
Synthesis and Structure of 6-Acetyl-2-Arylhydrazone Derivatives of Thiazolo[3,2-a]Pyrimidine

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
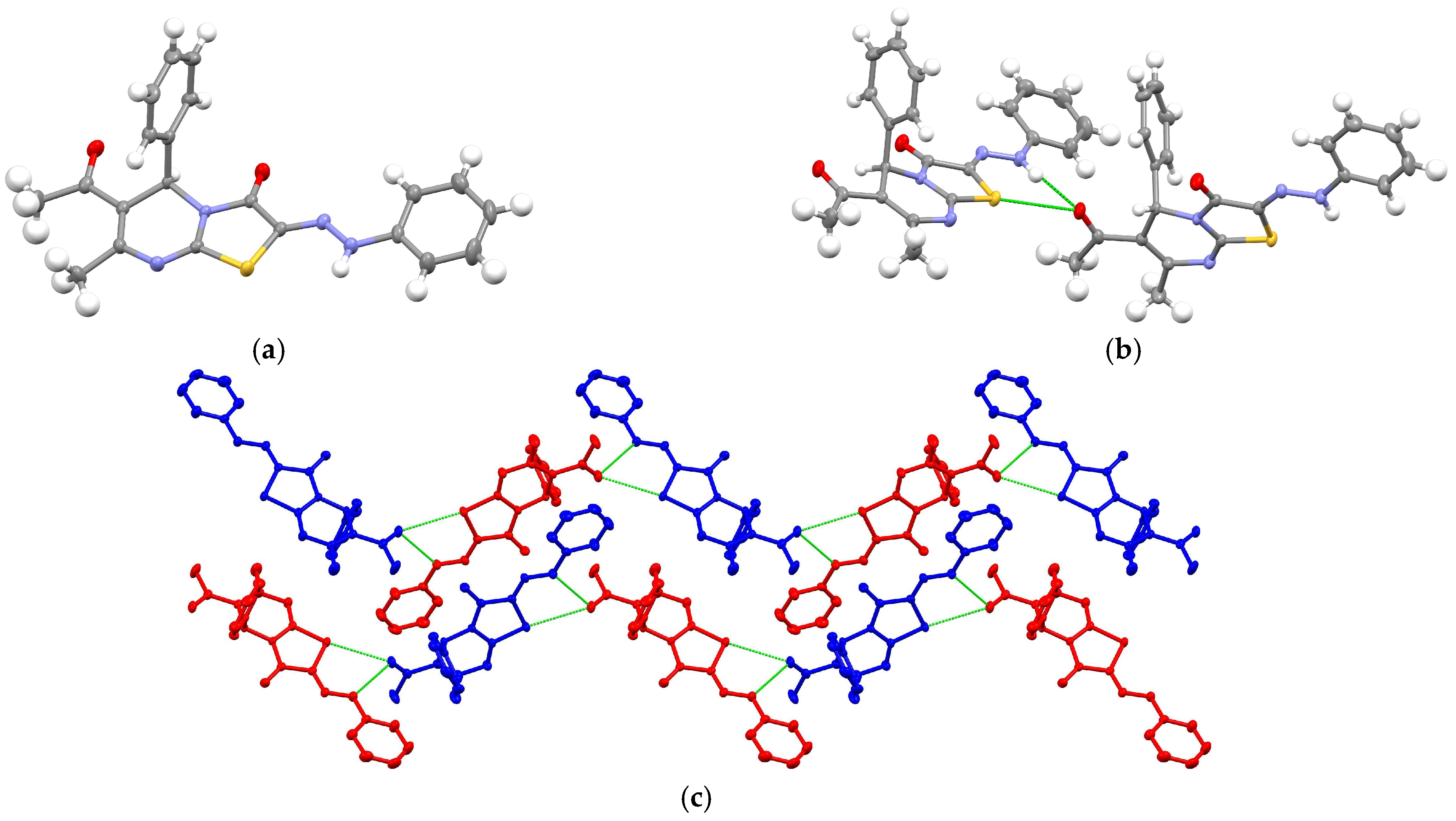
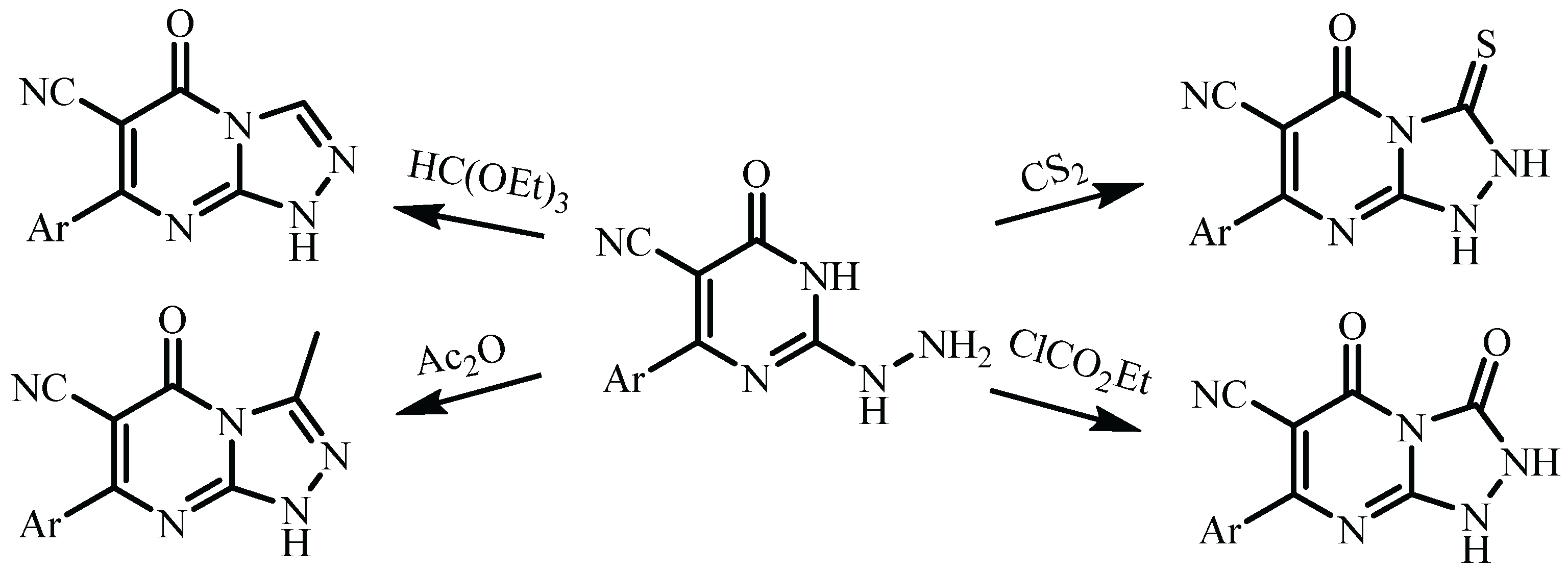
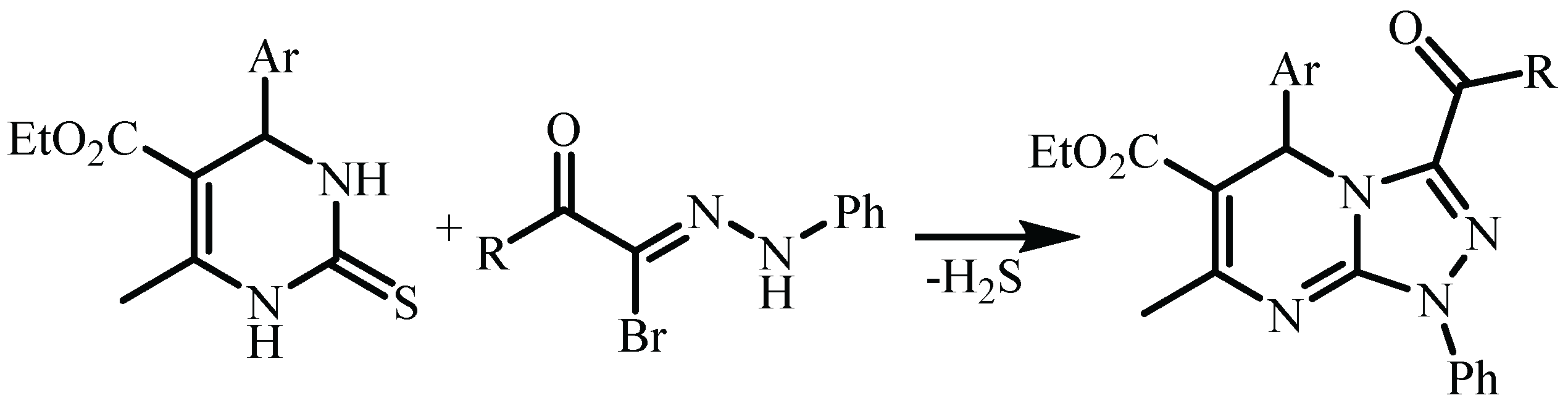
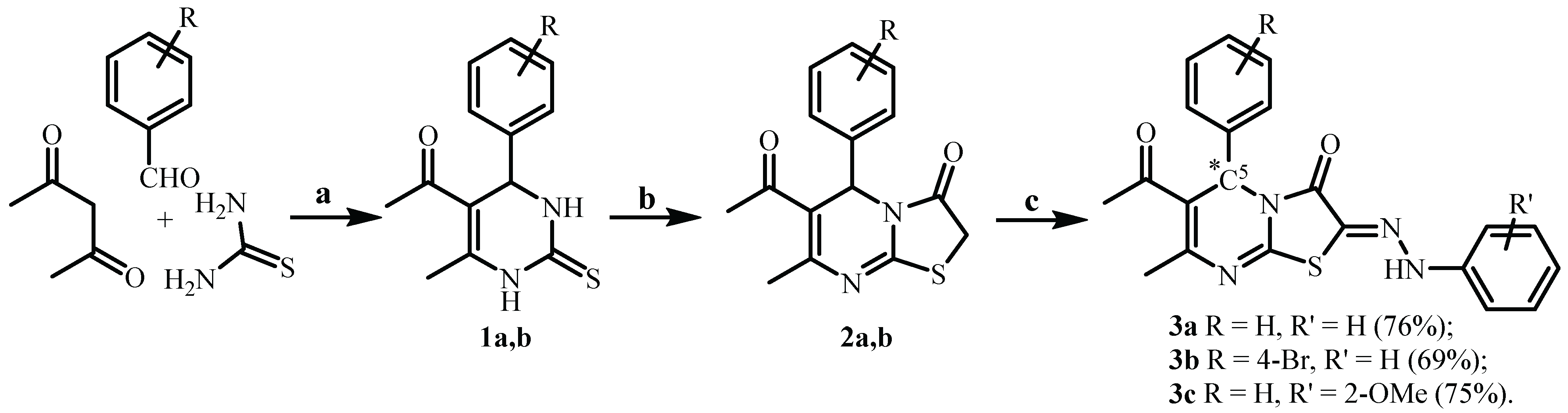
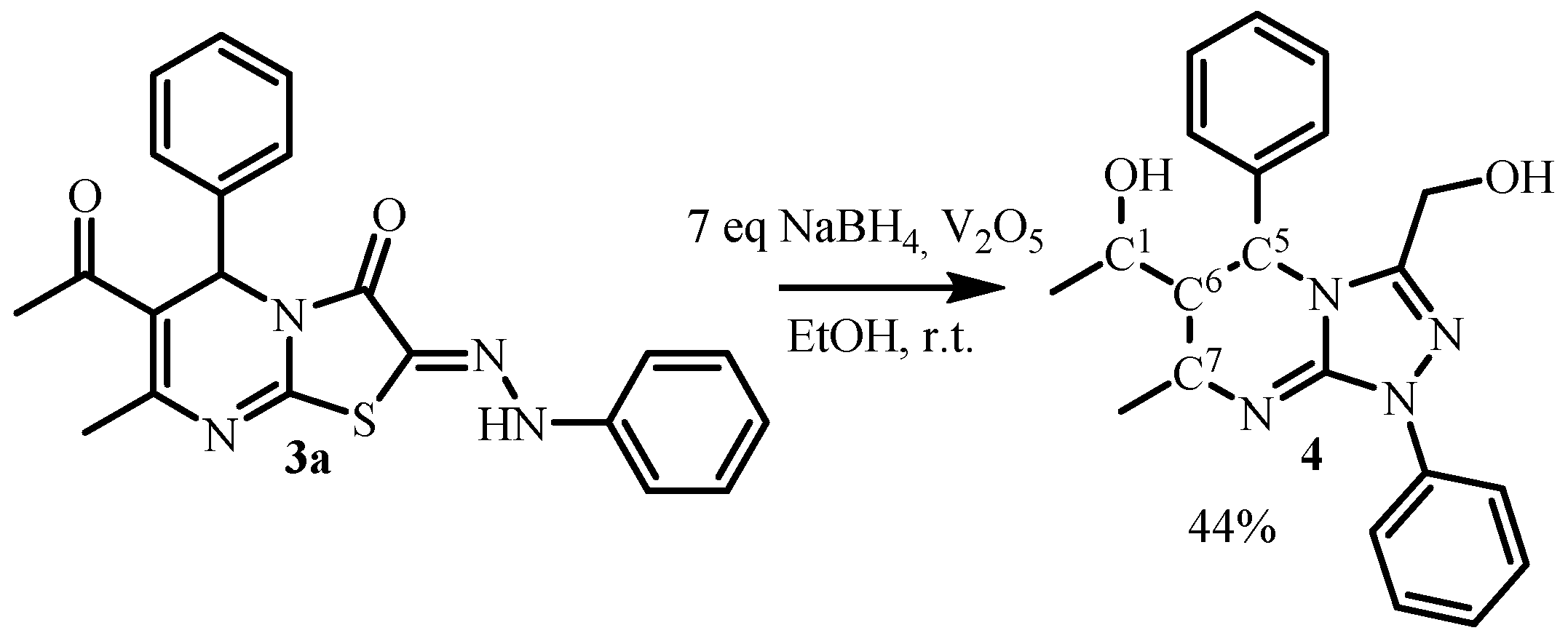
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, G. Recent progress in 1,2,4-triazolo [1,5-a] pyrimidine chemistry. Adv. Heterocycl. Chem. 2007, 95, 143–219. [Google Scholar]
- Gujjar, R.; Marwaha, A.; El Mazouni, E.; White, J.; White, K.L.; Creason, S.; Shackleford, D.M.; Baldwin, J.; Charman, W.N.; Buckner, F.S.; et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009, 52, 1864–1872. [Google Scholar] [PubMed] [Green Version]
- Yu, W.; Goddard, C.; Clearfield, E.; Mills, C.; Xiao, T.; Guo, H.; Morrey, J.D.; Motter, N.E.; Zhao, K.; Block, T.M.; et al. Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. J. Med. Chem. 2011, 54, 5660–5670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Shi, D.Q. Synthesis and herbicidal activity of O,O-dialkyl N-[2-(5,7-dimethyl-[1,2,4] triazolo [1,5-a] pyrimidin-2-yloxy) benzoxyl]-1-amino-1-substitutedbenzyl phosphonates. J. Heterocycl. Chem. 2010, 47, 162–166. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Z.M.; Chen, C.N.; Jiang, L.L.; Yang, G.F. Synthesis and Fungicidal Activities of New 1,2,4-Triazolo [1,5-a] pyrimidines. Chem. Biodivers. 2009, 6, 1254–1265. [Google Scholar] [CrossRef]
- Łakomska, I.; Wojtczak, A.; Sitkowski, J.; Kozerski, L.; Szłyk, E. Platinum (IV) complexes with purine analogs. Studies of molecular structure and antiproliferative activity in vitro. Polyhedron 2008, 27, 2765–2770. [Google Scholar] [CrossRef]
- Ashour, H.M.; Shaaban, O.G.; Rizk, O.H.; El-Ashmawy, I.M. Synthesis and biological evaluation of thieno [2′,3′:4,5] pyrimido [1,2-b][1,2,4] triazines and thieno [2,3-d][1,2,4] triazolo [1,5-a] pyrimidines as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2013, 62, 341–351. