
Diversity of Microbial Communities in Trade Wastes—Implications for Treatments and Operations

1
ARC Training Centre for the Transformation of Australia’s Biosolids Resource, RMIT University, Bundoora, VIC 3083, Australia
2
School of Science, RMIT University, Bundoora, VIC 3083, Australia
*
Author to whom correspondence should be addressed.
Appl. Microbiol. 2024, 4(2), 682-703; https://doi.org/10.3390/applmicrobiol4020047
Submission received: 4 April 2024
/
Accepted: 16 April 2024
/
Published: 19 April 2024
(This article belongs to the Special Issue Exclusive Papers Collection of Editorial Board Members and Invited Scholars in Applied Microbiology (2023, 2024))
Abstract
:Industrial wastewaters display a complex and diverse range of physicochemical properties that are measured, studied, and treated by businesses and water service providers. Less frequently measured are the microbial communities in these wastes, despite possible implications for health, equipment maintenance, and the environment. This study aimed to assess the microbial communities of eighteen raw and discharge-ready wastewaters across eleven industrial sites to compare the microbial compositions of these wastewaters across different industry sectors, on-site treatment levels, and other wastewater components. The potential for variance in the biomethane yield, depending on microbial communities, was also measured. Using targeted sequencing, a unique taxonomy was identified, including genera linked to animals (Acetitomaculum, Lactobacillus, NK4A214, Prevotella, and Shuttleworthia), cooling water (Bosea, Legionella, Methyloversatilis, and Reyranella), and extreme conditions (Alkalibacillus, Geobacillus, Halorubrum, and Pyrobaculum). However, the compositions of the microbial communities were not found to be directly correlated to industry sector or on-site treatment levels, nor were they found to have a direct effect on the biomethane potential. However, the presence of certain individual taxa is linked to the methane yield and treatment status and may be explained in the context of physicochemical properties while serving as potential markers for identifying, improving, or developing on-site processes.
1. Introduction
Wastewaters from different industrial processes display a complex and diverse range of physicochemical characteristics [1], which can change because of the effects of the waste treatment processes undertaken at a particular site. These characteristics include pH, temperature, dissolved solids, and metals and can be affected by pumping, stagnation, exposure to light, and the combining of streams. The growth of individual microorganisms in wastewater is influenced by all these conditions and can be further influenced/varied directly through disinfection [2] or biological treatment [3].
The microbial community contained in these wastes is consequential. For example, within the bounds of a site and sewerage network, Thiobacillus spp. [4] and Desulfobulbus spp. [5] are examples of bacteria that biologically mediate the corrosion of concrete and steel. Furthermore, the health and safety of workers and others in the vicinity can be affected by pathogens, including Mycobacterium [6] and Legionella [7], while areas downstream of discharge points have been found to have increased abundances of potential pathogens, like Arcobacter and Chlamydia [8], as well as altered biofilms with increased antimicrobial resistances [9]. It is also possible that microbes in wastewater could influence or indicate the suitability of certain treatment processes, particularly resource recovery and biological methods. For example, the phosphorus removal of municipal wastewaters is improved in anoxic/aerobic membrane bioreactors by non-sterile substrates [10], and nitrifiers in influents increase the stability and shorten the start-up times of activated sludge sequencing batch reactors that are treating municipal wastewaters [11]. Opportunities for exploiting such ecological knowledge are based on process optimisations without major capital expenditures for engineered solutions. However, research into the microbiome of industrial wastewater is limited, with large knowledge gaps remaining, preventing such process optimisations. Research usually focuses on individual biological treatment processes, such as the overloading of anaerobic digesters [12] or augmentation by selected bacteria [13]; municipal wastewater infrastructure, including temporal changes in sewer microbiota [14]; or end-of-pipe effects, like those on nitrogen-cycling bacteria in rivers near effluent discharge points [15]. The microbial diversity present in wastewater at the point of the source, as well as the effects that on-site treatment has on it, remain largely unknown, especially for industrial wastewater.
To improve our knowledge base, this study was undertaken to identify microbial communities of industrial wastewaters before and after on-site treatment, aiming to investigate any links between their microbiomes, levels of treatment, industries, and physicochemical properties. Hence, this work provides a novel inventory of microbial communities indigenous to selected treated and untreated industrial wastewaters, highlighting taxa of interest and links to undertaken treatments and resource recovery potential.
2. Materials and Methods
2.1. Sampling
Treated and untreated effluents were supplied from 12 de-identified industrial sites in Melbourne, Australia, as previously described [16]. Samples were stored at 4 °C before transport and physicochemical testing and frozen and stored at −18 °C for microbial analysis.
2.2. Physicochemical Analyses of Wastes
Standard gravimetric methods were used to calculate the total and volatile solids [17]. The pH was measured using a Hanna Instruments HI5221 pH meter (Hanna Instruments Australia, Keysborough, VIC, Australia), and the salinity was measured using a Horiba Scientific LAQUAtwin-Salt-11 salt meter (Horiba, Ltd., Kyoto, Japan). The chemical oxygen demand (COD), phosphate (PO43− by acid–persulphate digestion), total nitrogen (TN by persulphate digestion), nitrates (NO3-N by the chromotropic acid method), nitrites (NO2-N, by the diazotisation method), and ammonia (NH3-N, by the salicylate method) were measured using appropriate Hach Test ’N Tube methods with a Hach DR900 colorimeter and a DRB200-reactor heating block (Hach, Dandenong South, VIC, Australia) [18,19,20,21,22,23]. De-identified discharge data were supplied by Greater Western Water.
