
Sol-Gel Synthesized Antimony Anodes for Sodium-Ion Batteries: Identifying Key Parameters for Optimization

 , and
, and
Abstract
:1. Introduction
2. Results
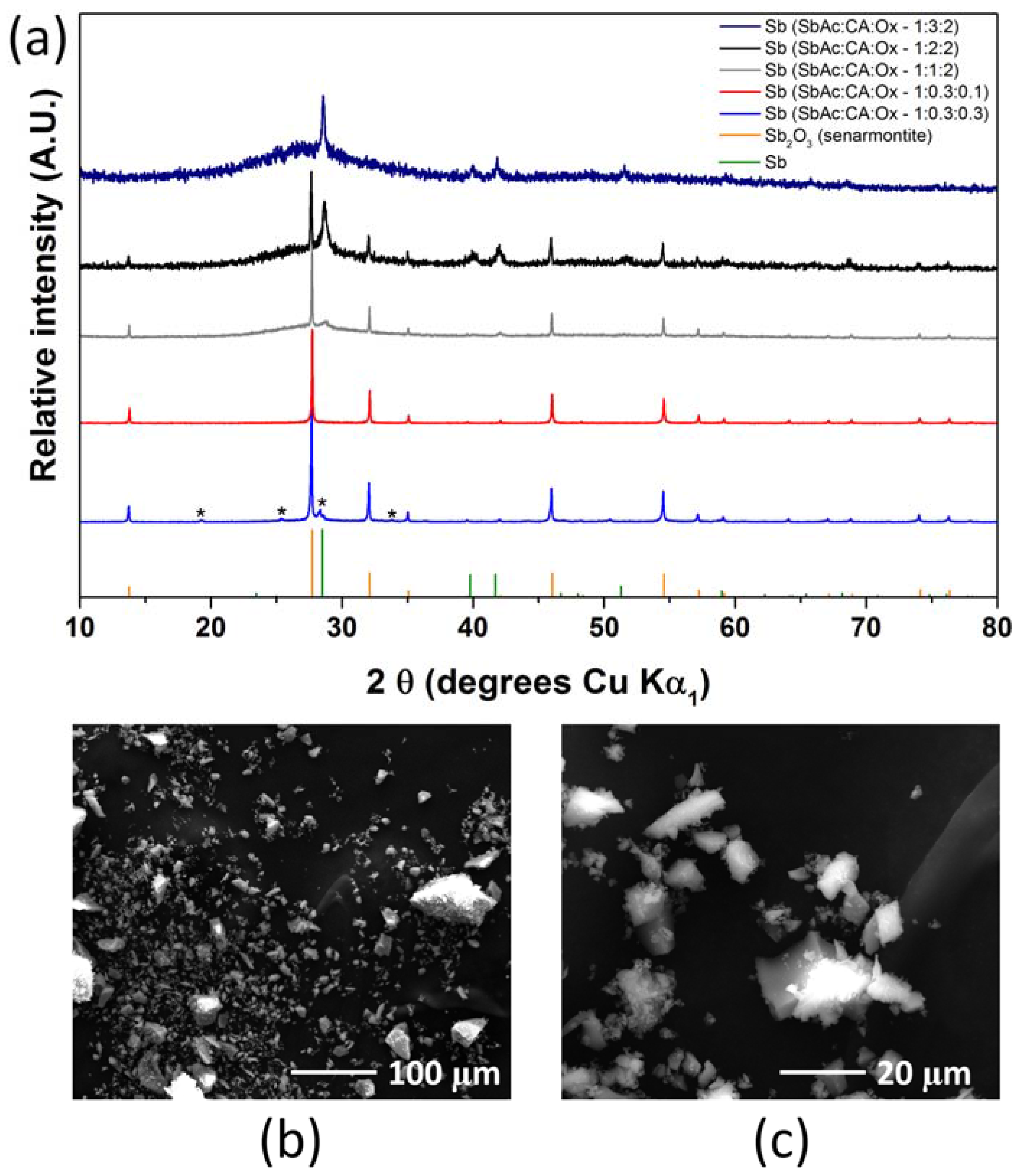
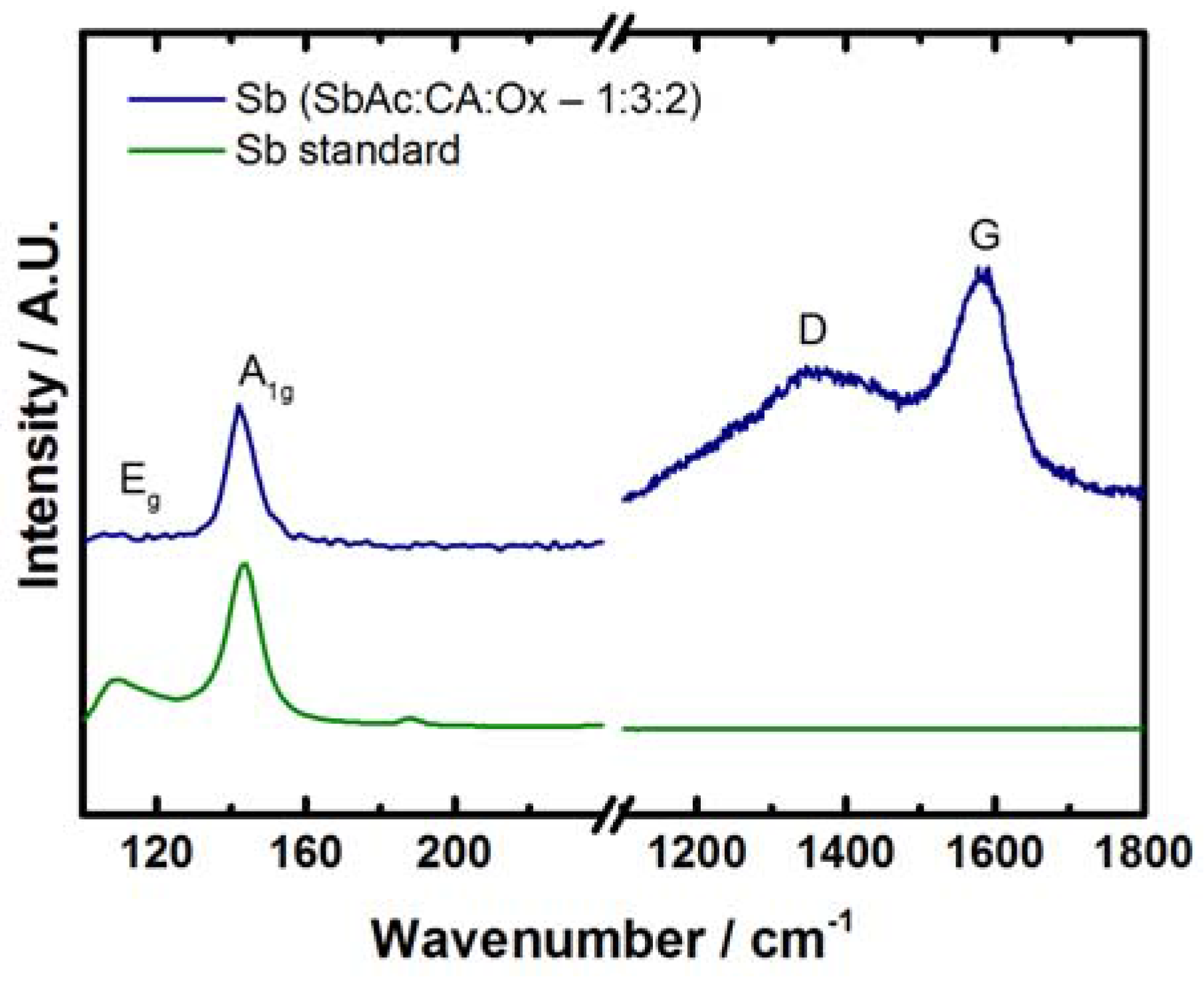
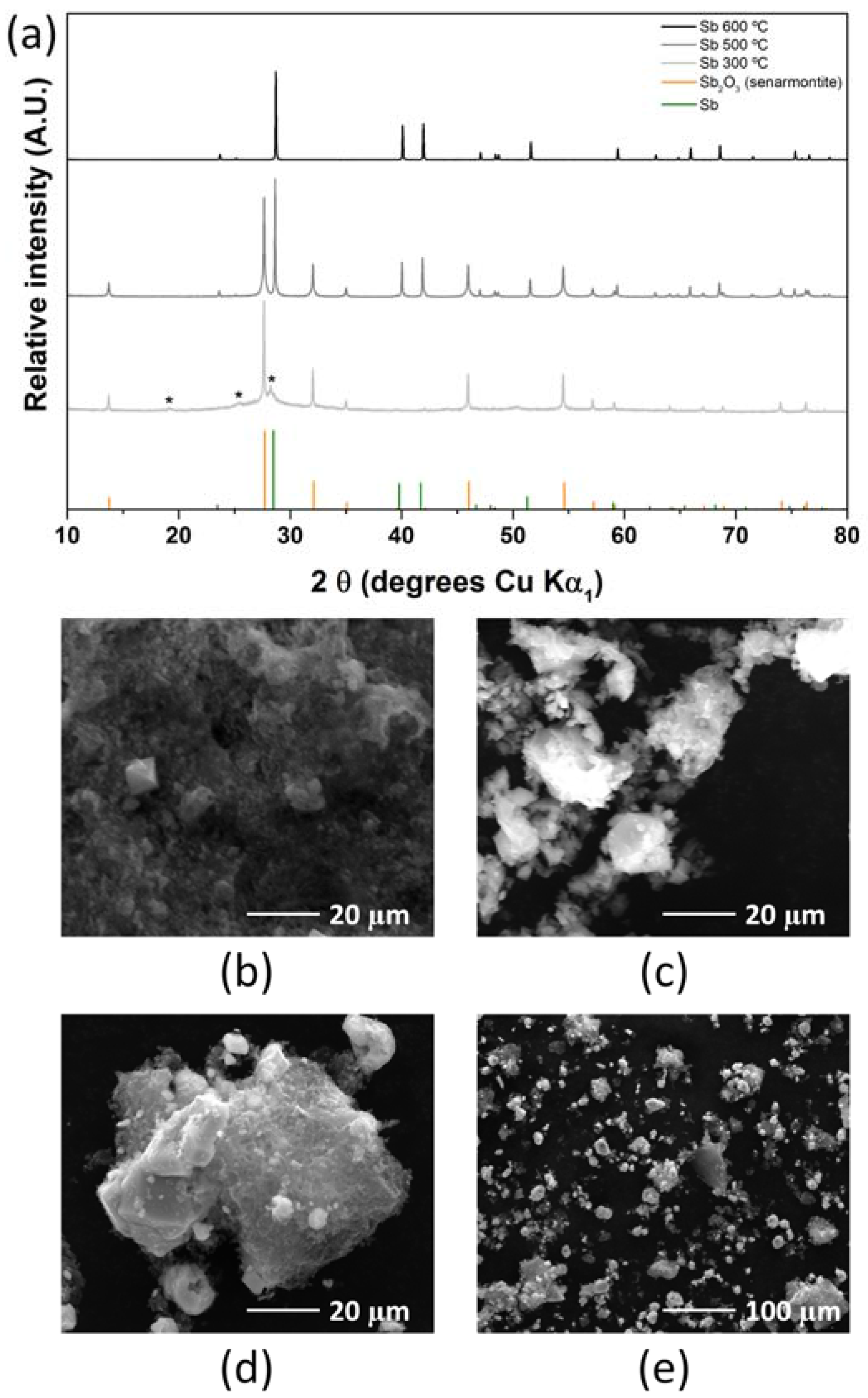
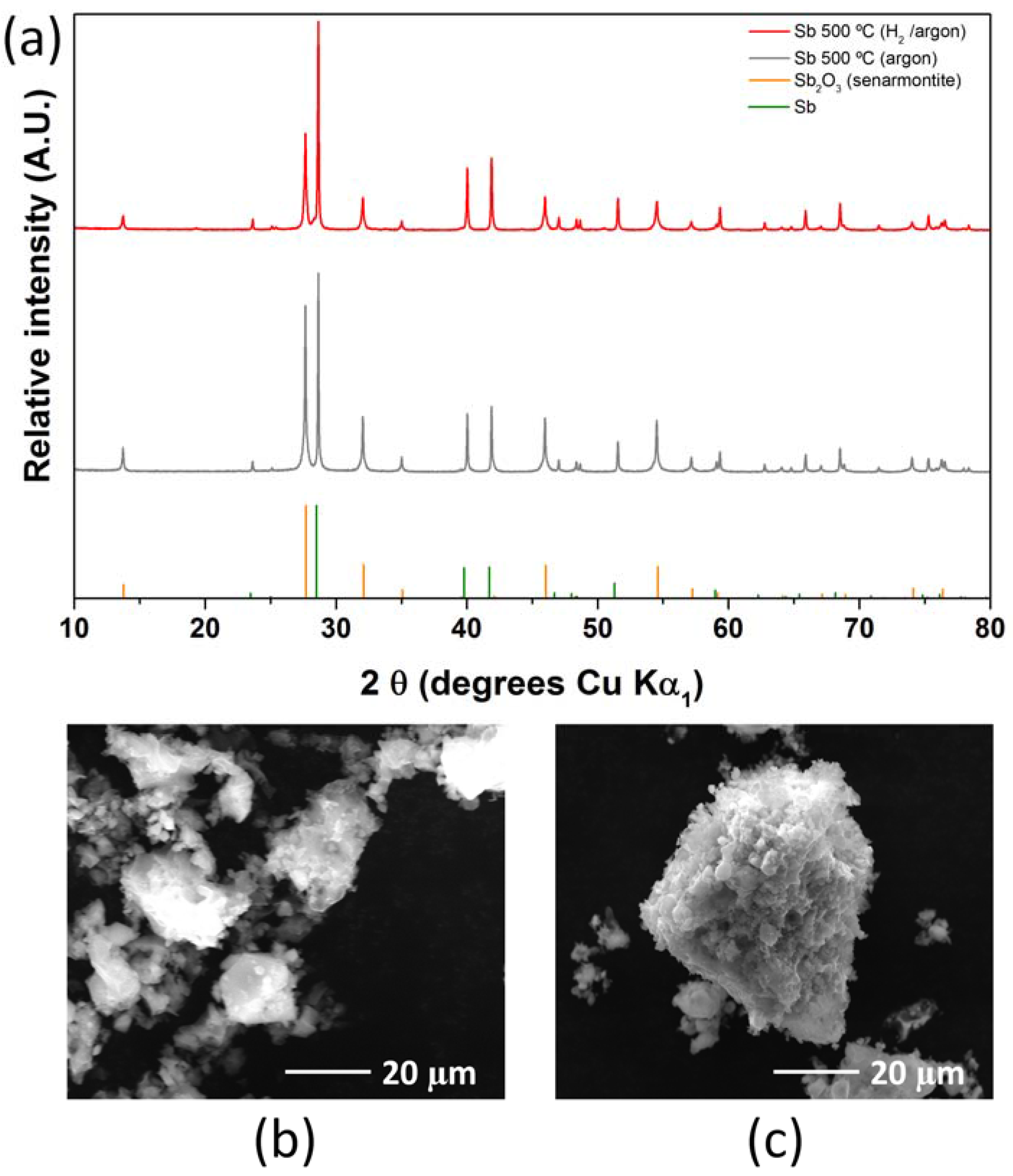
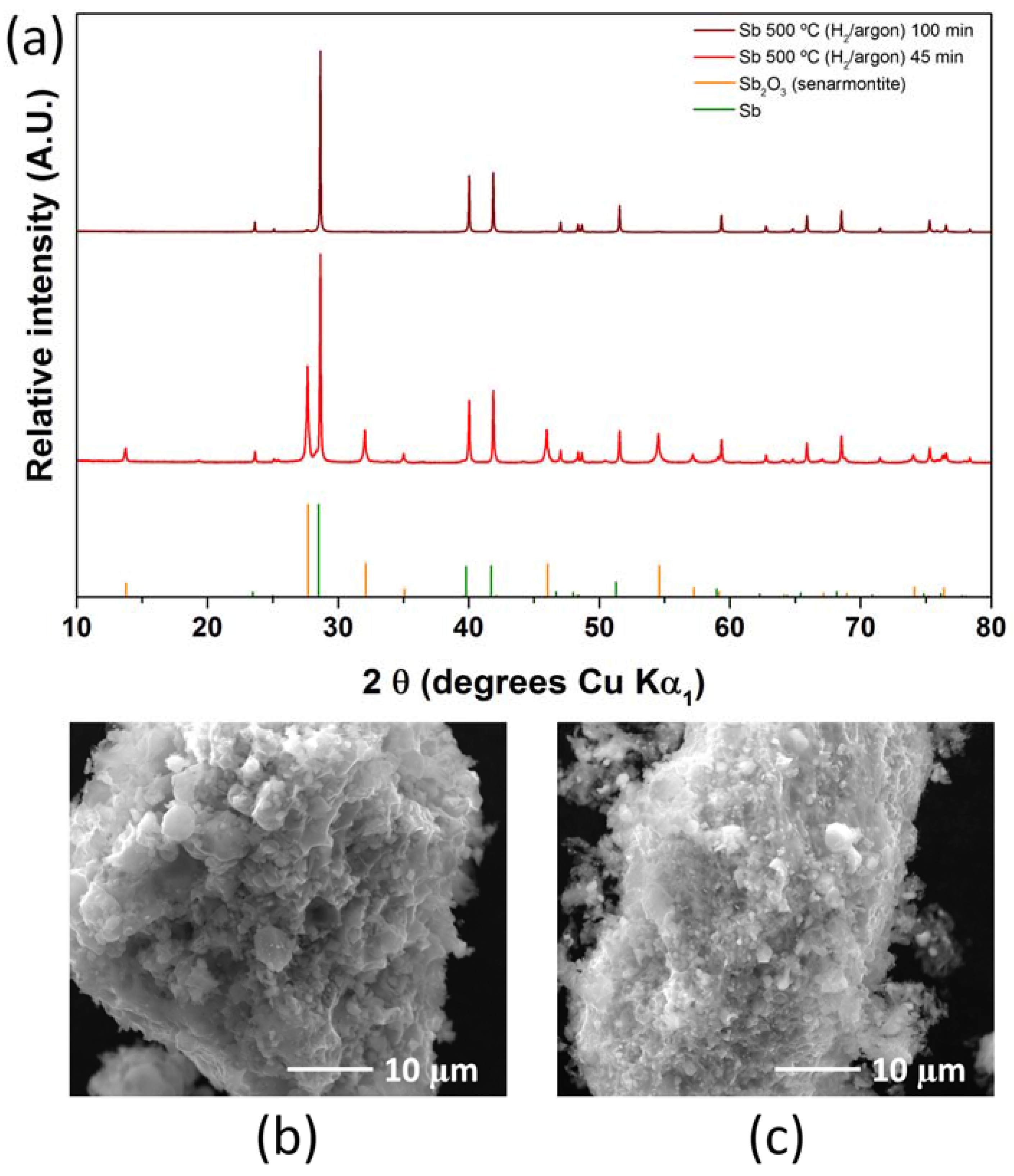
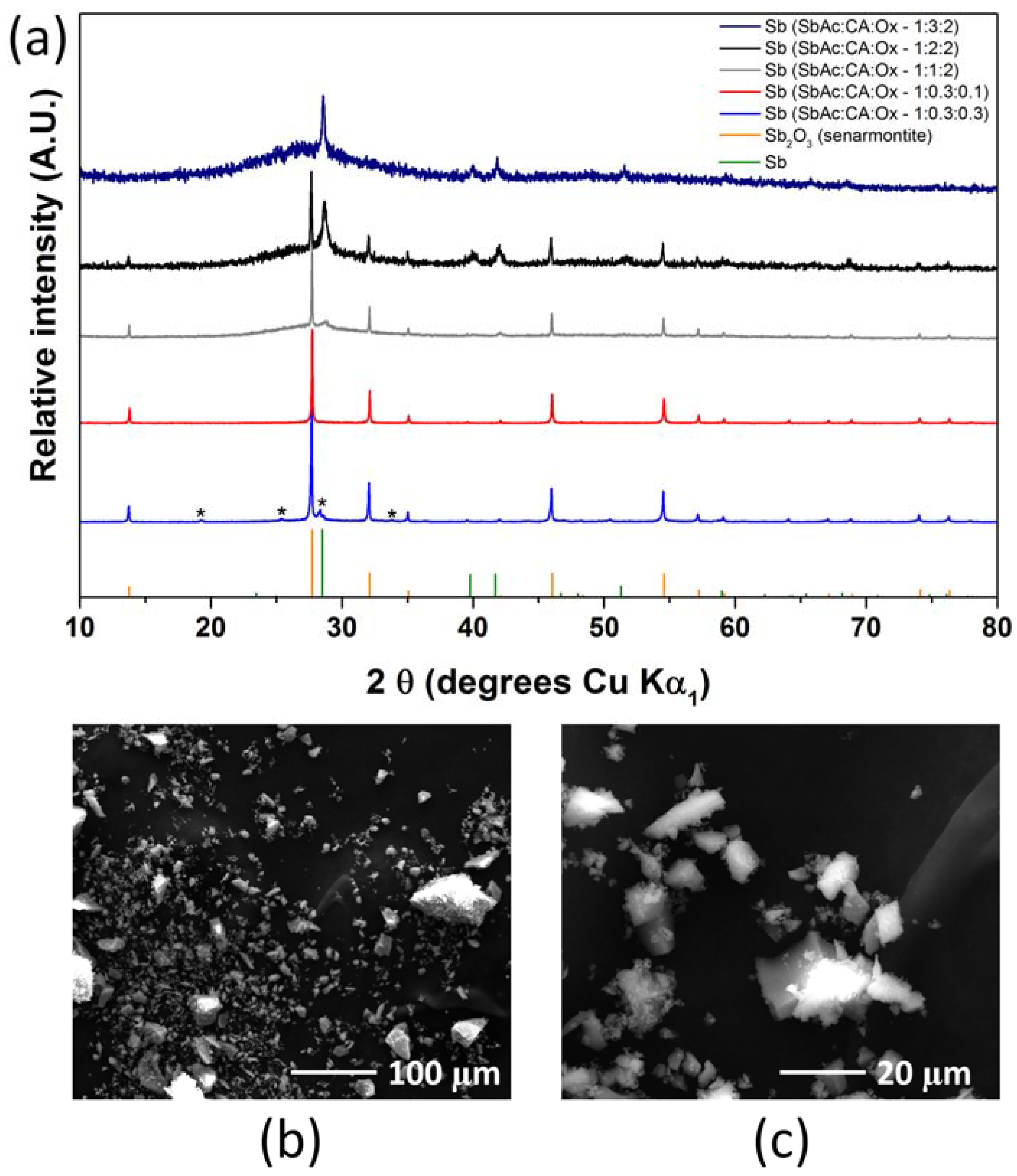
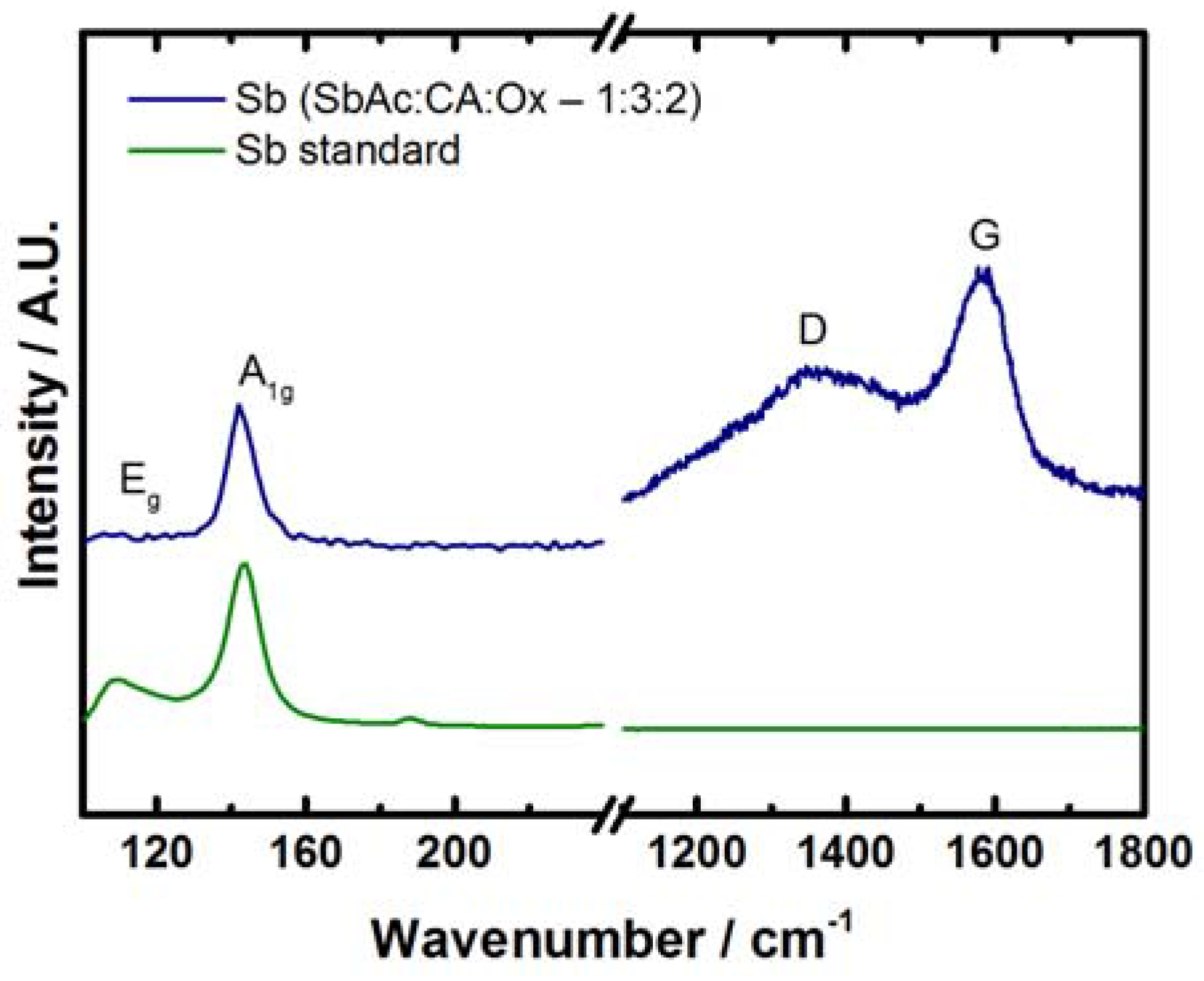
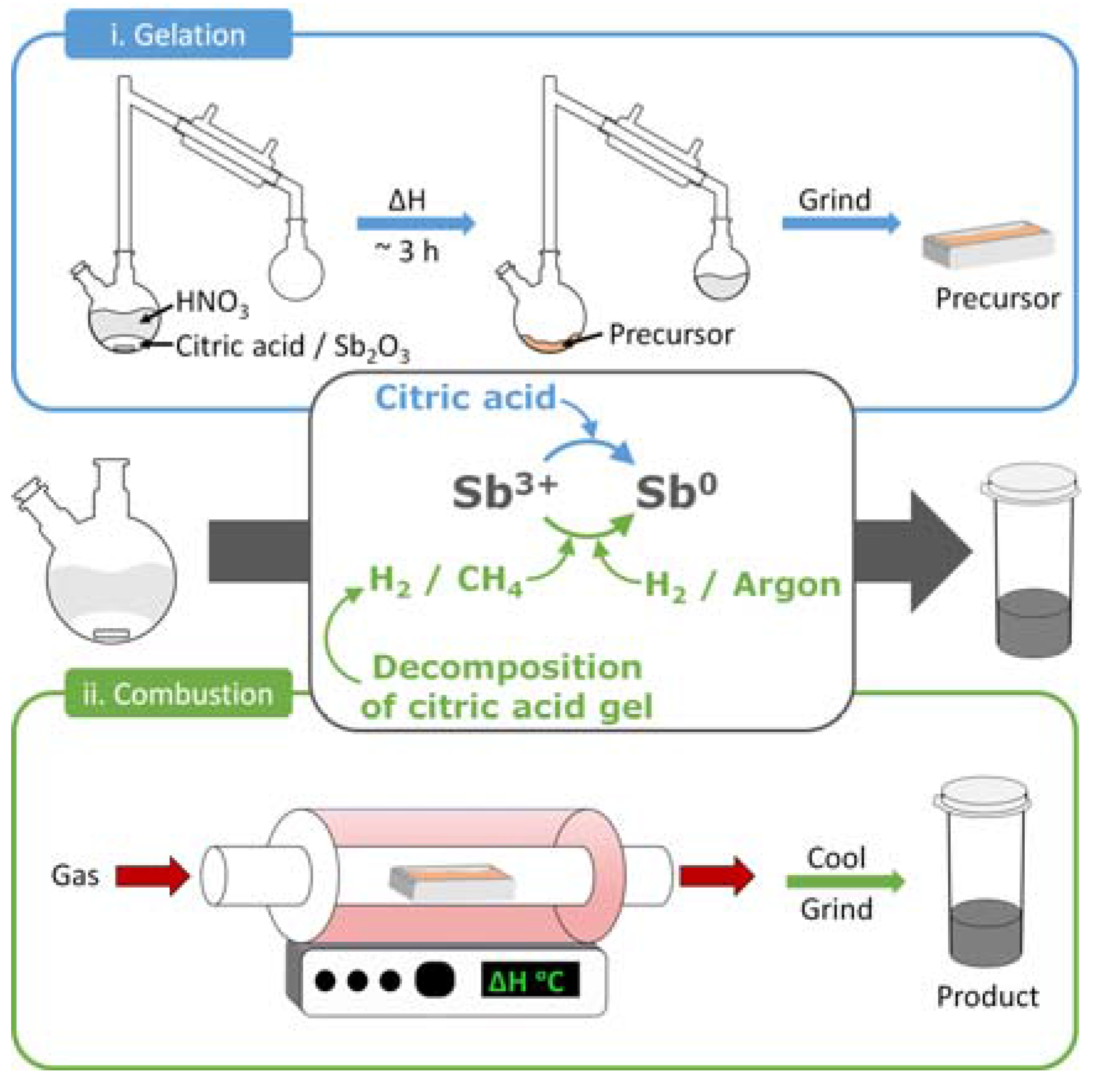
2.1. Antimony Acetate/Acetic Acid Synthetic Route
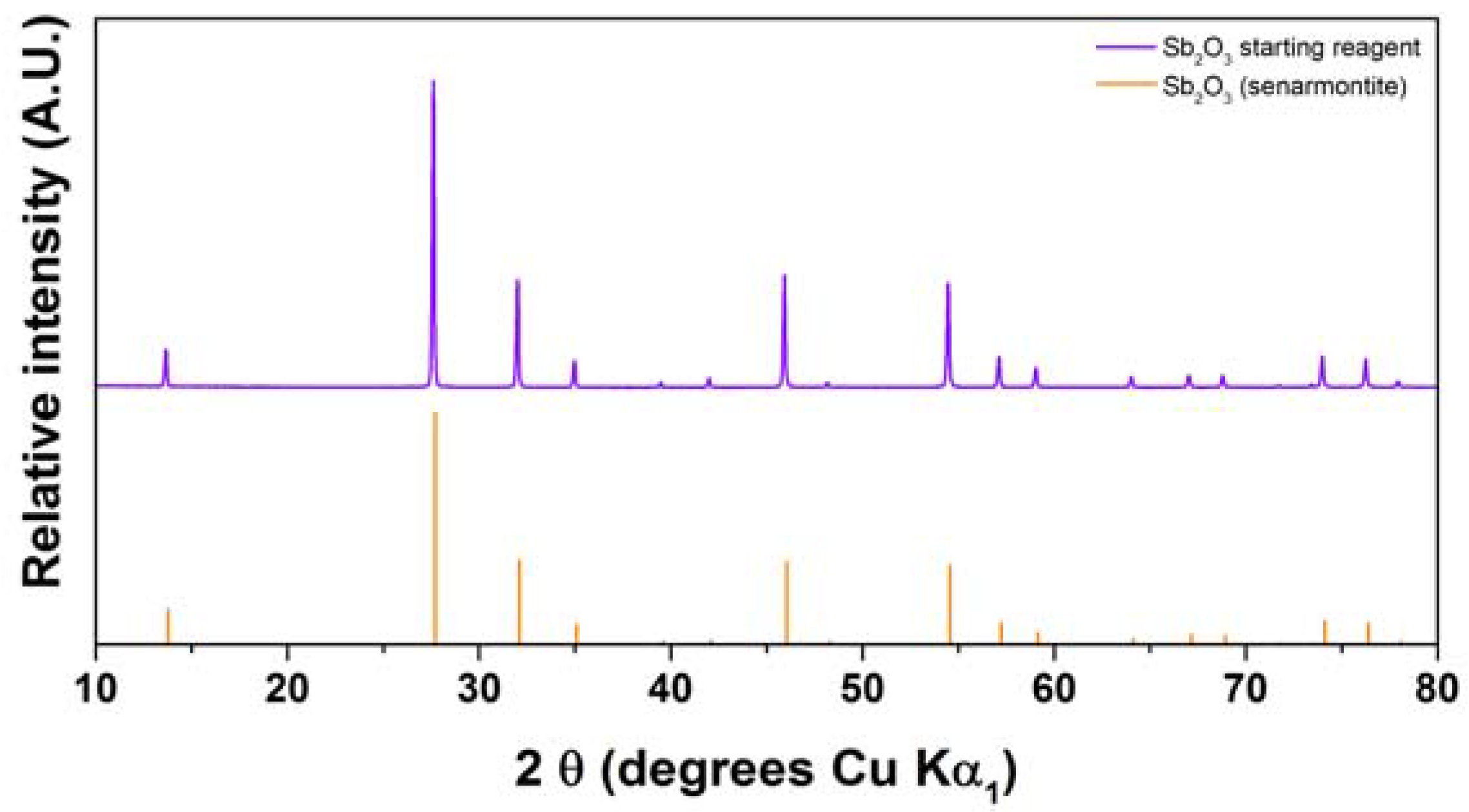
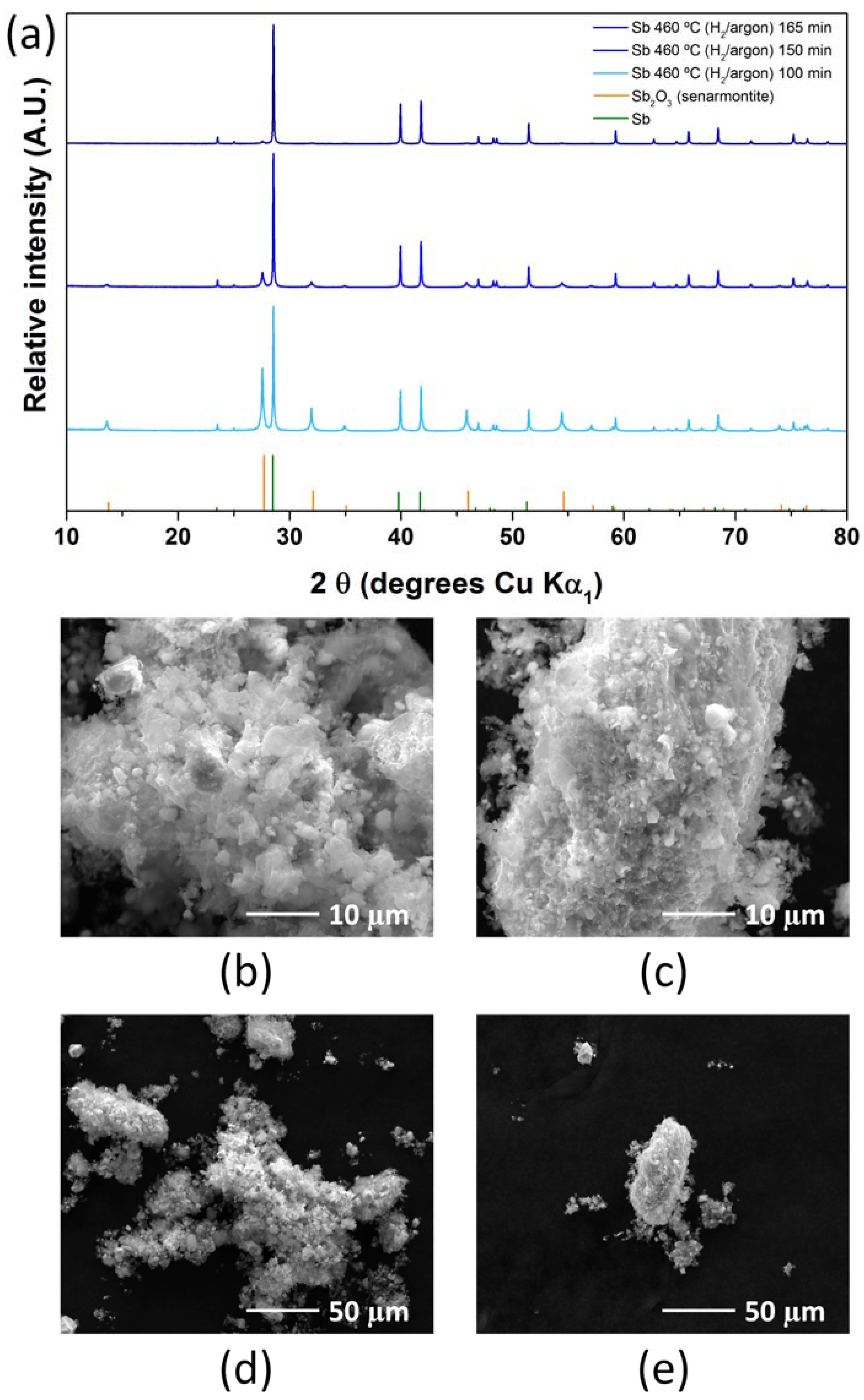
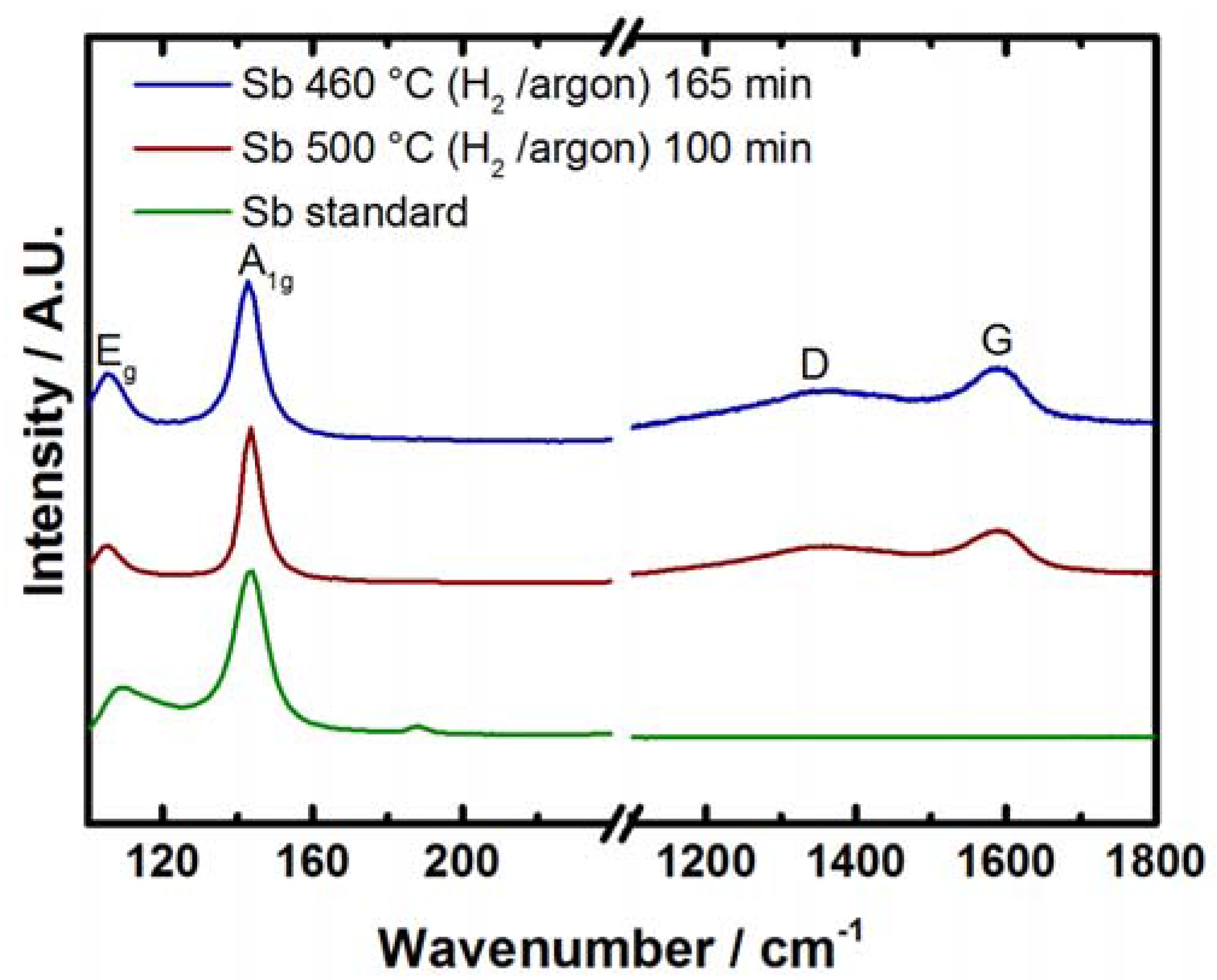
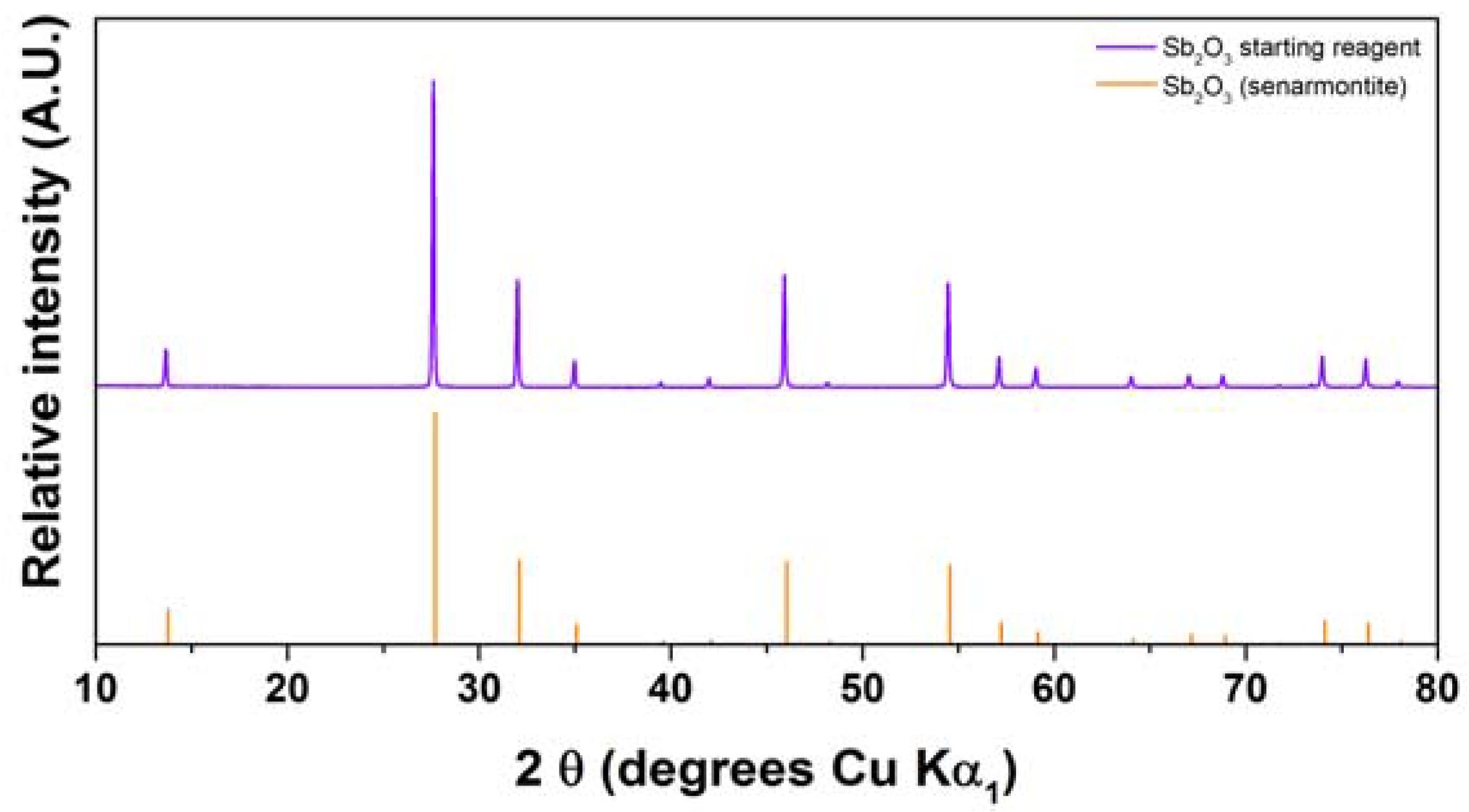
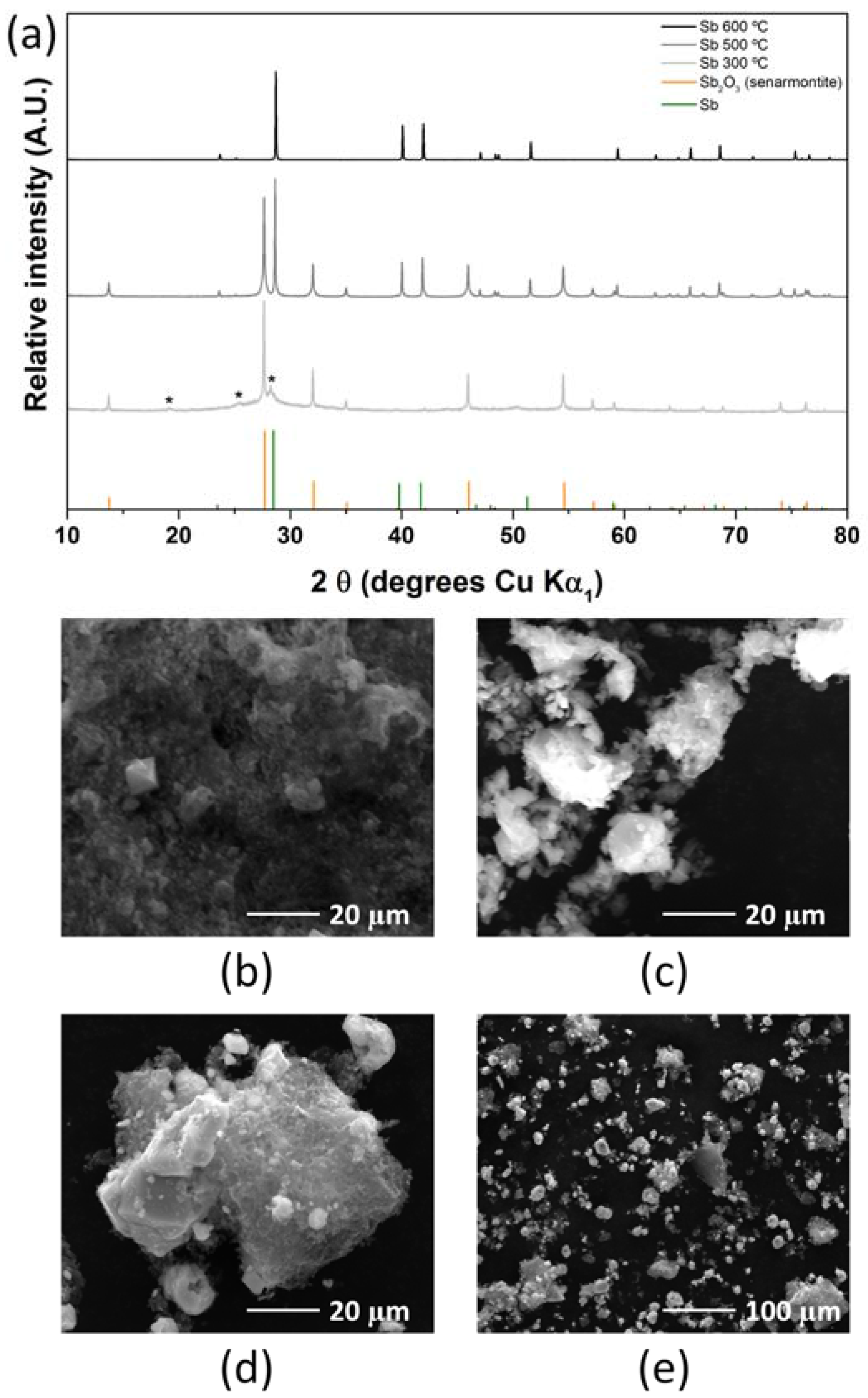
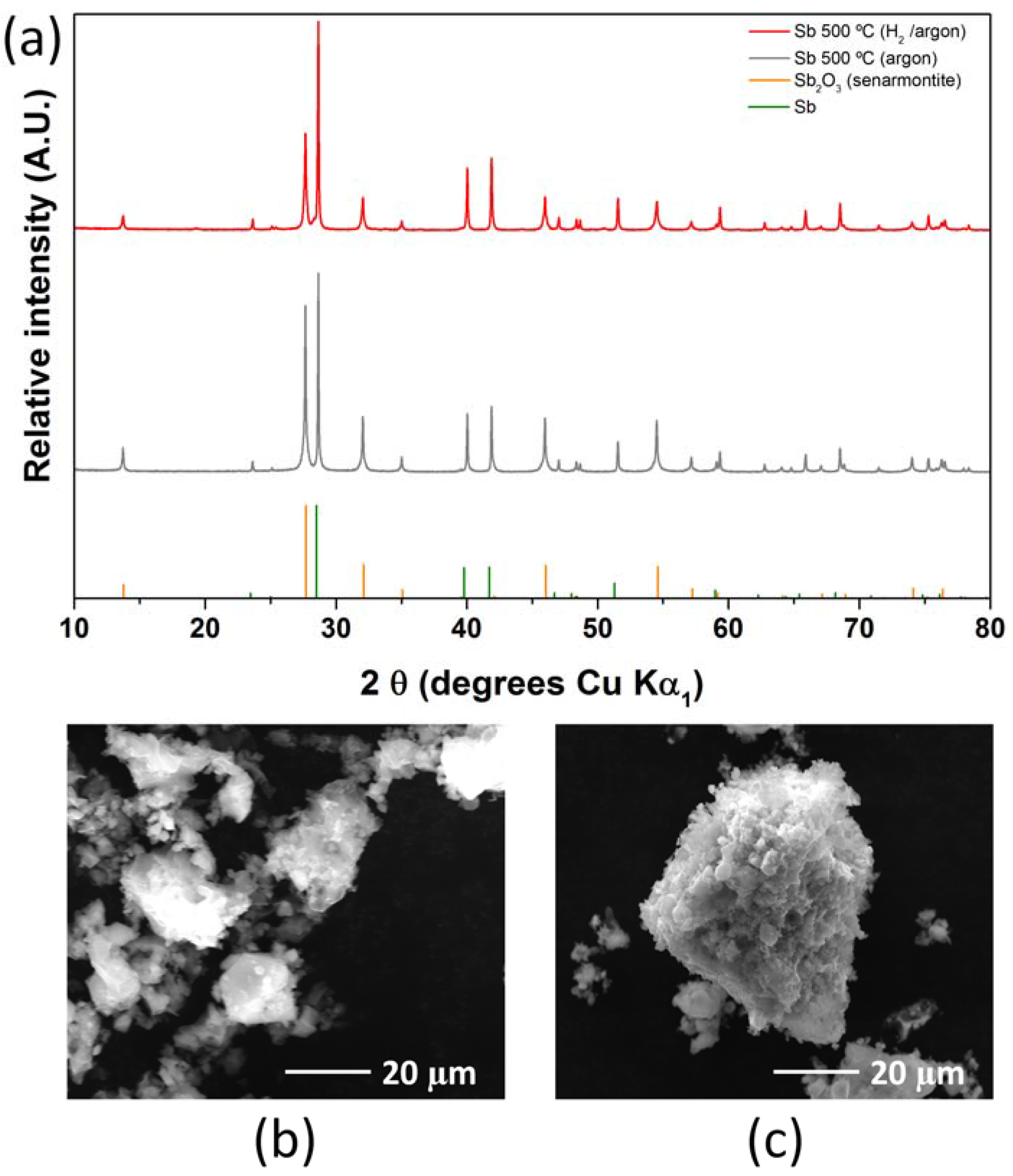
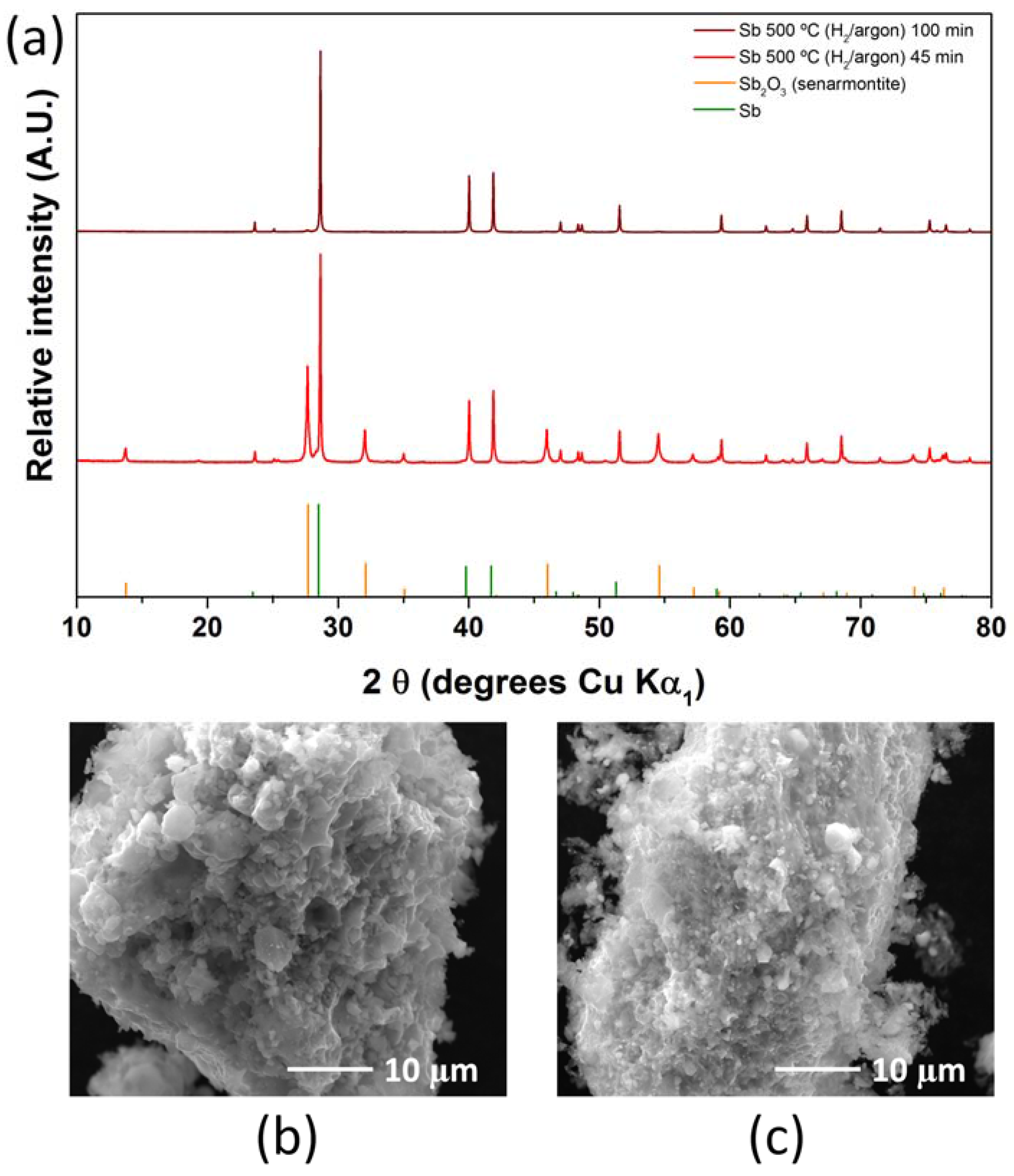
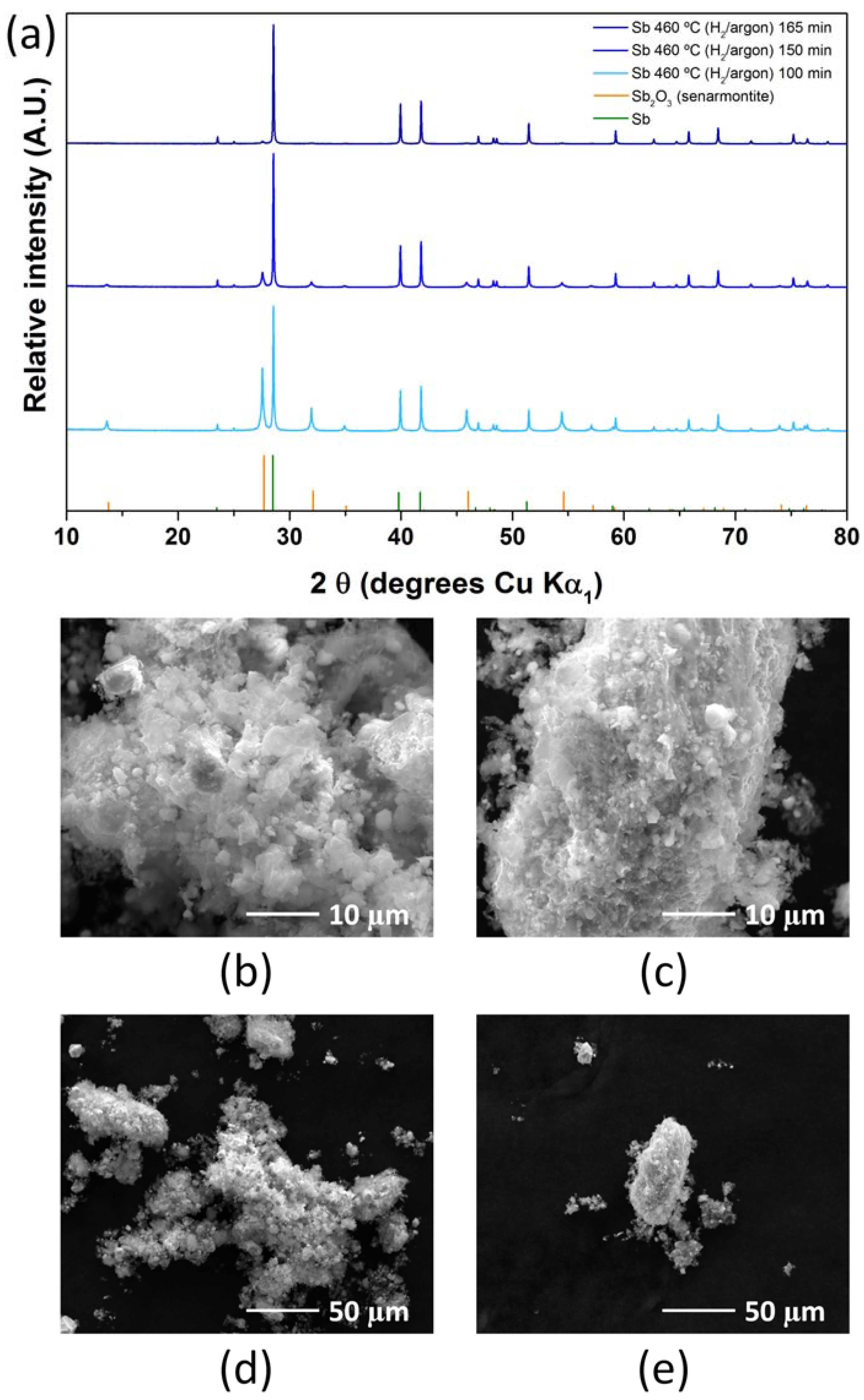
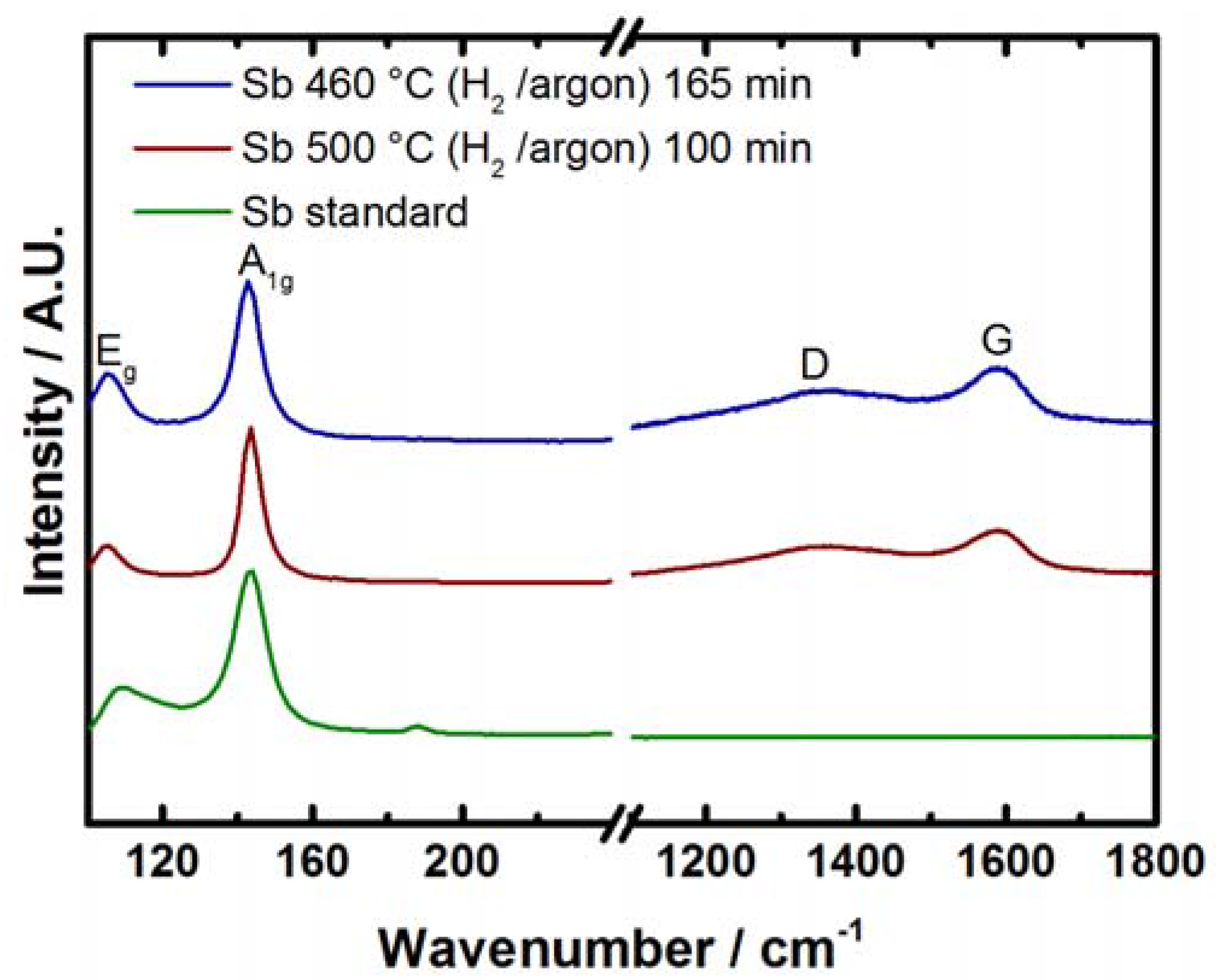
2.2. Antimony Oxide Synthetic Route
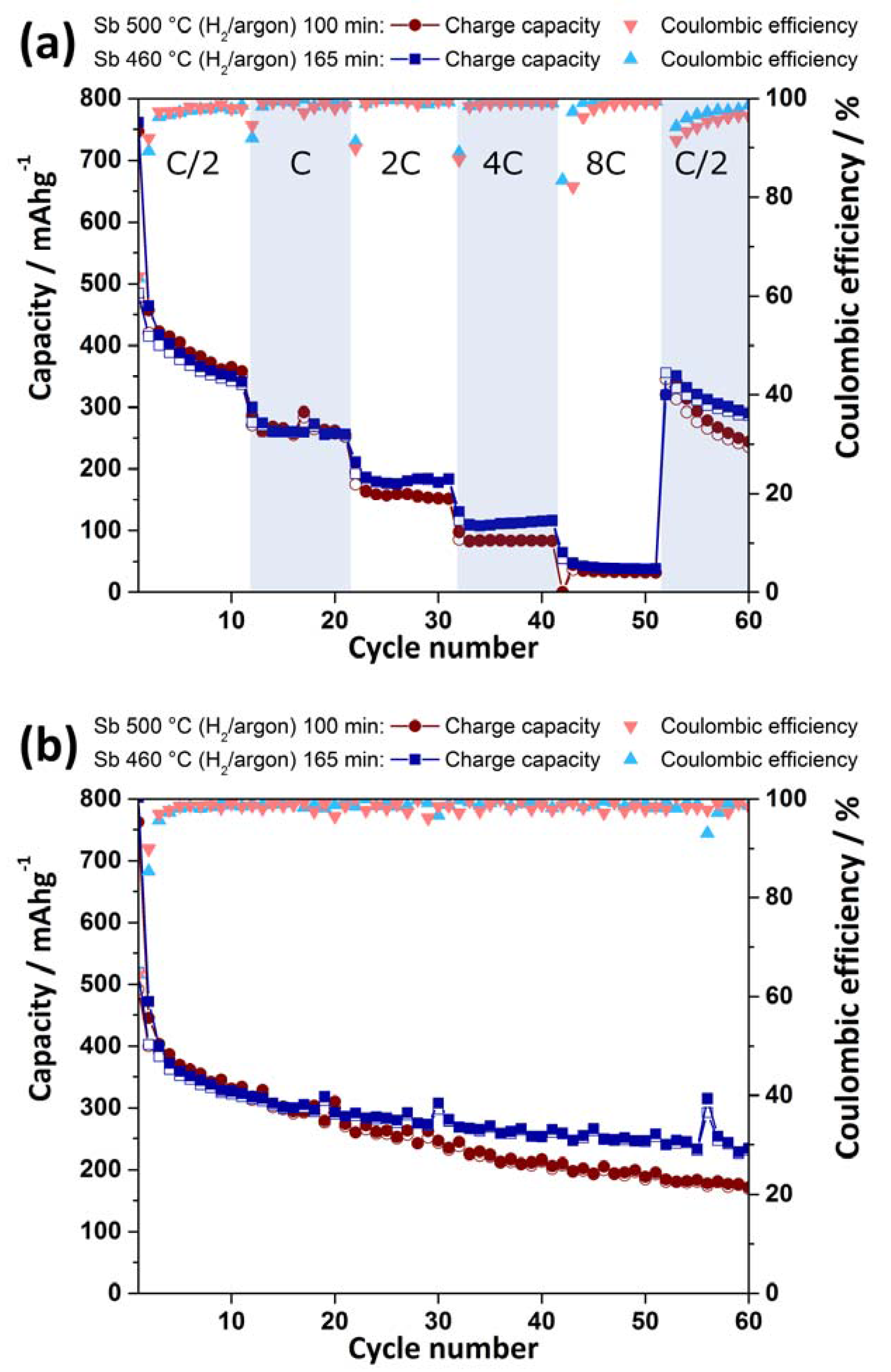
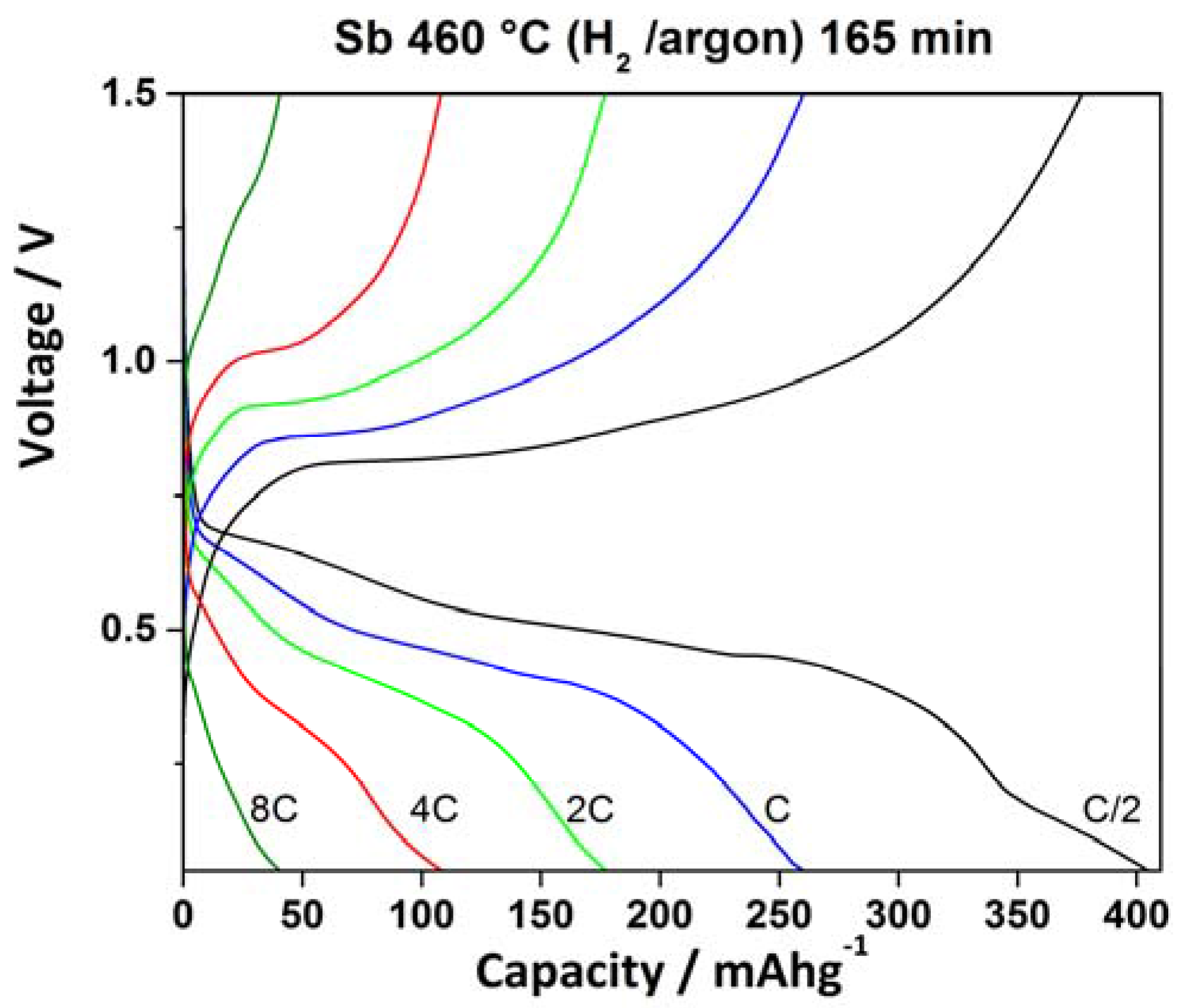
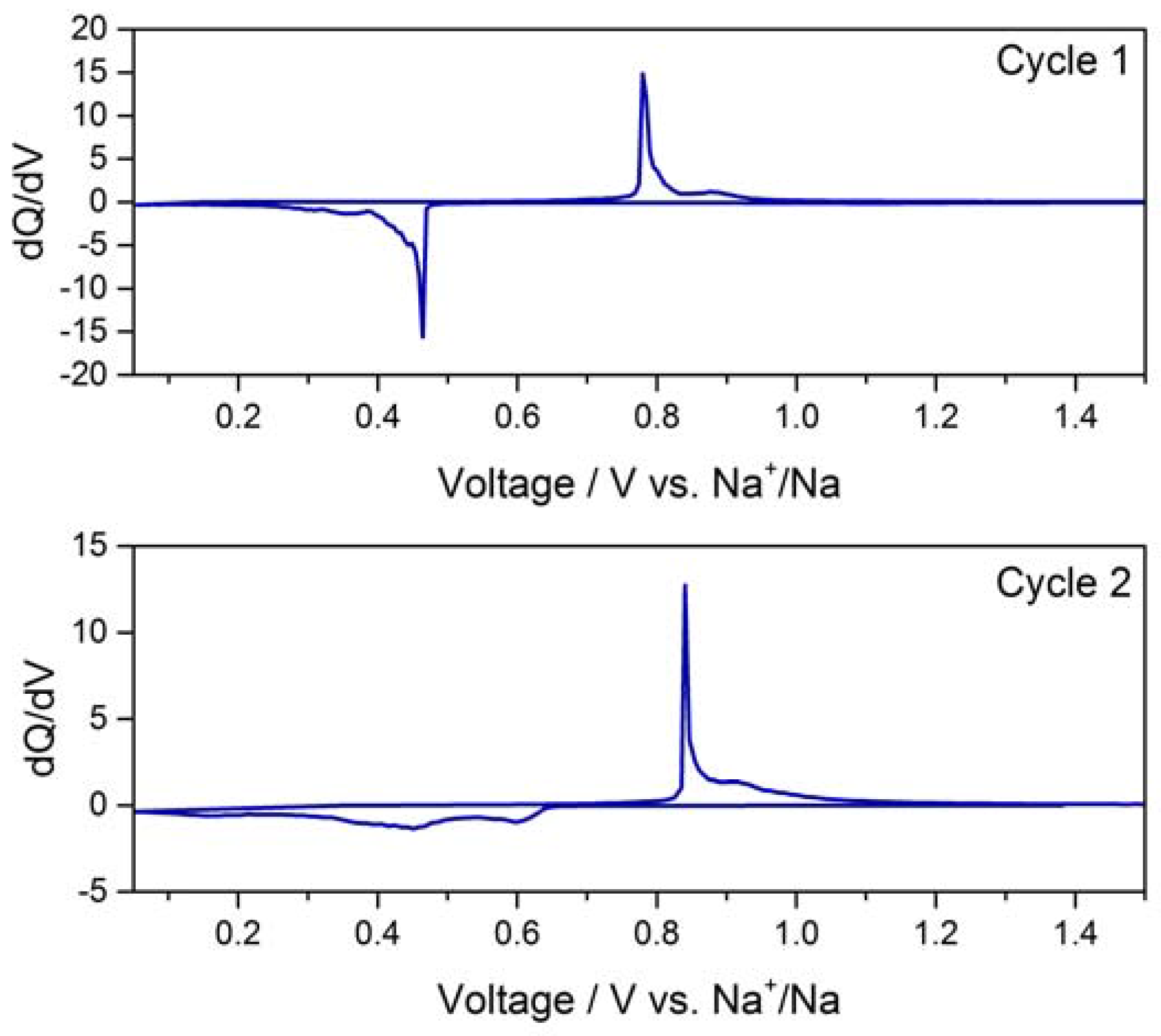
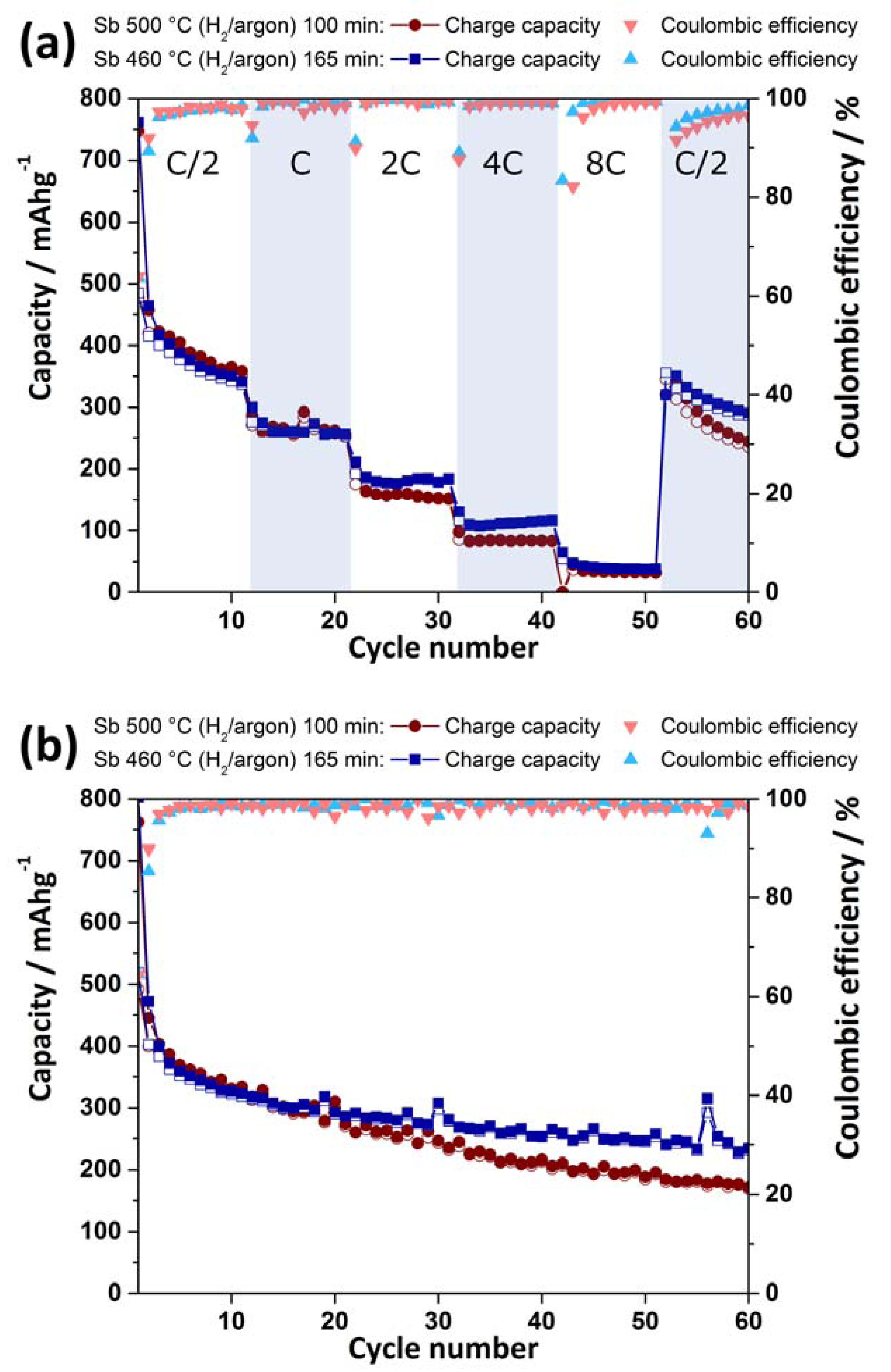
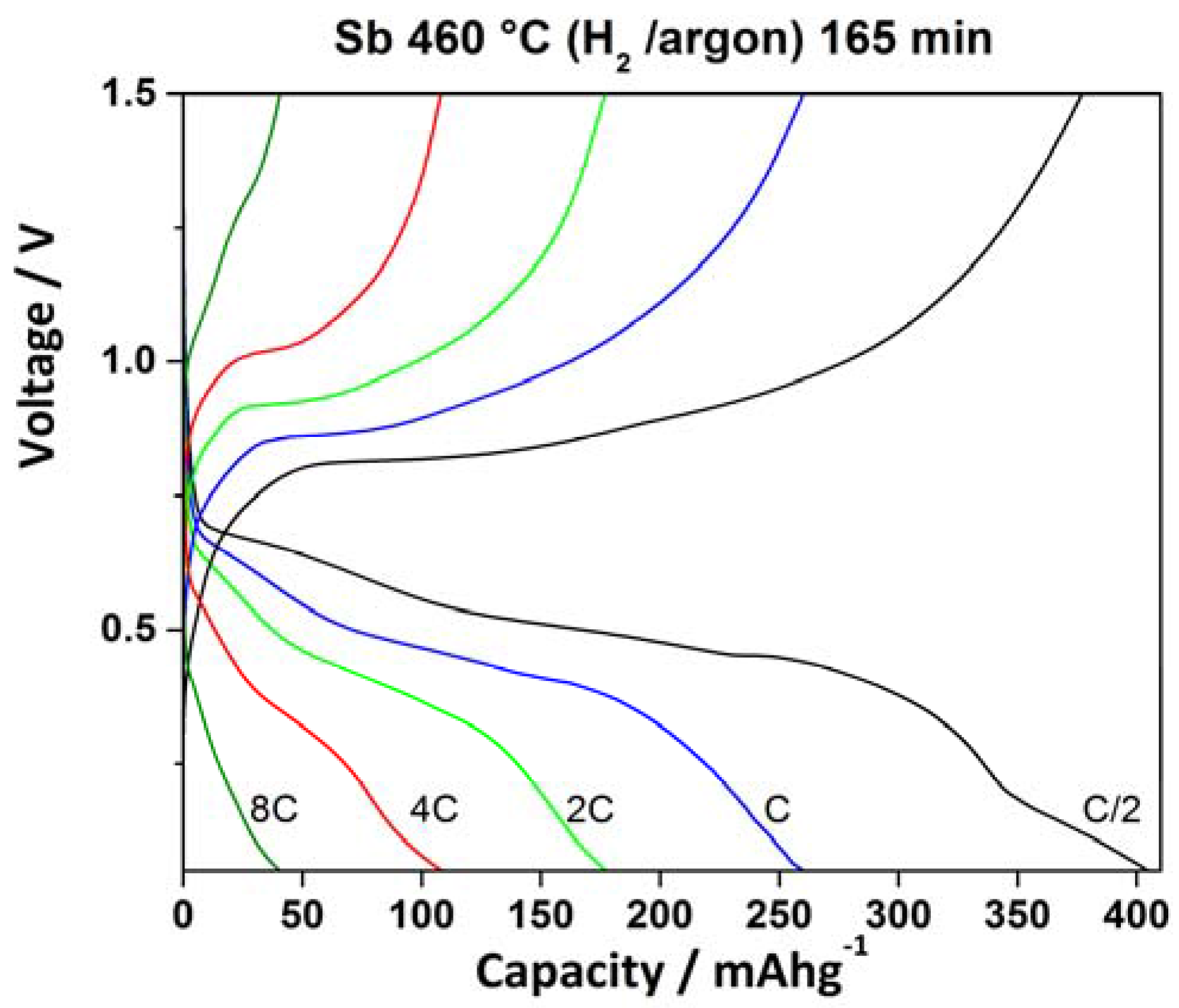
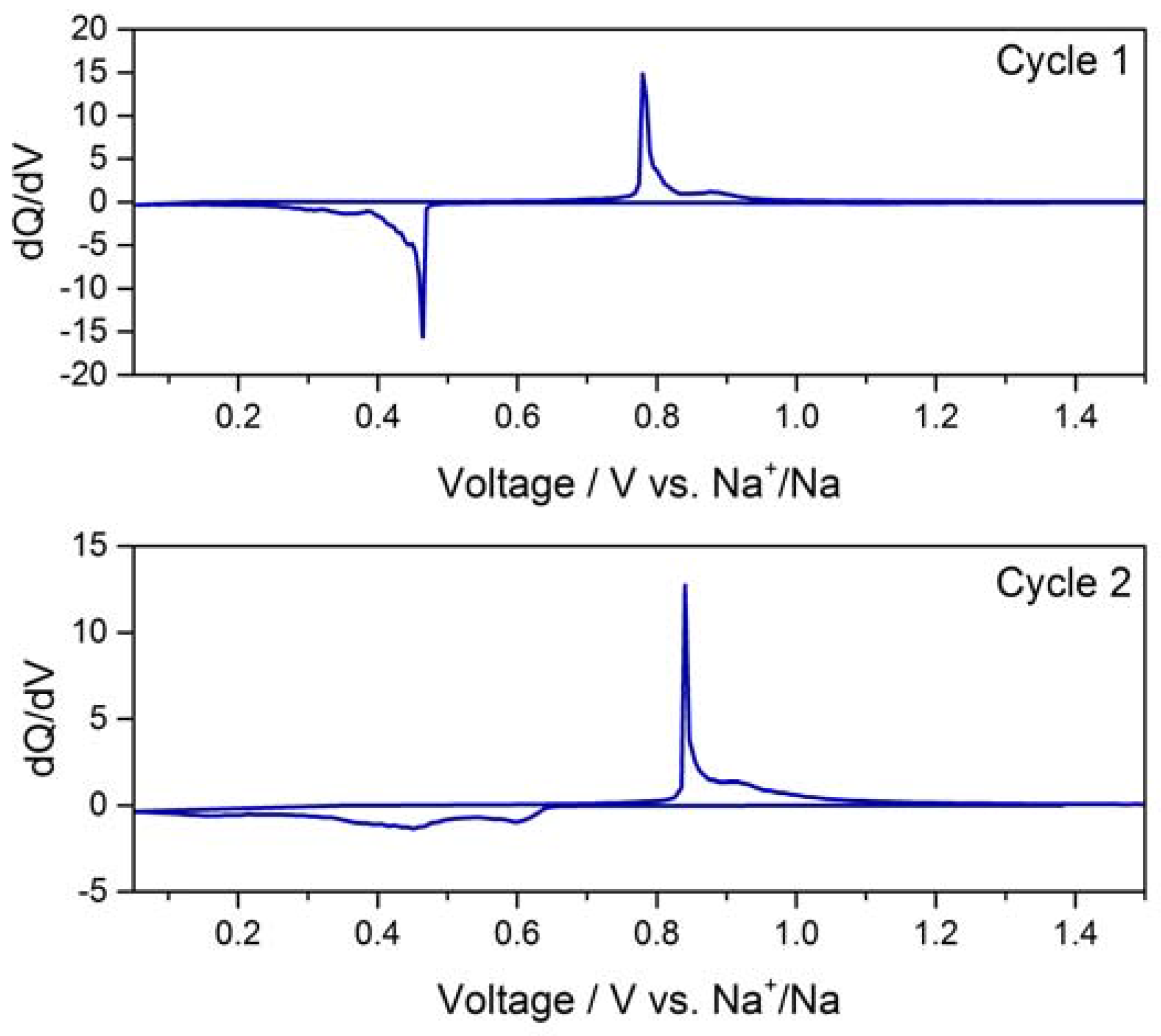
2.3. Electrochemical Characterization
3. Conclusions
4. Materials and Methods
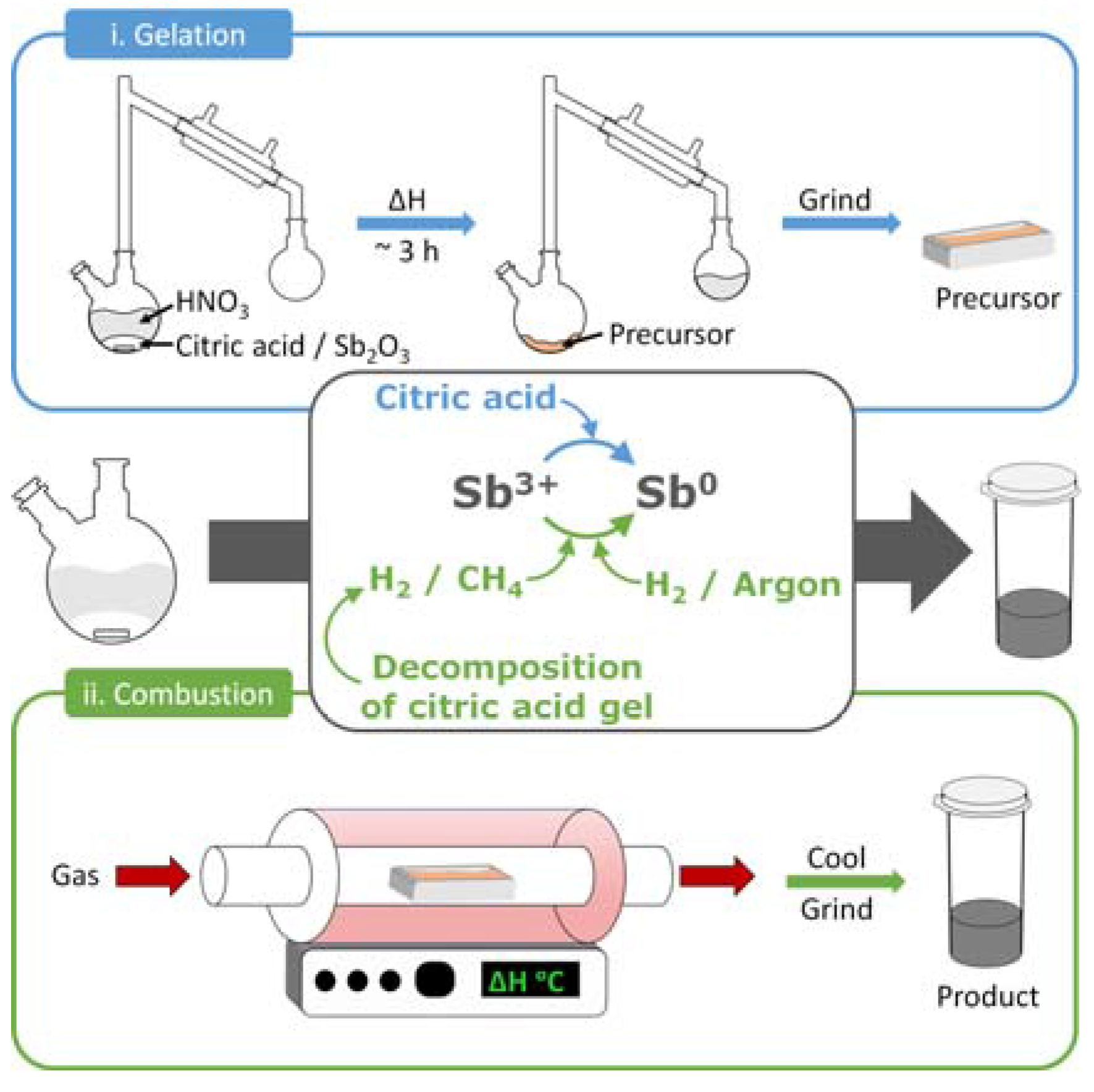