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, S.; Liu, Z.J.; Chen, W.; Fu, J.; Zeng, Q.F.; Zhu, H.L. Synthesis and antimicrobical evaluation of a novel class of 1,3,4-thiadiazole: Derivatives bearing 1,2,4-triazolo [1,5-a] pyrimidine moiety. Eur. J. Med. Chem. 2013, 64, 54–61. [Google Scholar] [CrossRef]
- Krezel, I. s-Triazolo[4,3-a] [1,3]diazacycloalkans. III: A novel synthesis of 2-aryl-3-oxo-2,3,5,6,7,8-hexahydro-s-triazolo[4,3-a] pyrimidines. Heterocycles 1986, 24, 93–100. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Khalil, A.K.; Abbass, E.M.; El-Naggar, A.M. Design, synthesis of new pyrimidine derivatives as anticancer and antimicrobial agents. Synth. Commun. 2017, 47, 1441–1457. [Google Scholar] [CrossRef]
- El-Zahar, M.I.; Abd El-Karim, S.S.; Haiba, M.E.; Khedr, M.A. Synthesis, antitumor activity and molecular docking study of novel benzofuran-2-yl pyrazole pyrimidine derivatives. Acta Pol. Pharm. Drug Res. 2011, 68, 357–373. [Google Scholar]
- Abdelhamid, A.O.; Sayed, A.R.; Zaki, Y.H. Reaction of Hydrazonoyl Halides 511: A Facile Synthesis of 5-Arylthiazoles and Triazolino[4,3-a]pyrimidines as Antimicrobial Agents. Phosphorus Sulfur Silicon 2007, 182, 1447–1457. [Google Scholar] [CrossRef]
