Generating CRISPR/Cas9-Derived Mutant Mice by Zygote Cytoplasmic Injection Using an Automatic Microinjector

Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
- C57BL/6Ntac female mice, age 4–6 weeks, for zygote collection
- F1 (CBA <WTSI>; C57BL/6J-Jax F1), mice for use as pseudopregnant females for embryo transfer post-zygote microinjection and vasectomized males.
![Mps 01 00005 i001]() CAUTION Experimental procedures involving animals must be carried out according to all relevant institutional and governmental regulations.
CAUTION Experimental procedures involving animals must be carried out according to all relevant institutional and governmental regulations. - Pregnant mare serum gonadotropin (PMSG; Intervet, Milton Keynes, Buckinghamshire, UK; Cat. no.: Folligon 5000 IU)
- Human chorionic gonadotropin (hCG; Intervet; Cat. no.: Chorulon 1500 IU)
- Potassium-supplemented simplex optimised medium (KSOM medium; AMS Biotechnology (Europe) Limited, Abingdon, Oxfordshire, UK; Cat. no.: GSM 5140)
![Mps 01 00005 i002]() CRITICAL STEP Store KSOM medium at −20 °C. After thawing, keep it at 4 °C and use it within 2 weeks. Use a fresh aliquot each day.
CRITICAL STEP Store KSOM medium at −20 °C. After thawing, keep it at 4 °C and use it within 2 weeks. Use a fresh aliquot each day. - Hyaluronidase (Sigma-Aldrich Company Ltd., Gillingham, Dorset, UK; Cat. no.: H3506)
- M2 medium (Sigma-Aldrich Company Ltd; Cat. no.: M7167)
- Flushing holding medium (FHM Media; AMS Biotechnology (Europe) Limited; Cat. no.: GSM 5130),
- Dulbecco’s phosphate-buffered saline 1× (DPBS 1×; Gibco Life Technologies Europe BV, Bleiswijk, South Holland, Netherlands; Cat. no.: 14190-094).


2.2. Equipment
- Inverted microscope with differential interference contrast (DIC)/Hoffman optics (Leica DMI 4000B, Leica Microsytems (UK) Ltd., Milton Keynes, Buckinghamshire, UK; or Zeiss Axiovert 200M, Carl Zeiss Ltd., Cambridge, Cambridgeshire, UK)
- Micromanipulator set (Eppendorf TransferMan 4r, Cell Tram Air; Eppendorf UK, Stevenage, Hertfordshire, UK)
- Microinjector (Eppendorf UK FemtoJet 4i)
- Microcentrifuge (Eppendorf UK 5415D)
- Holding pipette (Vacutip; Eppendorf UK; Cat. no.: 5175 108.000)
- Microinjection pipette (also referred to as microinjection needle or tip; Eppendorf UK, Femtotip Cat. no.: 5242 952.008)
- Microinjection pipette loading tips (microloader tips; Eppendorf UK; Cat. no.: 5242 956.003)
- CO2 incubator (New Brunswick Galaxy 48 R; Eppendorf UK)
- Stereomicroscope (Leica M125; Leica Microsytems (UK) Ltd.)
- Cavity Slide for microinjection of zygotes VWR International Ltd., Leighton Buzzard, Bedfordshire, UK; Cat. no.: 631-9475)
- One-well culture dishes (Falcon In Vitro Fertilisation (IVF) dish; Corning GmbH, Wiesbaden, Hesse, Germany; Cat. no.: 353653)
- 60 mm Petri dish, Falcon (60 mm × 15 mm; Corning GmbH, Cat. no.: 353004)
- Two pairs of Forceps Dumont #5 (Interfocus Ltd., Linton, Cambridgeshire, UK; Cat. no.: 91150-20)
- 1 mL Pipette and tips (Gilson, Dunstable, Bedfordshire, UK)
3. Procedure
3.1. Zygote Preparation. Time for Completion: 1 h
- Intraperitoneally (IP) inject 10 female C57BL/6NTac (or strain of choice) 4–6 weeks old mice with PMSG (5 IU) at 11.00 a.m.–1 p.m. on day 1.
- After 48 h (i.e., on day 3), IP inject female mice with hCG (5 IU). After the hCG injection, house female mice with C57BL/6NTac stud male mice overnight for mating.
![Mps 01 00005 i003]() CRITICAL STEP The most effective hormone regime to give maximal numbers of eggs will differ between strains and sub-strains and should be determined empirically.
CRITICAL STEP The most effective hormone regime to give maximal numbers of eggs will differ between strains and sub-strains and should be determined empirically. - 21–22 h post-mating, check female mice for the presence of a copulation plug.
- Euthanize the mice and collect zygote-cumulus mass complexes by dissecting the oviduct into prewarmed 37 °C PBS ensuring you do not cut the ampulla.
- Move an oviduct into 1.8 mL of pre-warmed M2 medium and using a stereomicroscope “pop” the oviduct using a pair of forceps at the swollen ampulla region to release the cumulus complex mass. Pin down the oviduct using one pair of forceps whilst gently tearing the ampulla with the other pair. Remove the “popped” oviduct and repeat the process for the remaining oviducts. See TROUBLESHOOTING, Appendix A (Table A1)
- Add 1 vial of thawed hyaluronidase (see Section 5) and gently swirl the dish to disperse cumulus cells. If necessary pipette the masses gently up and down several times with a 1 mL Gilson tip, and wait for the cumulus cells to fall away from the zygotes. This will take 30 s to a couple of minutes.