4.1. Synthetic Methods
4.2. Physcial and Chemical Characterisation
4.3. Electrochemical Characterisation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Divya, K.C.; Østergaard, J. Battery energy storage technology for power systems—An overview. Electr. Power Syst. Res. 2009, 79, 511–520. [Google Scholar] [CrossRef]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Bruce, P.G.; Freunberger, S.A.; Hardwick, L.J.; Tarascon, J.-M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 2012, 11, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Palomares, V.; Serras, P.; Villaluenga, I.; Hueso, K.B.; Carretero-González, J.; Rojo, T. Na-ion batteries, recent advances and present challenges to become low cost energy storage systems. Energy Environ. Sci. 2012, 5, 5884–5901. [Google Scholar] [CrossRef]
- Palomares, V.; Casas-Cabanas, M.; Castillo-Martínez, E.; Han, M.H.; Rojo, T. Update on Na-based battery materials. A growing research path. Energy Environ. Sci. 2013, 6, 2312–2337. [Google Scholar] [CrossRef]
- Lu, Q.; Chen, J.G.; Xiao, J.Q. Nanostructured electrodes for high-performance pseudocapacitors. Angew. Chemi. Int. Ed. Engl. 2013, 52, 1882–1889. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Park, K.-S. The Li-ion rechargeable battery: A perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Dahbi, M.; Yabuuchi, N.; Kubota, K.; Tokiwa, K.; Komaba, S. Negative electrodes for Na-ion batteries. Phys. Chem. Chem. Phys. 2014, 16, 15007–15028. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research development on Sodium-ion batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Komaba, S. Review—Practical issues and future perspective for Na-ion batteries. J. Electrochem. Soc. 2015, 162, A2538–A2550. [Google Scholar] [CrossRef]
- Luo, W.; Shen, F.; Bommier, C.; Zhu, H.; Ji, X.; Hu, L. Na-ion battery anodes: Materials and electrochemistry. Acc. Chem. Res. 2016, 49, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, M.; Buqa, H.; Hardwick, L.J.; Hahn, M.; Würsig, A.; Scheifele, W.; Novák, P.; Kötz, R.; Veit, C.; Petrat, F.-M. Nano silicon for lithium-ion batteries. Electrochim. Acta 2006, 52, 973–978. [Google Scholar] [CrossRef]
- Marcinek, M.; Hardwick, L.J.; Richardson, T.J.; Song, X.; Kostecki, R. Microwave plasma chemical vapor deposition of nano-structured Sn/C composite thin-film anodes for Li-ion batteries. J. Power Sources 2007, 173, 965–971. [Google Scholar] [CrossRef]