2.3. Biomethane Potential Assessment
The biomethane potential (BMP) was assessed, as previously described, using a custom lab-scale set-up [16] or “Nautilus BMP reactors” (Anaero Technology, cambridgeshire, UK) [24] as applicable. Briefly, digestion vessels were prepared with real industrial wastewater substrates and mature waste-activated sludge at an inoculum-to-substrate ratio (ISR) of 3.5 on a grams-per-VS basis. The digesters were sealed and held at 38 °C in a water bath. When the Nautilus was used, the gas flow volume was measured in near-real time through liquid displacement buckets and corrected for atmospheric temperature and pressure, followed by gas collection in 3 L Tedlar PLV gas-sampling bags with Thermogreen LB-2 septa (Sigma Aldrich, Saint Louis, MO, USA). Methane concentrations were measured using a calibrated portable multi-gas monitor with an infrared detector (MX6 iBrid, CH4 range 0–100%, ±1%, Industrial Scientific, Pittsburgh, PA, USA) Tests were continued until the daily methane production had decreased to less than 1% of the cumulative total for a period of at least one week.
2.4. Targeted 16S-rRNA Sequencing
Frozen genomics samples were thawed, and DNA was extracted using a DNeasy Powersoil Pro extraction kit (Qiagen, Hilden, Germany), as per the manufacturer’s instructions [25], and stored at −20 ℃. DNA concentrations were quantified using a Qubit 4.0 fluorometer (Invitrogen, Carlsbad, CA, USA). The 16S Metagenomic Sequencing Library Preparation Guide (Illumina, San Diego, CA, USA) was followed [26], using primers targeting the V3V4 region of the 16S-rRNA gene with primers Pro341FB (CCTACGGGNBGCWSCAG) and Pro805R (GACTACNVGGGTATCTAATCC) [27,28] prior to sequencing (Illumina MiSeq, 600 cycles) [26]. Blanks were included in the library preparation to assess contamination. A total of 20 libraries from this study were pooled together with 137 other 16S libraries from unrelated projects and then sequenced together at 8 pM on an Illumina MiSeq (2 × 300 bp), including 20% phiX, after library preparations using Nextera XT indices.
Processing of fastq files was conducted on the Research Cloud made available by the Australian Research Data Commons (ARDC). The combined processing of the sequences (all the sequenced libraries) included primer trimming with cutadapt [29] followed by pairing forward and reverse reads, denoising, filtering, and dereplicating the sequences using DADA2 [30] (error rates were trained on 277,158,882 bases in 1,015,234 reads from 53 samples) after truncating all the amplicon sequences to 273 and 220 nucleotides (forward and reverse reads, respectively) using the default settings of the QIIME2 [31] pipeline apart from relaxing the expected error from 2 to 6 for reverse reads.
This followed taxonomic classification (qiime feature-classifier classify-sklearn) [32] of amplicon sequence variants (ASVs) using the SILVA database after training the classifier (qiime feature-classifier fit-classifier-naïve-bayes) on taxonomy and sequence files [33,34]. The subset of samples for this study was then used for further analysis. Site 6 was removed from the analysis because of potential sample contamination.
2.5. Data Analysis and Visualisation
A total of 175,557 quality reads at a median frequency of 9873 reads per sample and 6506 ASVs were obtained. All the analyses and charting were performed in R, version 4.2.2 [35]. ASVs with fewer than five reads were filtered for quality. Abundances were normalised through rarefication to the minimum sample size prior to calculating the richness, Shannon diversity (H’) and Pielou evenness (J) using the phyloseq package [36]. Kruskal–Wallis tests were used to assess differences in alpha diversity between treated and untreated effluents. Prior to principal component analysis (PCA) analysis, relative abundances were transformed to centred log-ratios (clrs) using the microbiome package [37]. Phyloseq was then used for PCA ordinations of clr-transformed abundances to assess compositional differences between sites. Canonical correspondence analysis (CCA) was performed on phylum-level abundances (Bray–Curtis dissimilarities) using the vegan [38] package to assess associations between bacteria and effluent of different sites and their chemical properties. To detect any treatment or site-specific genera, differential abundance analysis was conducted using the ANCOM-BC package on clr-transformed abundances [39]. For treatment effects, sites were limited to those with both treated and untreated samples available.
3. Results and Discussion
3.1. Wastewater Properties
Wastewater samples were obtained from businesses in a broad range of industries, including edible fats (Site 1), brewing (Site 2), waste management/logistics (Sites 3 and 9, respectively), chemical production (Sites 4, 8, 10, and 11), food (Site 5), and animal products (Sites 7 and 12), with both untreated and treated samples available for Sites 1, 2, 3, 5, 9, and 12 (Table 1).