- Using a mouth pipette, pick up the embryos and place them into a fresh dish of 500 μL FHM followed by washing the embryos through at least 3 × 100 μL drops of FHM medium.
- Remove unfertilised embryos and place the remaining fertilised zygotes (see Figure 1 identifying the main structures of the fertilised mouse zygote) into pre-equilibrated KSOM medium at 37 °C in a 5% CO2 incubator until ready for injection (see Section 5).
![Mps 01 00005 i004]() CRITICAL STEP Long-term exposure to hyaluronidase can cause embryo degradation. The embryos should remain in hyaluronidase for the minimum time possible. Embryos must be washed through several times in FHM after exposure.
CRITICAL STEP Long-term exposure to hyaluronidase can cause embryo degradation. The embryos should remain in hyaluronidase for the minimum time possible. Embryos must be washed through several times in FHM after exposure.


3.2. Preparation of CRISPR/Cas9 Materials. Time for Completion: 30 min
- 9.
- 10.
- Briefly mix all the components and store them at −80 °C until required for microinjection.
![Mps 01 00005 i005]() PAUSE STEP CRISPR/Cas9 injection mix maybe stored at −80 °C for up to one year without any detrimental effect on activity. However, we do not recommend refreezing materials once thawed for microinjection.
PAUSE STEP CRISPR/Cas9 injection mix maybe stored at −80 °C for up to one year without any detrimental effect on activity. However, we do not recommend refreezing materials once thawed for microinjection.

3.3. Preparation for Microinjection. Time for Completion: 20 min
- 11.
- Place 20–30 embryos into a large drop of FHM in the well of a depression slide (100 to 500 μL depending on the size of the depression) or as many as you can comfortably inject in a 30 min period and move to the inverted microscope microinjection rig ready for injection (Figure 2).
![Mps 01 00005 i006]() CRITICAL STEP Our preference is to not use mineral oil to cover the drop. However, this means that after 30 min or so evaporation of the drop will start to take place and there can be a negative impact on the embryos due to osmotic effects. Change the media between each injection dish and do not place more embryos on the dish than you can inject in a 30 min period. If you start to notice embryos showing signs of osmotic stress such as shrinking or increased lysis rates on injection immediately change to a fresh dish.
CRITICAL STEP Our preference is to not use mineral oil to cover the drop. However, this means that after 30 min or so evaporation of the drop will start to take place and there can be a negative impact on the embryos due to osmotic effects. Change the media between each injection dish and do not place more embryos on the dish than you can inject in a 30 min period. If you start to notice embryos showing signs of osmotic stress such as shrinking or increased lysis rates on injection immediately change to a fresh dish. - 12.
- At 40× times magnification attach the holding pipette to the instrument holder and lower into the cavity slide microinjection dish.
- 13.
- Thaw the CRISPR/Cas9 injection mix and spin it for 1 min at 13,200 rpm in the microcentrifuge and store on ice ready for microinjection.
- 14.
- Load 2.5–3 μL of injection mix into a Femtotip using a microloader tip. Ensure the mix reaches the tip of the microinjection needle.
- 15.
- Attach the Femtotip to the instrument holder connected to the Femtojet and switch on the Femtojet and allow it to reach pressure.
- 16.
- Set a balance pressure by adjusting the PC (pressure compensation) range to ca. 40–60 hPa and lower the Femtotip into the microinjection dish until it is the same focal plane as the holding pipette.
- 17.
- Increase the magnification to 200–400× and move the Femtotip next to an embryo and ensure both injection tip and zygote are in focus and press ‘Clean’. If the CRISPR/Cas9 injection mix is flowing you should see a visible stream and the embryo will be pushed away by the pressure. See TROUBLESHOOTING, Appendix A (Table A1)
![Mps 01 00005 i007]() CRITICAL STEP: If the pressure compensation is set too high you will have an uncontrolled injection with high volumes of CRISPR materials delivered into the cytoplasm and the embryos will lyse shortly after.
CRITICAL STEP: If the pressure compensation is set too high you will have an uncontrolled injection with high volumes of CRISPR materials delivered into the cytoplasm and the embryos will lyse shortly after.


3.4. Injection of Zygotes. Time for Completion: 1 h
- 18.
- 19.