- Hayner, C.M.; Zhao, X.; Kung, H.H. Materials for rechargeable Lithium-ion batteries. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 445–471. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Xin, F.; Wang, X.; He, W.; Han, W. High capacity group-IV elements (Si, Ge, Sn) based anodes for lithium-ion batteries. J. Mater. 2015, 1, 153–169. [Google Scholar] [CrossRef]
- Kim, Y.; Ha, K.-H.; Oh, S.M.; Lee, K.T. High-capacity anode materials for Sodium-ion batteries. Chem. Eur. J. 2014, 20, 11980–11992. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Liu, Y.; Cao, K.; Zhao, Y.; Jiao, L.; Wang, Y.; Yuan, H. Update on anode materials for Na-ion batteries. J. Mater. Chem. A 2015, 3, 17899–17913. [Google Scholar] [CrossRef]
- Darwiche, A.; Marino, C.; Sougrati, M.T.; Fraisse, B.; Stievano, L.; Monconduit, L. Better cycling performances of bulk Sb in Na-ion batteries compared to Li-ion systems: An unexpected electrochemical mechanism. J. Am. Chem. Soc. 2012, 134, 20805–20811. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Kravchyk, K.; Walter, M.; Kovalenko, M.V. Monodisperse antimony nanocrystals for high-rate Li-ion and Na-ion battery anodes: Nano versus bulk. Nano Lett. 2014, 14, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Baggetto, L.; Ganesh, P.; Sun, C.-N.; Meisner, R.A.; Zawodzinski, T.A.; Veith, G.M. Intrinsic thermodynamic and kinetic properties of Sb electrodes for Li-ion and Na-ion batteries: Experiment and theory. J. Mater. Chem. A 2013, 1, 7985–7994. [Google Scholar] [CrossRef]