- Gomha, S.M.; Muhammad, Z.A.; Edrees, M.M. Ethyl 7-Methyl-1-(4-nitrophenyl)-5-phenyl-3-(thiophen-2-yl)-1,5-dihydro-[1,2,4]triazolo [4,3-a]pyrimidine-6-carboxylate. Molbank 2017, 2, M942. [Google Scholar] [CrossRef] [Green Version]
- Lashmanova, E.A.; Agarkov, A.S.; Rybakov, V.B.; Shiryaev, A.K. Rearrangement of thiazolo[3,2-a]pyrimidines into triazolo[4,3-a]pyrimidines induced by C=N bond reduction. Chem. Heterocycl. Compd. 2019, 55, 1217–1221. [Google Scholar] [CrossRef]
- Agarkov, A.S.; Konorov, G.V.; Nefedova, A.A.; Gabitova, E.R.; Islamov, D.R.; Ovsyannikov, A.S.; Shiryaev, A.K.; Solovieva, S.E.; Antipin, I.S. Synthesis and structure of new derivatives of triazolo[4,3-a]pyrimidine and 2-phenylhydrazones of thiazolo[3,2-a]pyrimidine. Butlerov Commun. 2021, 68, 122–128. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Mobinikhaledi, A.; Foroughifar, N.; Jirandehi, H.F. Investigation of the effects of some heat sinks in microwave-assisted synthesis of some biginelli compounds. Phosphorus Sulfur Silicon 2004, 179, 2259–2263. [Google Scholar] [CrossRef]