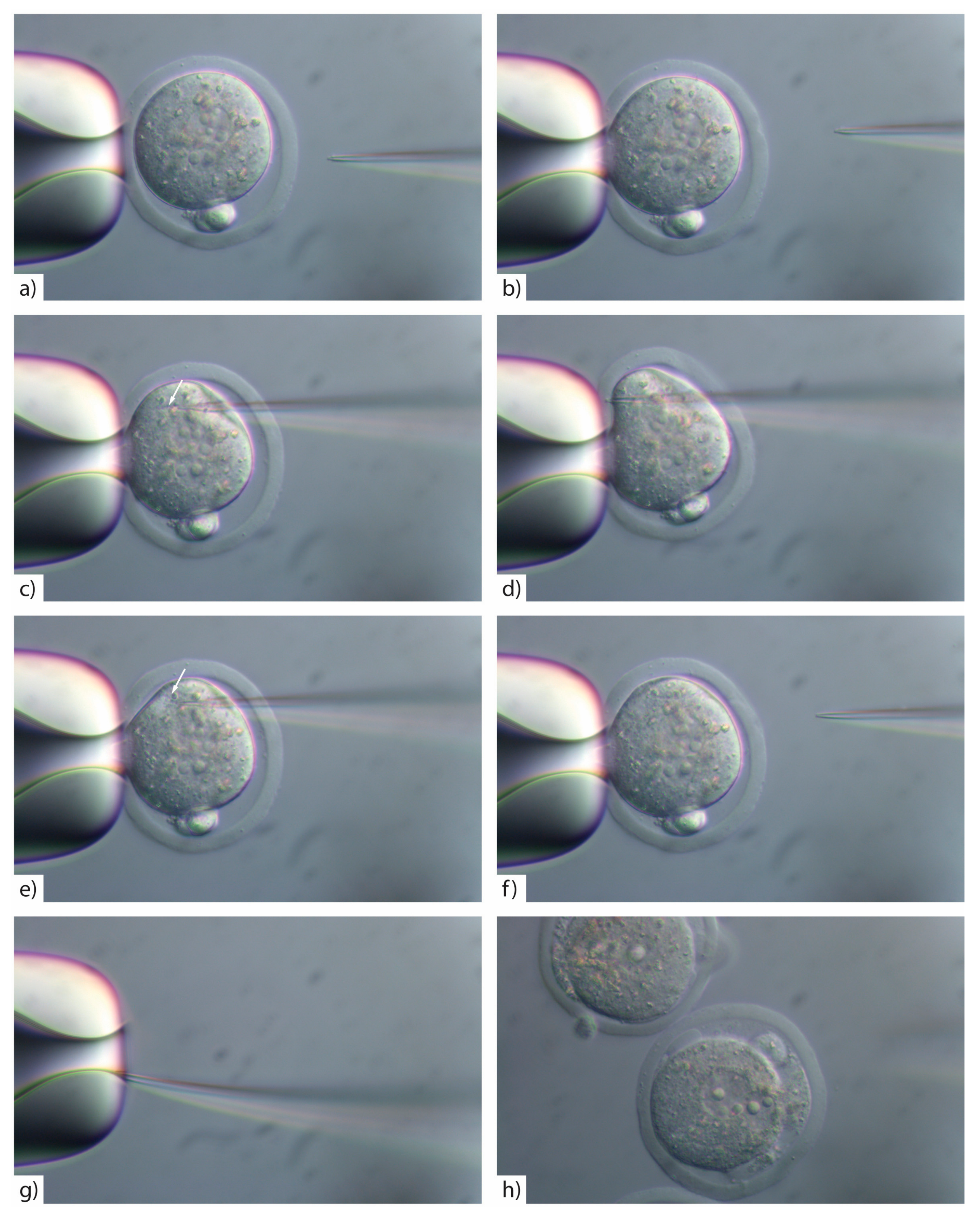
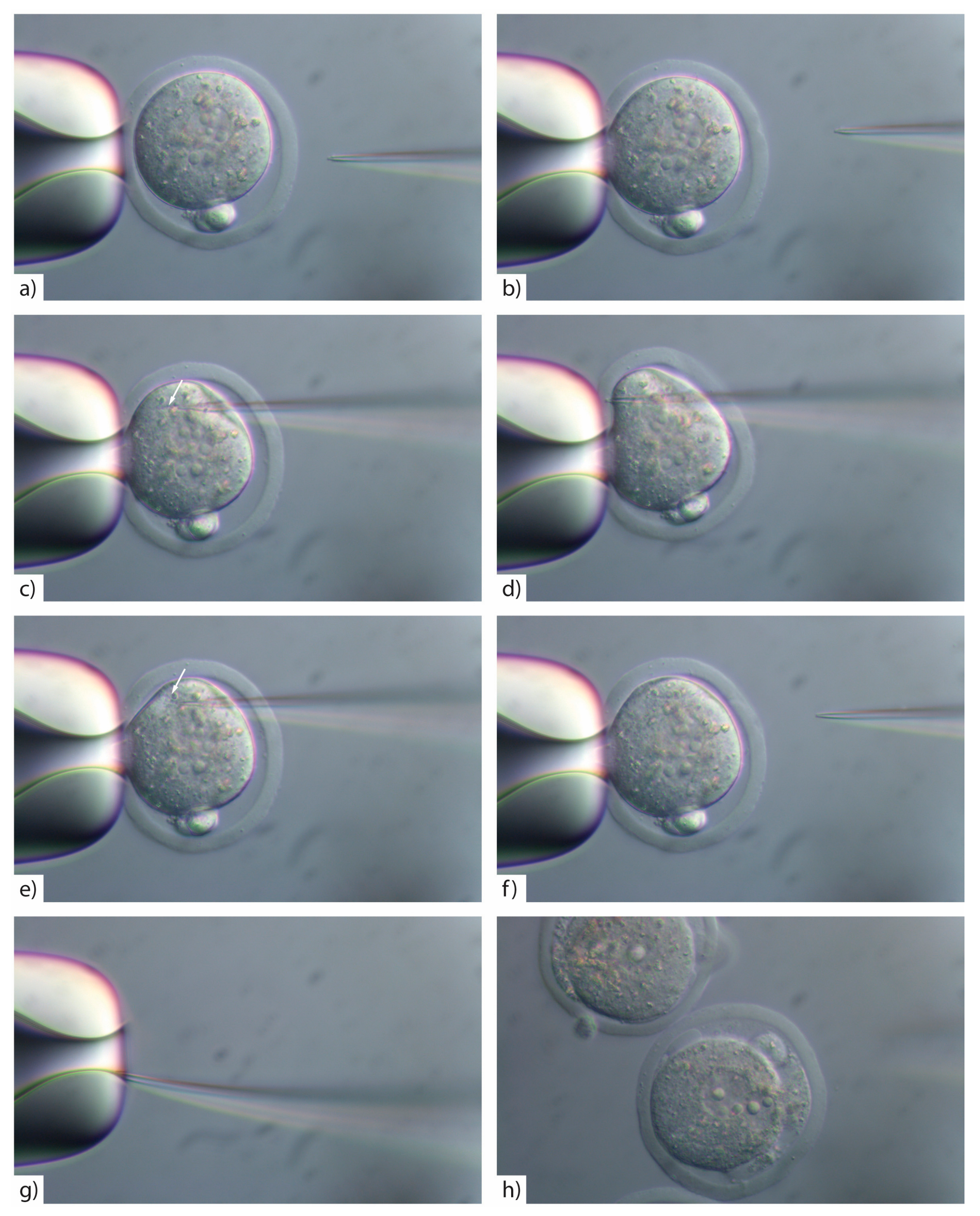
- Insert the injection tip into the zygote and pause briefly halfway inside the egg to see the formation of a small droplet around the injection tip. This shows the CRISPR/Cas injection mix is flowing (Figure 3c). See TROUBLESHOOTING, Appendix A (Table A1)
- 20.
- Push the pipette forward again until it reaches the opposite side of the oolemma. Pass the pipette tip gently through the oolemma to break the membrane and draw back into the embryo (Figure 3d).