- Hou, H.; Jing, M.; Yang, Y.; Zhu, Y.; Fang, L.; Song, W.; Pan, C.; Yang, X.; Ji, X. Sodium/Lithium storage behavior of antimony hollow Nanospheres for rechargeable batteries. ACS Appl. Mater. Interfaces 2014, 6, 16189–16196. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Jing, M.; Zhang, Y.; Chen, J.; Huang, Z.; Ji, X. Cypress leaf-like Sb as anode material for high-performance sodium-ion batteries. J. Mater. Chem. A 2015, 3, 17549–17552. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, X.; Zhang, Y.; Hou, H.; Jing, M.; Zhu, Y.; Fang, L.; Chen, Q.; Ji, X. Cathodically induced antimony for rechargeable Li-ion and Na-ion batteries: The influences of hexagonal and amorphous phase. J. Power Sources 2015, 282, 358–367. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, X.; Xu, Y.; Liu, Y.; Zheng, S.; Xu, K.; Hu, L.; Wang, C. Electrospun Sb/C fibers for a stable and fast Sodium-ion battery anode. ACS Nano 2013, 7, 6378–6386. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Jing, M.; Yang, Y.; Zhang, Y.; Song, W.; Yang, X.; Chen, J.; Chen, Q.; Ji, X. Antimony nanoparticles anchored on interconnected carbon nanofibers networks as advanced anode material for sodium-ion batteries. J. Power Sources 2015, 284, 227–235. [Google Scholar] [CrossRef]