Treatments differed between sites. Water treatment unit operations undertaken included removing particles through straining, screening, and settling; separating hydrophobic liquids in oil interceptors; cooling the wastewater stream; adjusting pH through chemical addition; controlling sulphide levels; and more complex procedures for coagulating, flocculating, and removing solids, like polymer addition and dissolved-air flotation (DAF) (Table 2).
Historical data (Supplementary Table S1) showed that the measured characteristics at discharge were unique to a site because of the interactions between the individual wastes produced and the treatment enacted. Although historical data should match the measured data of treated samples for a given site [40], this was not always the case. This may be affected by the long-term average composite nature of the data held by the water corporation. Consistent with the purpose of on-site treatment, the untreated samples were not reflected in the discharge data (Supplementary Table S1).
High COD (>9500 mg/L) and VS (>3500 mg/L) characterised the untreated streams of Sites 1, 2, 3, 5, and 9 [41] and the treated sample (7T), which also contained high measured nitrogen, sodium, and dissolved solid concentrations according to discharge data (Table 1). Otherwise, treated samples were lower in COD and VS, and Sample 4U was dilute in all the measured parameters. Sites 1, 5, and 10 contained high phosphate concentrations (>150 mg/L), consistent with other edible fat and food manufacturers [41,42,43] and certain chemical producers (Table 1). Most samples were in the pH range of 6–8; however, 3U and 9U had high pH values (>12), and 2U, 2T, 5U, and 9T had low pH values (<6). Low pH values in untreated food-manufacturing wastewater may be caused by microbial acidogenesis, where monomers, such as simple sugars, are converted to short-chain organic acids [44]. The pH in treated samples is predominantly the result of neutralisation treatments. The pH of the treated sample for Site 2 was much closer to that of the untreated sample than the historical data, indicating a potential treatment process upset. Confirmed by available discharge results, low total nitrogen concentrations were measured in samples from Sites 1, 4, 8, 10, and 11 and before treatment at Site 9 (Table 1). Discharge from Sites 3, 5, and 8 historically contained greater than 500 mg/L of oil and grease. Sites 4, 7, 8, and 10 generally discharged at a lower temperature (<20 °C) and 1, 2, and 9 at a higher temperature (>28 °C). High gas yields were measured from untreated streams at Sites 3, 5, 9, and 12 and the treated stream at Site 5. Medium yields were gained from post-treatment samples at Sites 2 and 9 (Table 1).
As expected, the chemical properties of the untreated effluents were diverse, reflecting the different production methods each site housed and the varying treatment processes undertaken. More surprisingly, despite general restrictions on the quality of the discharge [45], each of the treated samples also has unique chemical properties. Although in some instances, discrepancies between measured and historical data indicate a short-term fluctuation in treatment effectiveness, most differences between treated samples can be explained by the contents of the water before treatment and the relatively broad range of trade waste qualities allowed by guidelines or special agreements.
3.2. Richness and Diversity Assessments of Microbial Communities among Treated and Untreated Trade Wastes
To assess if the wastewater microbial diversity was different between treated and non-treated effluents or associated with high potential for biogas production, the richness (of observed taxa), Shannon diversity (H’), and Pielou’s evenness (J) were calculated, representing the number of organisms, number of effective (or abundant) organisms, and their distributions within each sample, respectively (Figure 1).
Across the 12 sites, both treatment and biogas yields were not associated with any of the diversity indices (Kruskal–Wallis, p > 0.05). The mean richness and diversity (H’) values appeared higher for treated samples (71.2 ± 53.7 and 3.24 ± 0.80, respectively) compared to untreated samples (52.3 ± 32.9 and 3.09 ± 0.73, respectively); however, the variability across the sites was too large (and sample sizes were too small) to confirm this observation. For sites with pre- and post-treatment samples available, no consistent trend was found. It was hypothesised that, generally, more concentrated and complex untreated wastewaters would support greater richness [46,47]. However, in some cases, inhibitors, like phenols or heavy metals, which cause the dominance of a limited number of species, are present in raw wastewater [48]. When these are removed by treatment, the growth of more diverse microbes is possible. Sample diversity here may also be shaped by the stability of the wastewater over time, resulting in more predictable microbial communities, as shown in long-term studies of parallel anaerobic digesters [49], meaning that a complex but consistent substrate does not necessarily translate to higher diversity. It is also likely that individual treatment vessels are home to unique microbial communities [50] and seed the wastewater as it is processed. Diversity may be changed in ways, on-site, that have no effect on BMP tests, including exposure to high temperatures or pressures [51]. Consequently, in future studies on industrial wastewater sites, an appropriate number of sites for sampling may need to be determined through power analysis to ensure sample-level diversity can differentiate between treated and untreated effluents.
3.3. Microbial Community Analysis of Microbial Communities among Trade Wastes at Different Levels of Treatment or BMP Yields
Using PCA to visualise the dominance of the taxa identified in the samples, some groupings were evident. Considering the first two principal components, a general group of Sites 1, 5, 7, 8, and 10 can be seen (Group A). Of the other samples, those from Sites 2, 3, and 9 form a cluster (Group B); the communities at Sites 4 and 11 (Group C) were different from those at the other sites, and Site 12 communities were different again (Group D) (Figure 2).