- 21.
- Look for movement inside the cytoplasm to signify the CRISPR/Cas injection mix has successfully been injected inside the embryo (Figure 3e). See TROUBLESHOOTING, Appendix A (Table A1)
- 22.
![Mps 01 00005 i008]() CRITICAL STEP: To avoid lysing the zygote, ensure that a minimal amount of CRISPR/Cas mix is injected, by rapidly withdrawing the pipette tip after injection.
CRITICAL STEP: To avoid lysing the zygote, ensure that a minimal amount of CRISPR/Cas mix is injected, by rapidly withdrawing the pipette tip after injection.- 23.
- If the pipette tip becomes blocked with cytoplasmic debris or the mRNA/protein prep contains debris/dust or is sticky due to the oligo prep, widen the tip size slightly by glancing the injection tip on the edge of the holding pipette (Figure 3g).
- 24.
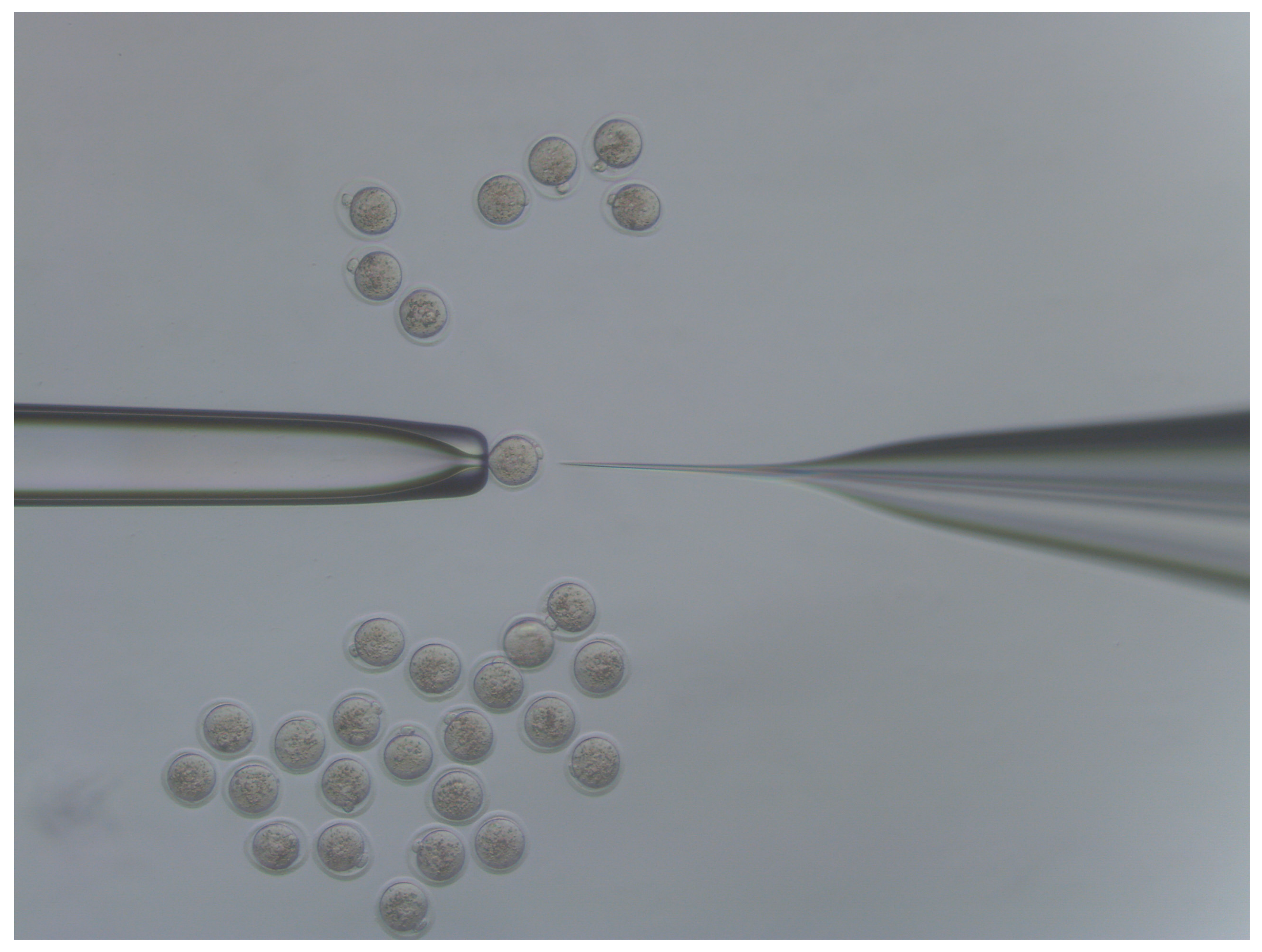
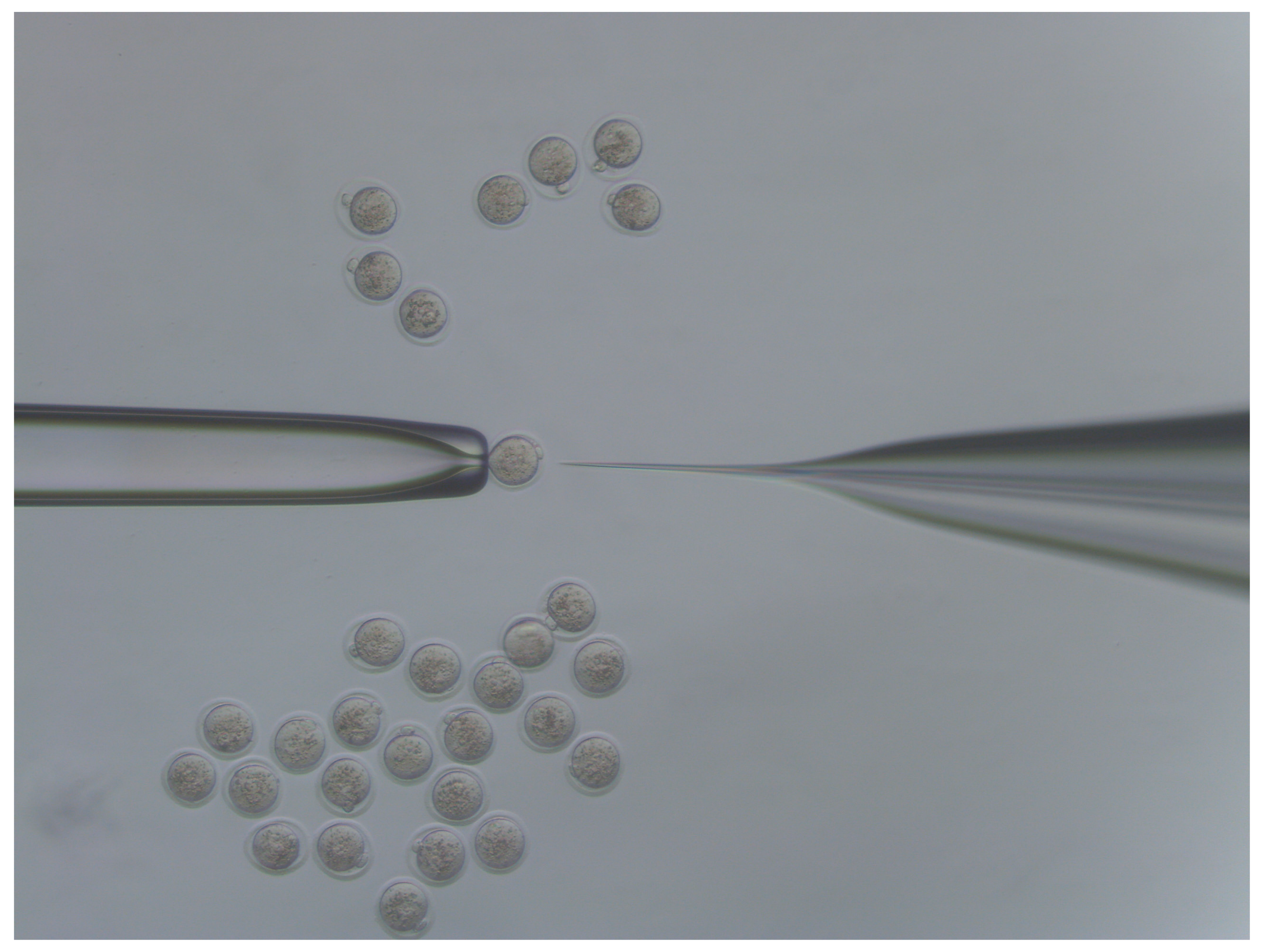
- Move injected embryos to the top of the dish and repeat the process for all embryos in the dish (Figure 4).