- Qian, J.; Chen, Y.; Wu, L.; Cao, Y.; Ai, X.; Yang, H. High capacity Na-storage and superior cyclability of nanocomposite Sb/C anode for Na-ion batteries. Chem. Commun. 2012, 48, 7070–7072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, Y.; Lu, Y.; Han, X.; Cheng, F.; Chen, J. Spherical nano-Sb@C composite as a high-rate and ultra-stable anode material for sodium-ion batteries. Nano Res. 2015, 8, 3384–3393. [Google Scholar] [CrossRef]
- Zhang, B.; Dugas, R.; Rousse, G.; Rozier, P.; Abakumov, A.M.; Tarascon, J.-M. Insertion compounds and composites made by ball milling for advanced sodium-ion batteries. Nat. Commun. 2016, 7, 10308. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, S.; Hua, Z.; Huang, H. Sol-gel autocombustion synthesis of metals and metal alloys. Angew. Chem. (Int. Ed. Engl.) 2009, 48, 8529–8531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, X.; Xu, Y.; Liu, Y.; Dai, Z.; Bao, J. An SbOx/reduced graphene oxide composite as a high-rate anode material for Sodium-ion batteries. J. Phys. Chem. C 2014, 118, 23527–23534. [Google Scholar] [CrossRef]
- Li, N.; Liao, S.; Sun, Y.; Song, H.W.; Wang, C.X. Uniformly dispersed self-assembled growth of Sb2O3/Sb@graphene nanocomposites on a 3D carbon sheet network for high Na-storage capacity and excellent stability. J. Mater. Chem. A 2015, 3, 5820–5828. [Google Scholar] [CrossRef]
