
Atypical Teratoid/Rhabdoid Tumor with Retained SMARCB1 (INI1) Expression and Rare SMARCA4 Gene Mutation: A Case Report of a Pediatric Patient
Abstract
:1. Introduction
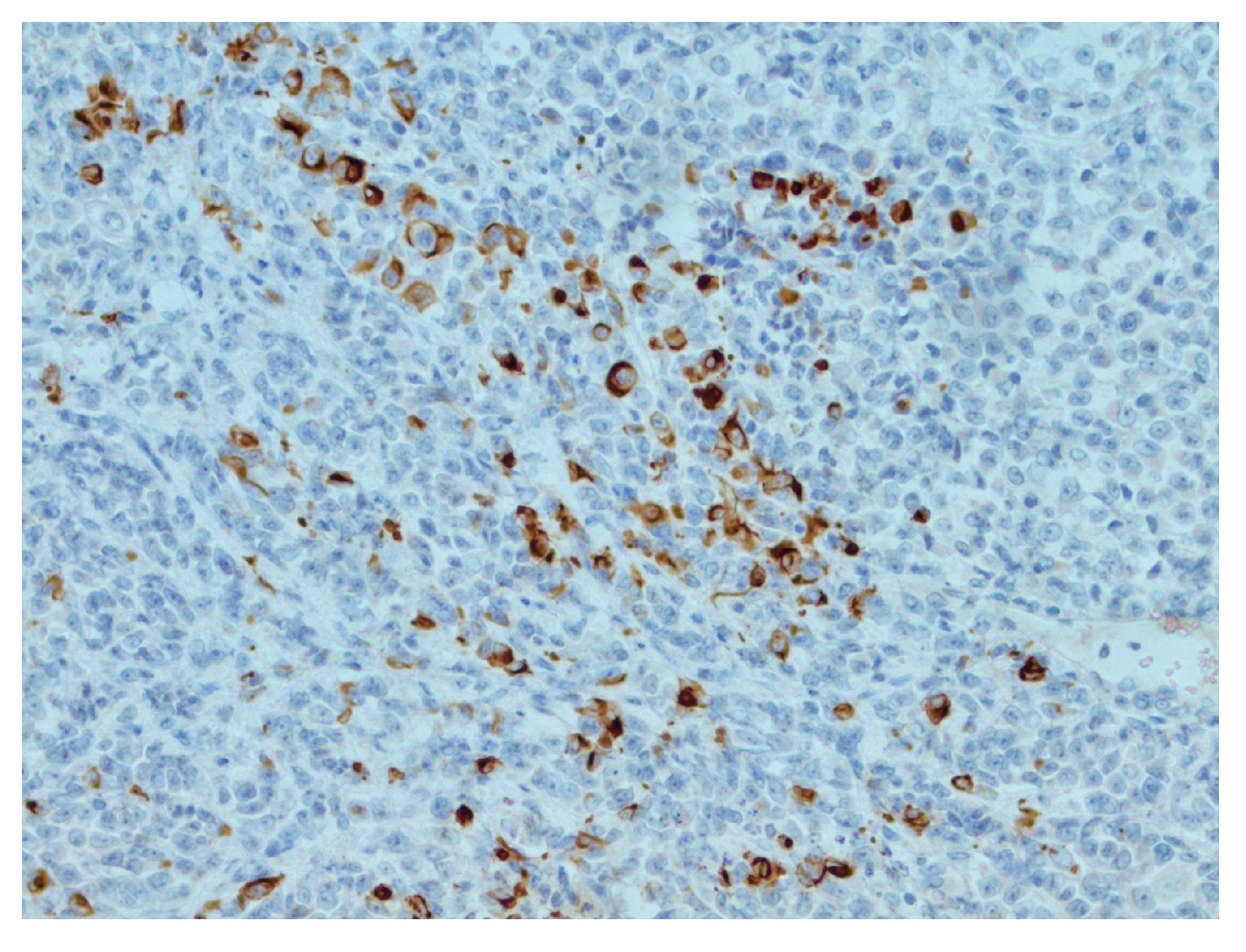
2. Detailed Case Description
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biswas, A.; Kashyap, L.; Kakkar, A.; Sarkar, C.; Julka, P.K. Atypical teratoid/rhabdoid tumors: Challenges and search for solutions. Cancer Manag. Res. 2016, 8, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Alva, E.; Rubens, J.; Chi, S.; Rosenberg, T.; Reddy, A.; Raabe, E.H.; Margol, A. Recent progress and novel approaches to treating atypical teratoid rhabdoid tumor. Neoplasia 2023, 37, 100880. [Google Scholar] [CrossRef] [PubMed]
- Nesvick, C.L.; Lafay-Cousin, L.; Raghunathan, A.; Bouffet, E.; Huang, A.A.; Daniels, D.J. Atypical teratoid rhabdoid tumor: Molecular insights and translation to novel therapeutics. J. Neurooncol. 2020, 150, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Bookhout, C.; Bouldin, T.W.; Ellison, D.W. Atypical teratoid/rhabdoid tumor with retained INI1 (SMARCB1) expression and loss of BRG1 (SMARCA4). Neuropathology 2018, 38, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Holdhof, D.; Johann, P.D.; Spohn, M.; Bockmayr, M.; Safaei, S.; Joshi, P.; Masliah-Planchon, J.; Ho, B.; Andrianteranagna, M.; Bourdeaut, F.; et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021, 141, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Thomas, C.; Miskiewicz, K.; Woltering, N.; Zin, F.; Nemes, K.; Bison, B.; Johann, P.D.; Hawes, D.; Bens, S.; et al. ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol. 2022, 143, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.W.; Biegel, J.A. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol. Ther. 2009, 8, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Mardinian, K.; Adashek, J.J.; Botta, G.P.; Kato, S.; Kurzrock, R. SMARCA4: Implications of an Altered Chromatin-Remodeling Gene for Cancer Development and Therapy. Mol. Cancer Ther. 2021, 20, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, C.; Shirai, R.; Kimura, Y.; Sioda, Y.; Osumi, T.; Kato, M.; Tomizawa, D.; Usami, K.; Ogiwara, H.; Aoki, H.; et al. ATRT-32. preparation of genetic testing system of AT/RT for pediatric cancer predisposition program. Neuro Oncol. 2018, 20 (Suppl. S2), i34. [Google Scholar] [CrossRef]
- Guo, G.; Zhuang, J.; Zhang, K.; Zhou, Z.; Wang, Y.; Zhang, Z. Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System in Children: Case Reports and Literature Review. Front. Surg. 2022, 9, 864518. [Google Scholar] [CrossRef]