- Zhang, G.P.; Tian, D.Y.; Shi, W.M. Efficient Catalytic Synthesis of 3,4-Dihydropyrimidin-2-ones/thiones via Little Acidic Ionic Liquid Combined with Rapid Heating Ways. J. Heterocycl. Chem. 2018, 55, 2522–2531. [Google Scholar] [CrossRef]
- Amrollahi, M.; Mobinikhaledi, A.; Forughifar, N. Synthesis of some new thiazolo pyrimidines using cyclocondensation reactions. Asian J. Chem. 2005, 17, 902–906. [Google Scholar]
- Agarkov, A.S.; Gabitova, E.R.; Galieva, F.B.; Ovsyannikov, A.S.; Voloshina, A.D.; Shiryaev, A.K.; Litvinov, I.A.; Solovieva, S.E.; Antipin, I.S. Structure and Biological Properties of 2-Phenylhydrazone Derivatives of Thiazolopyrimidines. Dokl. Chem. 2022, 503, 45–50. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
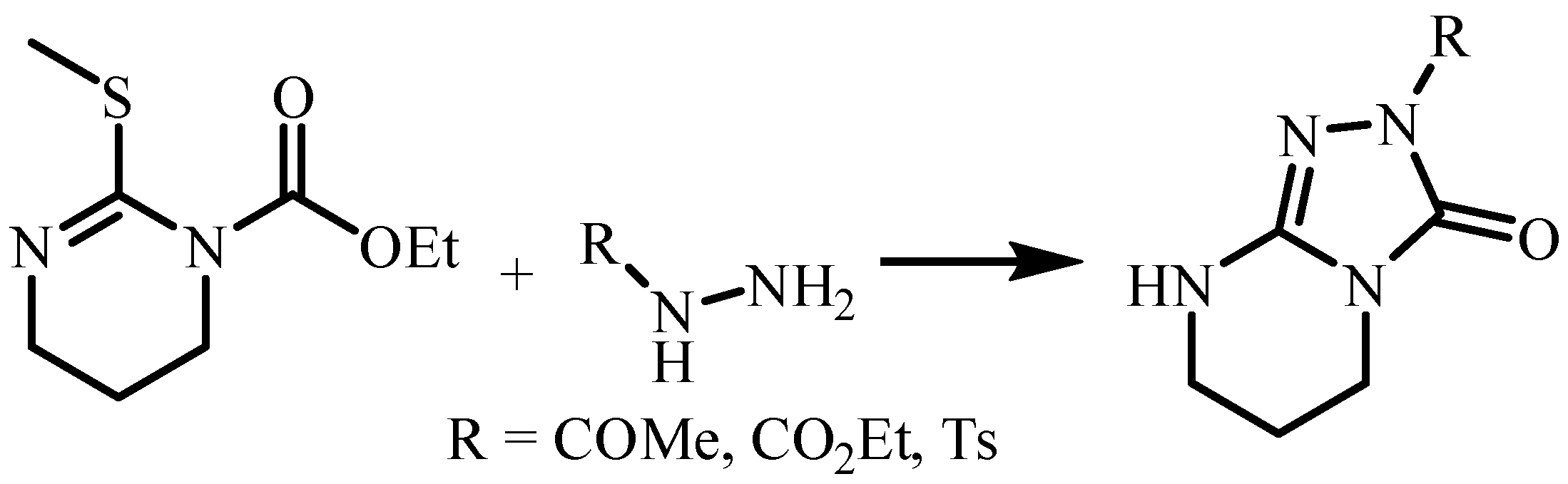{kind=link}
{kind=link}
{kind=link}
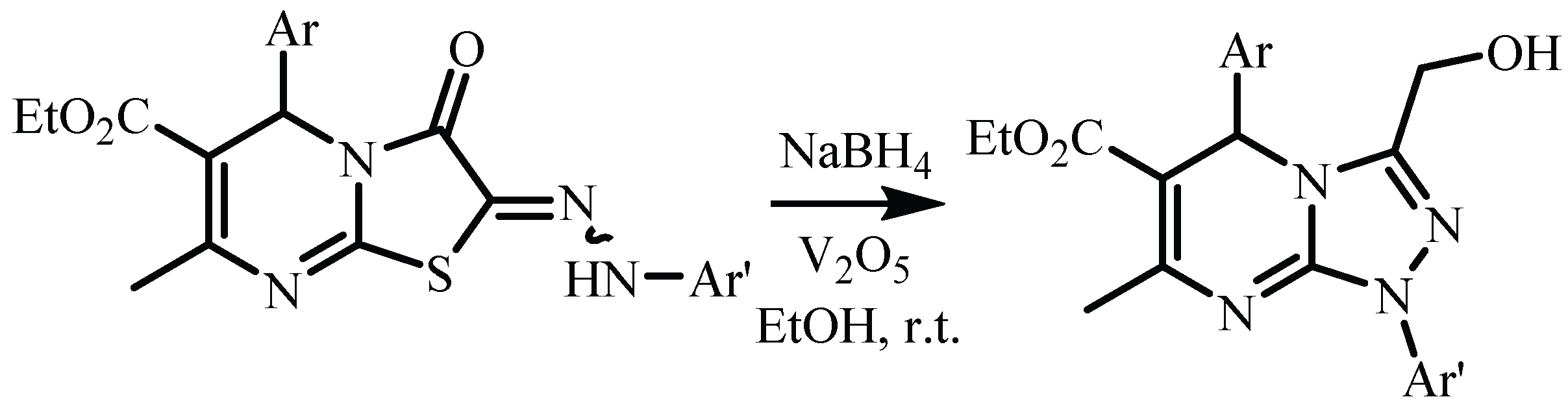{kind=link}
{kind=link}
{kind=link}
| Compound | 3a (from Ethanol) |
|---|---|