Group B consists of all the samples from the two logistics companies and the brewery. Both sites in Group C were chemical-manufacturing businesses, and Site 12 was in the animal product industry. Site 7, notable for high sodium concentrations, both measured and historically (Table 1, Supplementary Table S1), also shared very few taxa with other sites. None of treatment levels, measured water chemistry values, or BMP assay biogas yields (Table 1) predicted microbial community similarity between samples. Apart from logistics companies being confined to Group B, industry type also did not predict similarity. It was observed that some individual genera were more-or-less abundant, depending on the treatment level. Across all the samples with both treated and untreated samples available, Sulfurospirillum, Leuconostoc, Desulfovibrio, and Clostridium sensu stricto 13 were more abundant in the treated samples, while Chryseobacterium, Enterococcus, Selenomonas, and Acinetobacter were more abundant in the untreated samples (p < 0.05) (Figure 3).
Consistent with untreated wastewaters generally producing higher BMP yields, the latter two taxa were also significantly more abundant in samples with high BMP yields (p < 0.05), while Desulfovibrio, and Clostridium sensu stricto 13 were more abundant in lower-yielding samples (Figure 4).
Conversely, Sulfurospirillum had a small but significant increase in higher-yielding samples. This genus was present in Sample 5T, the only treated sample with a high gas yield. Bosea and Reyranella were also represented in low-yielding substrates, in Group C, the provenance of which is described further in Section 3.5 below. Prevotella, Lactobacillus, and NK4A214 are present in high-yielding samples from brewery, logistics, and food sites (Sites 2, 3, and 5, respectively). Members of the family Synergistaceae are implicated in higher biogas yields in anaerobic digestion [52], in agreement with an increase in high-yielding substrates in this study.
3.4. Taxonomic Abundances—Overall Trends
Comparisons were made to investigate which characteristics correlated to phylum abundance. At the phylum level, Proteobacteria were the most common across the samples (mean relative abundance = 0.519), followed by Firmicutes (0.224), Bacteroidota (0.133), and Campylobacterota (0.047) (Figure 5).
Campylobacterota were classified within the phylum Proteobacteria until recently [53]. These phyla were three of the four most abundant in a combined domestic/industrial sewer [14], and Proteobacteria and Firmicutes were the two most common phyla in anaerobic digesters treating municipal waste-activated sludge [54]. Within individual sites, taxonomic abundances changed markedly after treatment, including the almost total disappearance of Campylobacterota at Site 1 or the large increase in Bacteroidota at Site 3. On-site treatment is undertaken to reach discharge specifications, so differences between the measured chemistries of untreated samples and discharge wastewater compositions are expected. Therefore, variation in the bacteria that are present in each medium is evident. There was no consistent phylum-level community between all the treated and all the non-treated samples or between the high-, medium-, and low-biogas-yielding samples. This is evident in Figure 2, as there is no grouping of samples with the same treatment status or yield level in the ordination.
Figure 6 shows the canonical correspondence analysis (CCA) of the samples, phyla, and measured chemical data.
The most striking observation is the relationship between the sodium concentration at Site 7 and Halobacterota, a phylum containing many halophiles. The abundances of Firmicutes in this sample and the untreated wastewaters of Sites 5 and 9 are also represented along this axis. The samples containing larger abundances of Verrucomicrobiota, Planctomycetota, and Proteobacteria generally also contained lower concentrations of sodium. These plots also map the high abundance of Campylobacter and low concentrations of nitrite, nitrate, and ammonia in Samples 1U and 10T.
3.5. Microbial Community Phylogenetic Analysis—Individual Sites
The microbes present in each sample are a function of conditions and wastewater composition, so links can be found between abundance and effluent properties. Genus-level investigation can provide more information about likely optimal conditions than the more general Phylum-level analysis [55]. Taxa described in this section were detected in the relevant samples, as per Figure 7. Sites have been ordered by groups observed in Section 3.3. Group A can be seen as a general grouping. Samples from brewery and logistics sites made up the entirety of Group B. Group C was characterised by the presence of genera found in cooling water, along with high diversity (observed taxa > 60, H’ > 3.5, and J > 0.86). Group D, consisting of treated and untreated samples from Site 12, was highly diverse.
3.5.1. Group A, Site 1—Edible Fats
The untreated and treated samples at Site 1 were dominated by anaerobic or microaerobic microbes. Sulfurospirillum and Desulfovibrio both reduce sulphur compounds [56] and are found at very high abundances before on-site treatment, going against the overall trend for these genera described earlier. After treatment, which includes sulphide control, their numbers markedly decreased. Desulfovibrio is also potentially involved in the decomposition of fats, at this site, as a butyric acid oxidiser [57]. Pseudomonas maintains a consistent relative abundance before and after treatment. Members of the Pseudomonas genus are aerobic and strictly respiratory, but some species can grow under anaerobic conditions by reducing nitrates. Otherwise, they are widely distributed, with minimal nutrient requirements [58], as demonstrated by their position in the top ten most abundant ASVs in samples U1, T1, U2, 3T, 4U, 8T, 9T, 10T, and 11T. Treatment increases the relative abundance of Yersiniaceae, a family of spore-forming facultative anaerobes that include some pathogenic species [59], and of Obesumbacterium proteus, implicated in beer spoilage [60]. Acetobacter, found in fermented foods, is also present [56].