![Mps 01 00005 i009]() CRITICAL STEP: Accurate focus is crucial for several steps in zygote micromanipulation. The injection pipette, zygote and the holding pipette must be in the same horizontal plane.
CRITICAL STEP: Accurate focus is crucial for several steps in zygote micromanipulation. The injection pipette, zygote and the holding pipette must be in the same horizontal plane.![Mps 01 00005 i010]() CRITICAL STEP: If you widen the microinjection needle tip by glancing off the side of the holding pipette you must reduce the PC as larger volumes of reagents introduced into the cytoplasm will result in embryo lysis (Figure 3h).
CRITICAL STEP: If you widen the microinjection needle tip by glancing off the side of the holding pipette you must reduce the PC as larger volumes of reagents introduced into the cytoplasm will result in embryo lysis (Figure 3h).![Mps 01 00005 i011]() CRITICAL STEP: If lysis of embryos is observed after widening the pipette tip and reducing the PC, exchange the injection pipette for a new one.
CRITICAL STEP: If lysis of embryos is observed after widening the pipette tip and reducing the PC, exchange the injection pipette for a new one. - 25.
- Culture the injected zygotes in KSOM medium at 37 °C in a 5% CO2 incubator and 30 min later remove any that might have lysed. The surviving embryos can then be transferred immediately to the oviduct of a pseudopregnant 0.5 d.p.c. recipient or left to culture overnight and transferred the next day to a 0.5 d.p.c. pseudopregnant recipient as two-cell embryos. See TROUBLESHOOTING, Appendix A (Table A1).




3.5. Embryo Transfer and Production of Mice. Time for Completion: 3 Weeks
- 26.
- Prepare pseudopregnant foster mothers by mating estrus selected F1female mice or chosen strain with vasectomized male mice the day before injection and inspecting for a vaginal plug on the day of injection (0.5 d.p.c.).
- 27.
- Perform an embryo transfer of 10–15 one-cell embryos into the oviduct of 0.5 d.p.c. recipients. If you have insufficient recipients to transfer all of your microinjected embryos, culture the embryos overnight and transfer two-cell embryos into the oviduct of 0.5 d.p.c. recipients set up on the day of injection. Recipient mothers deliver pups at approximately 19.5 d.p.c.
![Mps 01 00005 i012]() CRITICAL STEP: Do not transfer more than 10–15 embryos to each oviduct (20–30/mouse in total). Survival rate of cytoplasmic CRISPR/Cas9-injected embryos can be high so transferring more embryos may result in pregnancy problems for the recipient mother. The vast majority of those embryos that survive microinjection will divide to two-cell embryos (over 90%) so culturing overnight is not advised or necessary.
CRITICAL STEP: Do not transfer more than 10–15 embryos to each oviduct (20–30/mouse in total). Survival rate of cytoplasmic CRISPR/Cas9-injected embryos can be high so transferring more embryos may result in pregnancy problems for the recipient mother. The vast majority of those embryos that survive microinjection will divide to two-cell embryos (over 90%) so culturing overnight is not advised or necessary. - 28.
- When pups reach two weeks of age ear clip pups and genotype. See TROUBLESHOOTING, Appendix A (Table A1).
- 29.
- Genotyped mice can be sexed and weaned at approximately three weeks after birth.

3.6. Summarized Time for Completion
- Steps 1–8 Zygote preparation: 1 h
- Steps 9–10 CRISPR/Cas9 preparation: 30 min
- Steps 11–17 Microinjection preparation: 20 min
- Steps 18–25 Injection of zygotes: 1 h
- Steps 26–29 embryo transfer and production of mice: 3 weeks
- Supplemental 1 Design and cloning of tyrosinase gRNAs: 1 week
4. Expected Results
5. Reagents Setup
- Thaw KSOM and filter through a 0.22 µm filter and aliquot in 5 mL aliquots and keep at 4 °C for up to two weeks.
- Place a single drop of 1 mL of KSOM medium in an IVF one well dish and fill the outer well with 2–3 mL PBS. Do not cover with mineral oil. Then place the dish into a 37 °C incubator preferably overnight or at least 2 h before use to equilibrate
- Thaw FHM and filter through a 0.22 µm filter and aliquot in 5 mL aliquots and keep at 4 °C for up to two weeks.
- Hyaluronidase. From a 100 mg stock powder make a 3 mg/μL working solution by dissolving the powder in 33.33 mL M2 media, filtering through a 0.22 µm filter, aliquoting in 100 μL aliquots in sterile Eppendorf tubes and freezing at −20 °C ready for use.