| Molecular formula | C21H18N4O2S |
| Formula | C21H18N4O2S |
| Formula Weight | 286.35 |
| Crystal System | monoclinic |
| Space group | P21/n |
| Cell parameters | a = 9.5262(8) Å, b = 12.1416(12) Å, c = 16.0525(16) Å; α = 90°, β = 90.248(4)°, γ = 90°. |
| V [Å3] | 1856.67 Å3 |
| Z and Z′ | 4 and 0 |
| D(calc) [g/cm3] | 1.397 |
| λ (Å) | (MoKα) 0.71073 |
| μ [/mm] | 0.200 |
| F(000) | 816 |
| Theta Min-Max [Deg] | 2.103–29.998° |
| Reflections measured | 58800 |
| Independent reflections | 5398 |
| Observed reflections [I > 2σ(I)] | 3596 |
| Goodness of fit | 1.035 |
| R [I > 2σ(I)] | R1 = 0.0492, wR2 = 0.1118 |
| R (all reflections) | R1 = 0.0936, wR2 = 0.1255 |
| Max. and Min. Resd. Dens. [e/Å−3] | 0.339 and −0.376 e Å−3 |
| Depositor number in CCDC | 2252794 |
| C1 C5 C6 C7 | Ph (at C5) | CH3-CH(OH)- (at C1) | CH3- (at C7) | E (kJ/mol) |
|---|---|---|---|---|
| RRRR/SSSS | e | e | e | 115.05 |
| RRRS/SSSR | e | e | a | 103.53 |
| RRSR/SSRS | a | e | a | 112.76 |
| RRSS/SSRR | a | e | e | 113.23 |
| RSRR/SRSS | a | e | e | 99.50 |