3.5.2. Group A, Site 5—Foods
Site 5 is a food-manufacturing business, and the microbes present in the raw wastewater are associated with animals. Shuttleworthia is present in the digestive systems of healthy chickens [61] and weaned calves [62]; NK4A214 has been isolated from yaks [63]. Acetitomaculum, Lactobacillus, and Prevotella are present in increased numbers in the rumen of cattle suffering from acidosis [64]. The likely mechanism of this is Acetitomaculum producing acetate, and Lactobacillus and Prevotella thriving in the subsequent low-pH environment. This tolerance is reflected in the relatively low pH of the waste stream. After treatment, the proportions of all these ASVs decreased, while Paludibacterium and other unclassified members of the Chromobacteriaceae family increased. Paludibacteria are facultative anaerobes [65], and the rest of the family members are either aerobes or facultative anaerobes [66]. Wastewater treatment at this site includes DAF, a process that clarifies water using air bubbles to remove suspended solids as a sludge [67], perhaps favouring aerobic species. DAF has been shown to separate algae and bacteria from water [67,68]. The effect of the treatment on the relative abundance correlates with neither typical cell sizes nor cell wall structures found in literature, despite DAF involving fluid dynamics and interactions between bubbles and solids. Surprisingly, despite these aerobic conditions, the methanogen Methanobrevibacter was only identified in the treated sample. This genus has been linked to stressed anaerobic digesters [12].
Figure 7.
Abundance plots at genus level, showing sites, treatment status, and level of gas yield from BMP tests. Plot is separated into groups (a–d) as identified in PCA (Figure 2). Genera are ordered by the highest relative abundance in the group.
Figure 7.
Abundance plots at genus level, showing sites, treatment status, and level of gas yield from BMP tests. Plot is separated into groups (a–d) as identified in PCA (Figure 2). Genera are ordered by the highest relative abundance in the group.

Site 5 was unique in this study in providing access to a source-separated wastewater stream: a relatively small volume discharged from cleaning a cooking vessel. Apart from Pseudomonas, Yersiniaceae, and other previously described taxa, thermophilic Pyrobaculum [69] and Geobacillus [70] were identified at low levels, reflecting the high temperatures involved in the individual unit’s operation. This sample showed low diversity (observed taxa = 19; H’ = 1.98; J = 0.67) but a high BMP yield. It is likely that the high-temperature conditions reduced the microbial diversity [71] in a way that was temporally limited and unmeasurable in the wastewater sample but may have served as a form of pre-treatment that increases the BMP yield [44].
3.5.3. Group A, Site 7—Animal Products
The extremely low gas yields of BMP assays for this substrate were linked to the high concentration of sodium found in discharge measurement data. High sodium levels are also reflected in the microbial community of the wastewater, which is dominated by halophiles.
Alkalibacillus and Haloterrigena require salt concentrations of 10% for optimal growth [55,72]. The optimal concentration is higher for Salicola, isolated from salterns and salt lakes, at 15% [73], and higher again for Halorubrum and Halobacterium, at over 20% [55,74]. Marinococcaceae inhabit high-salinity areas, such as the shores of salt lakes [75]. Corynebacterium has been identified in an anaerobic membrane bioreactor inoculated with intertidal wetland sediment for treating high-salinity wastewater [76]. Jeotgalicoccus is slightly halophilic and was first identified in a salted preserved seafood dish [77]. Dietzia has also been found in fish and fish-based food-processing water [78]. All the above-named bacteria are aerobes or facultative anaerobes. The genus Corynebacteria also contains facultative anaerobes and aerobes [78]. Conversely, Halanaerobacter is an obligate anaerobe [79]. The presence of obligate aerobes and anaerobes at this site and others indicates the presence of microniches of different conditions and potentially a temporal factor as operations progress. Another indicator of this is the presence, albeit at very low levels, of Methanobrevibacter and Methanosphaerula. Both genera are known to increase in relative abundance compared to other methanogens when anaerobic digesters are under chemical stress [12]. In this case, they may be the last surviving remnants of an individual process’s low-sodium, low-oxygen wastewater before the combination of streams.
3.5.4. Group A, Site 8—Chemicals
The wastewater at this site contained a very low concentration of extractable DNA. The majority of that which was extracted and sequenced was from the previously described Enterobacteriaceae family and Pseudomonas genus. This sample was shown to be a poor substrate for biomethane production, despite medium levels of COD and VS. Historical data for this site indicate testing for certain herbicides and other xenobiotic compounds, suggesting that wastewater at this site may contain chemicals that inhibit growth, both on-site and later if used as a digestion substrate. Pseudomonas have been found to be able to degrade herbicides [80], chlorinated aromatics [81], and coking wastewater [82], so they may be better suited to grow on this substrate.