- PMSG and HCG. Dissolve separately in 0.9% saline to a final concentration of 50 IU/mL and filter through a 0.22 µm filter and aliquot in 1 mL aliquots in sterile Eppendorf tubes and store at −80 °C
- Dilute Cas9 mRNA (Trilink, San Diego, California, USA; Cat. no.: L-7206) or Cas9 protein (LabOmics, Nivelles, Walloon Brabant, Belgium; Cat. no.: Cas9-TOO) to the required working concentration in RNase-free water. Add gRNA (Protocol S1) and if required a single-stranded oligodeoxynuceotide (ssODN) (Table 1)
- ssODN stock, 1 μg/μL (IDT Leuven, Flemish Brabant, Belgium). Resuspend ssODN in RNase-free water to a final concentration of 1 μg/μL. Store the stock at −20 °C.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 5 | Cumulus cells sticks to forceps when removing from oviduct. | Withdraw the forceps from the M2 and surface tension will pull the cumulus encased zygotes into the drop. | |
| 17 | No injection mix is flowing from the pipette | Cas9 preparation has caused injection needle blockage | Spin injection mix at max speed for 1 min prior to injection |
| You have not cleared the injection needle prior to injection | Press ‘Clean’ on the Femtojet to deliver a high pressured burst of air to the needle tip | ||
| Needle is blocked | Gently break off a small piece of the injection needle tip or change needle | ||
| 19 | No droplet is visible around injection tip when tip is in the cytoplasm | Pressure compensation (PC) is insufficient to produce a flow | Increase balance pressure to 40–60 hPa or until you get a flow |
| Needle is blocked | Repeat step 17 and press ‘Clean’. If the issue persists gently break off a small piece of the injection needle tip or change needle | ||
| 21 | No movement is visible inside the cytoplasm post-injection | Oolemma has not been broken. | Withdraw the injection needle part way back into the cytoplasm and then reattempt penetration of the membrane at a different point. |
| Optics are not focused correctly on cytoplasm | Adjust optics so you can see clearly the needle tip inside the cytoplasm. | ||
| No movement is visible inside the cytoplasm post-injection despite repeated attempts at penetration. | Oolemma has not been broken. | Change needle as it has become blunt. | |
| Inject the membrane at the point at which the embryo is held by the holding pipette. This point of the membrane is held under negative pressure making penetration easier. | |||
| It’s difficult to control the flow of CRISPR material during injection | PC is too high | Decrease the balance pressure until flow is controlled | |
| Injection tip size is too large | Change to a fresh needle | ||
| 22 | Cytoplasmic membrane attaches to the needle on exiting embryo post injection | Needle is sticky with cytoplasmic/nucleolar material | Change needle |
| 25 | High lysis rate post injection | PC is too high | Reduce balance pressure so that flow of CRISPR/Cas material is delivered in a controlled manner |
| Pipette tip is large post breaking the tip | Change needle | ||
| Injection needle is left in the cytoplasm too long | Remove the injection needle rapidly post-injection | ||
| Injection needle and zygote not in the same horizontal plane | Adjust the injection needle so that it is in the same focal plane as the zygote. | ||
| Osmotic effects making zygotes more sensitive to injection | Change to a fresh drop immediately. | ||
| Zygotes appear shrunken prior to injection | Evaporation of media from dish | Change to a fresh drop immediately | |
| 28 | Low mutant rates | CRISPR materials have not been delivered to the cytoplasm | Ensure the oolemma has been broken and not just invaginated around the pipette. |
| CRISPR/Cas9 materials at wrong concentration | Make sure that concentrations are correct and sufficient to generate mutations. |
References
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, J.; Wu, H.; Wang, J.; Ma, K.; Li, Z.; Zhang, X.; Zhang, P.; Huang, X. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013, 23, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.R.; Tibbit, C.; Ponting, C.P.; Liu, J.-L. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Zhou, H.; Bi, H.; Fromm, M.; Yang, B.; Weeks, D.P. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013, 41, e188. [Google Scholar]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Brinster, R.L.; Chen, H.Y.; Trumbauer, M.E.; Yagle, M.K.; Palmiter, R.D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc. Natl. Acad. Sci. USA 1985, 82, 4438–4442. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Barg-Kues, B.; Broll, S.; Bode, J.; Niemann, H.; Kues, W. Cytoplasmic injection of circular plasmids allows targeted expression in mammalian embryos. Biotechniques 2009, 47, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Sumiyama, K.; Kawakami, K.; Yagita, K. A simple and highly efficient transgenesis method in mice with the Tol2 transposon system and cytoplasmic microinjection. Genomics 2010, 95, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Dunlap-Brown, M.; Butler, S.P.; Velander, W.H.; Gwazdauskas, F.C. Murine embryo development following cytoplasmic injection of linear and condensed DNA. Open J. Anim. Sci. 2012, 2, 244–252. [Google Scholar] [CrossRef]