- Li, D.; Yan, D.; Ma, J.; Qin, W.; Zhang, X.; Lu, T.; Pan, L. One-step microwave-assisted synthesis of Sb2O3/reduced graphene oxide composites as advanced anode materials for sodium-ion batteries. Ceram. Int. 2016, 42, 15634–15642. [Google Scholar] [CrossRef]
- Wang, G.-Z.; Feng, J.-M.; Dong, L.; Li, X.-F.; Li, D.-J. Porous graphene anchored with Sb/SbOx as sodium-ion battery anode with enhanced reversible capacity and cycle performance. J. Alloys Compd. 2017, 693, 141–149. [Google Scholar] [CrossRef]
- Li, W.; Wang, K.; Cheng, S.; Jiang, K. A two-dimensional hybrid of SbOx nanoplates encapsulated by carbon flakes as a high performance sodium storage anode. J. Mater. Chem. A 2017, 5, 1160–1167. [Google Scholar] [CrossRef]
- Degtyareva, O.; Struzhkin, V.V.; Hemley, R.J. High-pressure Raman spectroscopy of antimony: As-type, incommensurate host-guest, and bcc phases. Solid State Commun. 2007, 141, 164–167. [Google Scholar] [CrossRef]
- Allan, P.K.; Griffin, J.M.; Darwiche, A.; Borkiewicz, O.J.; Wiaderek, K.M.; Chapman, K.W.; Morris, A.J.; Chupas, P.J.; Monconduit, L.; Grey, C.P. Tracking Sodium-Antimonide phase transformations in Sodium-ion anodes: Insights from operando pair distribution function analysis and solid-state NMR spectroscopy. J. Am. Chem. Soc. 2016, 138, 2352–2365. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Sb (SbAc:CA:Ox—1:3:2) |
