Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin. Fluids 2018, 3, 36. https://doi.org/10.3390/fluids3020036
Tsermentseli SK, Kontogiannopoulos KN, Papageorgiou VP, Assimopoulou AN. Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin. Fluids. 2018; 3(2):36. https://doi.org/10.3390/fluids3020036
Chicago/Turabian StyleTsermentseli, Stella K., Konstantinos N. Kontogiannopoulos, Vassilios P. Papageorgiou, and Andreana N. Assimopoulou. 2018. "Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin" Fluids 3, no. 2: 36. https://doi.org/10.3390/fluids3020036
APA StyleTsermentseli, S. K., Kontogiannopoulos, K. N., Papageorgiou, V. P., & Assimopoulou, A. N. (2018). Comparative Study of PEGylated and Conventional Liposomes as Carriers for Shikonin. Fluids, 3(2), 36. https://doi.org/10.3390/fluids3020036