| RSRS/SRSR | a | e | a | 118.68 |
| RSSR/SRRS | e | e | a | 111.95 |
| RSSS/SRRR | e | e | e | 113.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarkov, A.S.; Mingazhetdinova, D.O.; Nefedova, A.A.; Ovsyannikov, A.S.; Shiryaev, A.K.; Litvinov, I.A.; Solovieva, S.E.; Antipin, I.S. Synthesis and Structure of 6-Acetyl-2-Arylhydrazone Derivatives of Thiazolo[3,2-a]Pyrimidine. Organics 2023, 4, 438-446. https://doi.org/10.3390/org4030031
Agarkov AS, Mingazhetdinova DO, Nefedova AA, Ovsyannikov AS, Shiryaev AK, Litvinov IA, Solovieva SE, Antipin IS. Synthesis and Structure of 6-Acetyl-2-Arylhydrazone Derivatives of Thiazolo[3,2-a]Pyrimidine. Organics. 2023; 4(3):438-446. https://doi.org/10.3390/org4030031
Chicago/Turabian StyleAgarkov, Artem S., Dilyara O. Mingazhetdinova, Anna A. Nefedova, Alexander S. Ovsyannikov, Andrey K. Shiryaev, Igor A. Litvinov, Svetlana E. Solovieva, and Igor S. Antipin. 2023. "Synthesis and Structure of 6-Acetyl-2-Arylhydrazone Derivatives of Thiazolo[3,2-a]Pyrimidine" Organics 4, no. 3: 438-446. https://doi.org/10.3390/org4030031