- Garrels, W.; Talluri, T.R.; Ziegler, M.; Most, I.; Forcato, D.O.; Schmeer, M.; Schleef, M.; Ivics, Z.; Kues, W.A. Cytoplasmic injection of murine zygotes with Sleeping Beauty transposon plasmids and minicircles results in the efficient generation of germline transgenic mice. Biotechnol. J. 2016, 11, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, D.; Fujihara, Y.; Satouh, Y.; Miyata, H.; Isotani, A.; Ikawa, M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 2013, 3, 3355. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Doe, B.; Ajduk, A.; Ward, M.A. Genomic DNA damage in mouse transgenesis. Biol. Reprod. 2007, 77, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Arai, Y.; Yamazaki, M.; Morita, S.; Kimura, M.; Itoh, M.; Abe, Y.; Hatada, I. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 2014, 4, 4513. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014, 9, 1956–1968. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Takemoto, T. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Sci. Rep. 2015, 5, 11315. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Dion, S.L.; Kutny, P.M.; Zhang, Y.; Cheng, A.W.; Jillette, N.L.; Malhotra, A.; Geurts, A.M.; Chen, Y.G.; Wang, H. Efficient CRISPR/Cas9-Mediated Genome Editing in Mice by Zygote Electroporation of Nuclease. Genetics 2015, 200, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Page, R.L.; Butler, S.P.; Subramanian, A.; Gwazdauskas, F.C.; Johnson, J.L.; Velander, W.H. Transgenesis in mice by cytoplasmic injection of polylysine/DNA mixtures. Transgenic Res. 1995, 4, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; de Angelis, M.H.; Wurst, W.; Kühn, R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 2010, 107, 15022–15026. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.H.; Baek, I.J.; Kim, D.H.; Jeon, J.; Lee, J.; Lee, K.; Jeong, D.; Kim, J.S.; Lee, H.W. Knockout mice created by TALEN-mediated gene targeting. Nat. Biotechnol. 2013, 31, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Shen, X.H.; Zheng, Z.; Wang, Z.D.; Liu, Z.H.; Jin, L.H.; Lei, L. Cytochalasin B treatment of mouse oocytes during intracytoplasmic sperm injection (ICSI) increases embryo survival without impairment of development. Zygote 2012, 20, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.M.; Moore, M.W. Towards an encyclopaedia of mammalian gene function: The International Mouse Phenotyping Consortium. Dis. Model. Mech. 2012, 5, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Boroviak, K.; Doe, B.; Banerjee, R.; Yang, F.; Bradley, A. Chromosome engineering in zygotes with CRISPR/Cas9. Genesis 2016, 54, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Boroviak, K.; Zhang, Q.; Assadi, G.; Kempster, S.L.; Sewell, G.W.; Saveljeva, S.; Ashcroft, J.W.; Clare, S.; Mukhopadhyay, S.; et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 2016, 17, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Scavizzi, F.; Ryder, E.; Newman, S.; Raspa, M.; Gleeson, D.; Wardle-Jones, H.; Montoliu, L.; Fernandez, A.; Dessain, M.L.; Larrigaldie, V.; et al. Blastocyst genotyping for quality control of mouse mutant archives: An ethical and economical approach. Transgenic Res. 2015, 24, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Behringer, R.; Gertsenstein, M.; Nagy, K.V.; Nagy, A. Manipulating the Mouse Embryo: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2014. [Google Scholar]


| Aim | Working Concentrations | Components |
|---|---|---|
| Indels by NHEJ | Cas9 (50 ng/μL) + gRNA (25 ng/μL) | Cas9 and one gRNA in 30 μL of H2O |
| Point mutation, small tag insertion or insertion of loxP | Cas9 (50 ng/μL) + gRNA (25 ng/μL) + ssODN (100 ng/μL) | Cas9, one (for point mutation) to four (insertion of loxP) gRNAs, ssODN (120–200 bp) in 30 μL of H2O |
| deletion of critical exons or rearrangement of larger fragments | Cas9 (50 ng/μL) + gRNA5a (6.5 ng/μL) + gRNA5b (6.5 ng/μL) + gRNA3a (6.5 ng/μL) + gRNA3b (6.5 ng/μL) + ssODN (100 ng/μL) | Cas9, two to four gRNAs, ssODN (120–200 bp) in 30 μL of H2O |
| Desired Allele | No. Genes | No. Embryos Microinjected (MI) | No. Embryos Survived MI (%) | No. Embryos Transferred | No. Embryos Born (%) | Total No. Mutants Born (%) * | No. Mutants with Desired Allele (%) |
|---|---|---|---|---|---|---|---|
| CE | 328 | 15,613 | 12,908 (83) | 12,304 | 3776 (31) | 2470 (65) | 1455 (39) |
| Point Mutation (SNP) | 29 | 2744 | 2280 (83) | 2140 | 727 (34) | 504 (69) | 277 (38) ** |
| Large deletions (9, 376–1, 151, 853 bp) | 5 | 1342 | 1093 (81) | 1063 | 273 (26) | 53 (19) *** | 17 (6) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doe, B.; Brown, E.; Boroviak, K. Generating CRISPR/Cas9-Derived Mutant Mice by Zygote Cytoplasmic Injection Using an Automatic Microinjector. Methods Protoc. 2018, 1, 5. https://doi.org/10.3390/mps1010005
Doe B, Brown E, Boroviak K. Generating CRISPR/Cas9-Derived Mutant Mice by Zygote Cytoplasmic Injection Using an Automatic Microinjector. Methods and Protocols. 2018; 1(1):5. https://doi.org/10.3390/mps1010005
Chicago/Turabian StyleDoe, Brendan, Ellen Brown, and Katharina Boroviak. 2018. "Generating CRISPR/Cas9-Derived Mutant Mice by Zygote Cytoplasmic Injection Using an Automatic Microinjector" Methods and Protocols 1, no. 1: 5. https://doi.org/10.3390/mps1010005
APA StyleDoe, B., Brown, E., & Boroviak, K. (2018). Generating CRISPR/Cas9-Derived Mutant Mice by Zygote Cytoplasmic Injection Using an Automatic Microinjector. Methods and Protocols, 1(1), 5. https://doi.org/10.3390/mps1010005