|---|---|
| D90 (µm) | 94.8 ± 2.3 |
| D50 (µm) | 39.0 ± 0.7 |
| D10 (µm) | 3.9 ± 0.2 |
| D[4,3] (µm) | 47.2 |
| Parameter | Sb 460 °C (H2/argon) 165 min | Sb 500 °C (H2/argon) 100 min |
|---|---|---|
| D90 (µm) | 12.0 ± 0.5 | 19.5 ± 1.6 |
| D50 (µm) | 4.2 ± 0.1 | 5.9 ± 0.2 |
| D10 (µm) | 1.0 ± 0.1 | 1.5 ± 0.1 |
| D[4,3] (µm) | 5.6 | 8.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drewett, N.E.; Gómez-Cámer, J.L.; Acebedo, B.; Galceran, M.; Rojo, T. Sol-Gel Synthesized Antimony Anodes for Sodium-Ion Batteries: Identifying Key Parameters for Optimization. Batteries 2017, 3, 20. https://doi.org/10.3390/batteries3030020
Drewett NE, Gómez-Cámer JL, Acebedo B, Galceran M, Rojo T. Sol-Gel Synthesized Antimony Anodes for Sodium-Ion Batteries: Identifying Key Parameters for Optimization. Batteries. 2017; 3(3):20. https://doi.org/10.3390/batteries3030020
Chicago/Turabian StyleDrewett, Nicholas E., Juan Luis Gómez-Cámer, Begoña Acebedo, Montserrat Galceran, and Teófilo Rojo. 2017. "Sol-Gel Synthesized Antimony Anodes for Sodium-Ion Batteries: Identifying Key Parameters for Optimization" Batteries 3, no. 3: 20. https://doi.org/10.3390/batteries3030020