3.5.5. Group A, Site 10—Chemicals
Telmatospirillum, Enterobacteriaceae, Pseudomonas, Desulfovibrio, and Macellibacteroides have all been shown to increase in relative abundance in an anaerobic sludge after exposure to selenate [46], and some halorespiring Sulfurospirillum species can use selenate as an electron acceptor when chlorinated compounds are not available [83]. These bacteria were present at high relative abundances in treated wastewater at this site, suggesting the presence of selenate. Somewhat discounting the theory of high selenate levels is the alternate link between Desulfovibrio and Sulfurospirillum and the presence of sulphur compounds, as indicated in discharge data [84,85], and the presence of Bacteroides, which are negatively affected by selenate [46]. Selenium was found at low concentrations by the water service provider at this site. Another parameter that is in the discharge-testing data is phosphorus, a major elemental component of phosphates. Candidatus accumulibacter, the only identified genus of the Rhodocyclaceae family at this site, can accumulate phosphate and is often found in phosphate removal trains at wastewater treatment plants [86]. This is compatible with the high phosphate concentration measured in the wastewater sample.
3.5.6. Group B, Site 2—Brewery
The predominant genera found in raw wastewater at Site 2 were Aeromonas and Lactobacillus. Aeromonas have been found in malting processes [87] and the treatment of brewing wastewater [88], along with Zymophilus [82]. Lactobacillus backii and Lactobacillus coryniformis, both present, are beer spoilage microorganisms [89,90] that show sensitivity to α-acid compounds in hops. The previously mentioned beer-spoiling Obesumbacteria species were not found. Despite both samples being of low pH, the obligate acidophile Acidocella [56] was found in the treated wastewater. At this site, treatment was found to increase the abundances of Prevotella oryzae and Prevotella paludivivens, slightly acidophilic anaerobes first isolated from irrigated rice-fields [91,92], and Clostridium and Leuconostoc, both of which have been found in sludge granules of UASB reactors treating distillery and brewery wastewaters [93]. Leuconostoc requires a rich growth medium for culturing [72]. Yersiniaceae, selected by treatment at Site 1, conversely, decreased in abundance following treatment at this site. Site 2 was also an outlier in previous BMP studies, showing a higher biogas yield for the treated substrate. The factors in Leuconostoc and Yersiniaceae abundances before and after treatments may also influence measured biogas yields.
3.5.7. Group B, Site 3—Waste Management/Logistics
The untreated effluent of Site 3, a logistics company, contained high proportions of Acinetobacter, Lactobacillus, Enterococcus, and members of the Enterobacteriaceae and Yersiniaceae families that could not be further classified. This is consistent with trends for untreated samples described earlier. Although precise species and strains could not be identified, members of each of these families and genera are known to be potentially pathogenic to humans, animals, and/or plants [58,59,94,95]. This is rare and opportunistic for Lactobacillus species, which are usually known as having probiotic effects [96,97]. After treatment, the proportions of all the above taxa were decreased, except for Yersiniaceae. The most common species identified in the treated stream was Dysgonomonas alginatilytica. D. alginatilytica is a facultative anaerobe that can metabolise alginate [98]. Alginate is a gelling agent for food products [99] and can be produced by Pseudomonas and Azotobacter [100]. As the increase in the abundance of D. alginatilytica occurs after treatment, it is unlikely that alginate is present from the main industrial process at this site. Azotobacter was not identified in these samples, and Pseudomonas were only identified at low levels, so D. alginatilytica is likely to be utilising a different substrate. Microvirgula aerodenitrificans, also found at higher levels post treatment, is an aerobic denitrifier shown to be able to remove nitrogen from wastewaters. Perhaps coincidentally, M. aerodenitrificans can be successfully seeded into activated sludge flocs, using alginate beads as a carrier [101]. Caproiciproducens, containing species capable for producing caproic acid from various substrates [102], including xylose [103], and Bacteroides graminisolvens, a xylanolytic species previously found in an anaerobic digester treating cattle waste [104], were also found at higher relative abundances in the treated stream.
3.5.8. Group B, Site 9—Waste Management/Logistics
Pectobacteriaceae are a family of facultatively anaerobic bacteria often implicated as plant pathogens [59]. Together with Yersiniaceae and Enterobacteriaceae, the order Enterobacterales make up a large proportion of the microbes identified in the treated effluent of Site 9, with Pseudomonas and Aeromonas also present. Before treatment, unidentified genera of the Micrococcaceae family were the most common. Like at Site 3, Lactobacillus and Acinetobacter were also present, confirming abundance trends. The presence of Proteiniphilum, found in methanogenic environments, to accelerate propionate degradation [105], suggests a link to the high biogas yield found for this sample. A higher number of unique taxa and greater Pielou evenness were observed before treatment (67 and 0.860) versus after treatment (52 and 0.770), respectively.
3.5.9. Group C, Site 4—Chemicals
The most common ASV found in the untreated wastewater of Site 4 was Legionella, a potential pathogen found in cooling water [7]. The presence of Legionella, Reyranella (which has also been isolated from cooling towers [106]) and Bosea, all of which can grow within amoebae [106], as well as Methyloversatilis, a prey organism of Legionella [107], indicates that cooling water can be a significant contribution to an enterprise’s wastewater. Apart from the immediate influx of certain microbes, this is also a source of treatment chemicals, such as microbicides, phosphates, or heavy metals [108], which may have downstream effects. Rhodococcus erythropolis [109], identified from this sample, and Pseudoxanthomonas [110], Acidivorax [111], and Immundisolibacter [112] species have been reported to degrade hydrocarbons, and Parvibaculum has been shown to oxidise linear alkylbenzene sulphonate, a commercial surfactant [113]. The presence of these ASVs may reflect the products of this chemical industry site.