- Yang, M.; Chen, X.; Wang, N.; Zhu, K.; Hu, Y.Z.; Zhao, Y.; Shu, Y.; Zhao, M.L.; Gu, W.Z.; Tang, H.F. Primary atypical teratoid/rhabdoid tumor of central nervous system in children: A clinicopathological analysis and review of literature in China. Int. J. Clin. Exp. Pathol. 2014, 7, 2411–2420. [Google Scholar] [PubMed]
- Karim, A.; Shaikhyzada, K.; Suleimenova, A.; Ibraimov, B.; Nurgaliev, D.; Poddighe, D. Case report: Atypical teratoid/rhabdoid tumor of the lateral ventricle in a male adolescent (case-based review and diagnostic challenges in developing countries). Front. Oncol. 2022, 12, 985862. [Google Scholar] [CrossRef] [PubMed]
- Bartelheim, K.; Nemes, K.; Seeringer, A.; Kerl, K.; Buechner, J.; Boos, J.; Graf, N.; Dürken, M.; Gerss, J.; Hasselblatt, M.; et al. Improved 6-year overall survival in AT/RT—Results of the registry study Rhabdoid 2007. Cancer Med. 2016, 5, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- GPOH. The Society for Paediatric Oncology and Haematology. European Rhabdoid Registry.EU-RHAB. Available online: https://www.gpoh.de/kinderkrebsinfo/content/e1676/e9032/e1758/e83294/download84621/EU-RHABProtokoll_2021_08_ger.pdf (accessed on 1 December 2023).
- Jaunmuktane, Z.; Capper, D.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Dutt, M.; Suraweera, N.; Pfister, S.M.; von Deimling, A.; Brandner, S. Methylation array profiling of adult brain tumours: Diagnostic outcomes in a large, single centre. Acta Neuropathol. Commun. 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.A.; Ho, B.; Huang, A. Atypical Teratoid Rhabdoid Tumour: From Tumours to Therapies. J. Korean Neurosurg. Soc. 2018, 61, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Hilden, J.M.; Meerbaum, S.; Burger, P.; Finlay, J.; Janss, A.; Scheithauer, B.W.; Walter, A.W.; Rorke, L.B.; Biegel, J.A. Central Nervous System Atypical Teratoid/Rhabdoid Tumor: Results of Therapy in Children Enrolled in a Registry. J. Clin. Oncol. 2004, 22, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Beaugrand, A.; Le Deley, M.C.; Bourdeaut, F.; André, N.; Leblond, P.; Bertozzi, A.-I.; Frappaz, D.; Rialland, X.; Fouyssac, F.; et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system. Cancer 2012, 118, 3812–3821. [Google Scholar] [CrossRef] [PubMed]
- Lafay-Cousin, L.; Hawkins, C.; Carret, A.S.; Johnston, D.; Zelcer, S.; Wilson, B.; Jabado, N.; Scheinemann, K.; Eisenstat, D.; Fryer, C.; et al. Central nervous system atypical teratoid rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium experience. Eur. J. Cancer 2012, 48, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Nemes, K.; Bens, S.; Bourdeaut, F.; Johann, P.; Kordes, U.; Siebert, R.; Frühwald, M.C. Rhabdoid Tumor Predisposition Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017; 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK469816/ (accessed on 23 January 2024).