3.5.10. Group C, Site 11—Chemicals
The microbial community of this wastewater is similar to that of the untreated effluent at Site 4. The presence of Parvibaculum, Rhodococcus, Immundisolibacter, Sulfuritalea [114], and Acidivorax, as well as other unknown Comamonadaceae species, suggests the presence of hydrocarbons and surfactants, while Reyranella, Legionella, Bosea, and Methyloversatilis all indicate that cooling-tower water is being sent to treatment. Rurimicrobium has been shown to increase in growth and lead to fouling in membrane bioreactors exposed to sunlight [115], conditions that are also possible in cooling towers or open trade-waste pits.
3.5.11. Group D, Site 12—Animal Products
Both treated and untreated wastewaters at this site had high richness (177 and 128), Shannon index (4.79 and 4.17), and Pielou evenness (0.92 and 0.86) values, respectively. Taxa identified from the untreated wastewater indicate complex inputs. Dechloromonas species, of the family Rhodocyclaceae, have been isolated from wastewater treatment processes [56], where they reduce nitrate and accumulate polyphosphate [116]. Brachymonas is an aerobe, with species that have been isolated from soybean waste sludge [117] and waste stabilisation ponds [118], and have applications in nitrogen removal [13]. Reflecting the high methane yields obtained in BMP assays, this wastewater supported populations of methanogens Methanocorpusculum [119], Methanospirillum [55], and Methanomicrobiales [120], as well as Synergistaceae, which are correlated with higher methane contents from anaerobic digestion [52]. Anaerocella was found in a methanogenic reactor treating rice-straw residue from cattle farm waste [121]. Revealing the industry sector at this site, the wider Rikenellaceae family contains mostly anaerobic bacteria frequently found in the digestive tracts of animals [74]. Clostridium sensu stricto 1 is also present in the digestive systems of pigs and can increase in abundance when the animal has been infected with pathogens [122]. Arcobacter is an animal- and food-borne pathogen that can grow at low temperatures [123]. Williamwhitmania, a genus including psychrophiles [124], may degrade long-chain fatty acids at low temperatures [125], a characteristic shared with WCHB1-41. At the other end of the optimal-growth-temperature scale, Thermobrachium is a thermophile that can grow very quickly in certain microniches [126], revealing the range of conditions under which wastewater is produced at this site.
All the above-mentioned taxa decreased in relative abundance after treatment as the overall measured diversity increased. Members of Rhizobiaceae, a highly diverse family [127], were present in the post-treatment sample, but further classification was not possible. As a settling process takes place as a part of the treatment at this site, the decrease in Arcobacter contrasts with the effects of settlers found at wastewater treatment plants [128]. Of the taxa showing increased abundance post treatment, Giesbergia species have been found in wastewater aeration tanks [129], and Lentimicrobium have been found in UASB treating high-strength wastewater [130]. The presence of bacteriolytic Phaselicystis, which exhibit antibiotic resistance to aminoglycoside antibiotics [131], and Desulfobulbus, which have implications for steel corrosion due to sulphate reduction byproducts [5], may give early warnings to operators concerned about maintenance and discharge quality issues.
4. Conclusions
This study investigated the taxonomic compositions of eighteen treated and untreated wastewater samples across eleven industrial sites. The novel inventories that were produced show that microbial communities in industrial wastewaters are as many and varied as the wastewaters themselves and provide significant information for the classification of trade-waste streams. In general, Proteobacteria was the most common phylum, while Pseudomonas was the genus that was present in the most samples. The connection between the conditions and a small number of identified taxa was apparent in some circumstances, such as the high sodium concentration at Site 7 favouring halophiles; however, there were limited links between the overall microbiome and the basic measurements recorded in the historical water chemistry data. Clostridium sensu stricto 13, Desulfovibrio, Leuconostoc, and Sulfurospirillum were positively correlated with treatments, and Acinetobacter, Enterococcus, Selenomonas, and Chryseobacterium were associated with untreated samples. Among others, Acinetobacter, Prevotella, and Lactobacillus were found to be more common in substrates with high BMPs, while Clostridium sensu stricto 13, Rhodococcus, and Flavobacterium were more abundant in low-yielding wastewaters. The presence of some organisms may allow for speculation on certain internal sources of wastewater or indicate certain parameters for closer inspection, like hydrocarbons.
The richness and diversity values of the treated wastewater group were not significantly different from those of the untreated group and do not predict a substrate’s suitability for anaerobic digestion. Where the cause of the decreased diversity is temporary, such as exposure to high temperatures, the BMP is not affected. Where the cause of the decreased diversity is carried within the wastewater, e.g., microbicidal compounds, the BMP is affected.
This work provides an original and valuable reference point for understanding the microbial communities present in industrial wastewaters and the information that they can provide to operators and water service providers.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/applmicrobiol4020047/s1: Table S1: Historical Data.