- Del Baldo, G.; Carta, R.; Alessi, I.; Merli, P.; Agolini, E.; Rinelli, M.; Boccuto, L.; Milano, G.M.; Serra, A.; Carai, A.; et al. Rhabdoid Tumor Predisposition Syndrome: From Clinical Suspicion to General Management. Front. Oncol. 2021, 11, 586288. [Google Scholar] [CrossRef]
- Calandrelli, R.; Massimi, L.; Pilato, F.; Verdolotti, T.; Ruggiero, A.; Attinà, G.; Gessi, M.; Colosimo, C. Atypical Teratoid Rhabdoid Tumor: Proposal of a Diagnostic Pathway Based on Clinical Features and Neuroimaging Findings. Diagnostics 2023, 13, 475. [Google Scholar] [CrossRef]
- Ginn, K.F.; Gajjar, A. Atypical teratoid rhabdoid tumor: Current therapy and future directions. Front. Oncol. 2012, 2, 114. [Google Scholar] [CrossRef] [PubMed]
- Buscariollo, D.L.; Park, H.S.; Roberts, K.B.; Yu, J.B. Survival outcomes in atypical teratoid rhabdoid tumors for patients undergoing radiotherapy in s surveillance, epidemiology, and end results analysis. Cancer 2012, 118, 4212–4219. [Google Scholar] [CrossRef] [PubMed]
- Seeringer, A.; Bartelheim, K.; Kerl, K.; Hasselblatt, M.; Leuschner, I.; Rutkowski, S.; Timmermann, B.; Kortmann, R.D.; Koscielniak, E.; Schneppenheim, R.; et al. Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors—Experience of the EU-RHAB registry. Klin. Padiatr. 2014, 226, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Alapetite, C.; Dieckmann, K.; Kortmann, R.-D.; Lassen-Ramshad, Y.; Maduro, J.H.; Albiac, M.R.; Ricardi, U.; Weber, D.C. ESTRO-SIOPE guideline: Clinical management of radiotherapy in atypical teratoid/rhabdoid tumors (AT/RTs). Radiother. Oncol. 2024, 110227. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Goyal, S.; Puri, T.; Das, P.; Sarkar, C.; Julka, P.K.; Bakhshi, S.; Rath, G.K. Atypical teratoid rhabdoid tumor of the brain: Case series and review of literature. Childs Nerv. Syst. 2009, 25, 1495–1500. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Gesk, S.; Oyen, F.; Rossi, S.; Viscardi, E.; Giangaspero, F.; Giannini, C.; Judkins, A.R.; Frühwald, M.C.; Obser, T.; et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am. J. Surg. Pathol. 2011, 35, 933–935. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Characteristics | ||
|---|---|---|
| Age at Diagnosis | 10 months | |
| Gender | Female | |
| Clinical Manifestation | Postprandial vomiting Nausea Neurological deficiency | |
| Tumor location | Supratentorial | |
| Immunohistochemical staining | INI1 | Positive |
| EMA | Negative | |
| SYN | Positive | |
| GFAP | Negative | |
| S-100 | Negative | |
| BRG1 | Negative | |
| IDH 1 | Negative | |
| SMARCA4 pathogenic variant | c.2693del, p.(Asn898Thrfs*12) | |
| Methylation class | AT/RT-SHH | |
| Survival | 9 months | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mališkina, A.M.; Franckeviča, I.; Višņevska-Preciniece, Z.; Grūtupa, M.; Kovaļova, Ž. Atypical Teratoid/Rhabdoid Tumor with Retained SMARCB1 (INI1) Expression and Rare SMARCA4 Gene Mutation: A Case Report of a Pediatric Patient. Reports 2024, 7, 28. https://doi.org/10.3390/reports7020028
Mališkina AM, Franckeviča I, Višņevska-Preciniece Z, Grūtupa M, Kovaļova Ž. Atypical Teratoid/Rhabdoid Tumor with Retained SMARCB1 (INI1) Expression and Rare SMARCA4 Gene Mutation: A Case Report of a Pediatric Patient. Reports. 2024; 7(2):28. https://doi.org/10.3390/reports7020028
Chicago/Turabian StyleMališkina, Anna Marija, Ivanda Franckeviča, Zelma Višņevska-Preciniece, Marika Grūtupa, and Žanna Kovaļova. 2024. "Atypical Teratoid/Rhabdoid Tumor with Retained SMARCB1 (INI1) Expression and Rare SMARCA4 Gene Mutation: A Case Report of a Pediatric Patient" Reports 7, no. 2: 28. https://doi.org/10.3390/reports7020028