Author Contributions
Conceptualisation, J.A.K.E. and A.S.B.; data curation, J.A.K.E.; formal analysis, J.A.K.E.; funding acquisition, A.S.B.; investigation, J.A.K.E.; methodology, J.A.K.E. and C.K.; project administration, A.S.B.; resources, A.S.B.; supervision, A.S.B.; visualisation, J.A.K.E.; writing—original draft preparation, J.A.K.E. and C.K.; writing—review and editing, J.A.K.E., C.K. and A.S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Greater Western Water. The authors acknowledge the support received for research through the provision of an Australian Government Research Training Program Scholarship.
Data Availability Statement
Data are contained within the article and supplementary materials. The data presented in this study are available on request from the corresponding author and approval by the sample suppliers. The data are not publicly available because of the privacy of the sample suppliers.
Acknowledgments
The authors acknowledge the support of GWW and the sample suppliers.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fanchiang, J.M.; Tseng, D.H.; Guo, G.L.; Chen, H.J. Ozonation of complex industrial park wastewater: Effects on the change of wastewater characteristics. J. Chem. Technol. Biotechnol. 2009, 84, 1007–1014. [Google Scholar] [CrossRef]
- Gutierrez-Sarabia, A.; Fernandez-Villagomez, G.; Martinez-Pereda, P.; Rinderknecht-Seijas, N.; Poggi-Varaldo, H.M. Slaughterhouse wastewater treatment in a full-scale system with constructed wetlands. Water Environ. Res. 2004, 76, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, L.; Deeb, R.; Sawaya, C.; El Khoury, C.; Wazne, M.; Harb, M. Anaerobic membrane bioreactor-based treatment of poultry slaughterhouse wastewater: Microbial community adaptation and antibiotic resistance gene profiles. Biochem. Eng. J. 2023, 192, 108847. [Google Scholar] [CrossRef]
- Parker, C.D. Species of sulphur bacteria associated with the corrosion of concrete. Nature 1947, 159, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Anandkumar, B.; George, R.P.; Maruthamuthu, S.; Palaniswamy, N.; Dayal, R.K. Corrosion behavior of srb desulfobulbus propionicus isolated from an Indian petroleum refinery on mild steel. Mater. Corros. 2011, 63, 355–362. [Google Scholar] [CrossRef]
- Murat, J.B.; Grenouillet, F.; Reboux, G.; Penven, E.; Batchili, A.; Dalphin, J.C.; Thaon, I.; Millon, L. Factors influencing the microbial composition of metalworking fluids and potential implications for machine operator’s lung. Appl. Environ. Microbiol. 2012, 78, 34–41. [Google Scholar] [CrossRef]
- Bonetta, S.; Bonetta, S. Editorial comments to the special issue: “Legionella contamination in water environment”. Pathogens 2020, 9, 1017. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Bu, Y.; Zhang, X.X.; Huang, K.; He, X.; Ye, L.; Shan, Z.; Ren, H. Metagenomic analysis of bacterial community composition and antibiotic resistance genes in a wastewater treatment plant and its receiving surface water. Ecotoxicol. Environ. Saf. 2016, 132, 260–269. [Google Scholar] [CrossRef]
- Haenelt, S.; Richnow, H.H.; Muller, J.A.; Musat, N. Antibiotic resistance indicator genes in biofilm and planktonic microbial communities after wastewater discharge. Front. Microbiol. 2023, 14, 1252870. [Google Scholar] [CrossRef]
- Xu, R.; Fan, F.; Lin, Q.; Yuan, S.; Meng, F. Overlooked ecological roles of influent wastewater microflora in improving biological phosphorus removal in an anoxic/aerobic MBR process. Environ. Sci. Technol. 2021, 55, 6270–6280. [Google Scholar] [CrossRef]
- Yu, L.; Li, R.; Delatolla, R.; Zhang, R.; Yang, X.; Peng, D. Natural continuous influent nitrifier immigration effects on nitrification and the microbial community of activated sludge systems. J. Environ. Sci. 2018, 74, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Khafipour, A.; Jordaan, E.M.; Flores-Orozco, D.; Khafipour, E.; Levin, D.B.; Sparling, R.; Cicek, N. Response of microbial community to induced failure of anaerobic digesters through overloading with propionic acid followed by process recovery. Front. Bioeng. Biotechnol. 2020, 8, 604838. [Google Scholar] [CrossRef] [PubMed]
- Leta, S.; Assefa, F.; Dalhammar, G. Enhancing biological nitrogen removal from tannery effluent by using the efficient Brachymonas denitrificans in pilot plant operations. World J. Microbiol. Biotechnol. 2005, 21, 545–552. [Google Scholar] [CrossRef]
- Guo, B.; Liu, C.; Gibson, C.; Frigon, D. Wastewater microbial community structure and functional traits change over short timescales. Sci. Total Environ. 2019, 662, 779–785. [Google Scholar] [CrossRef]
- Martinez-Santos, M.; Lanzen, A.; Unda-Calvo, J.; Martin, I.; Garbisu, C.; Ruiz-Romera, E. Treated and untreated wastewater effluents alter river sediment bacterial communities involved in nitrogen and sulphur cycling. Sci. Total Environ. 2018, 633, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.A.K.; Ball, A.S.; Shah, K. Investigations into valorisation of trade wastewater for biomethane production. Heliyon 2023, 9, e13309. [Google Scholar] [CrossRef]
- Baird, R.B.; Eaton, A.D.; Rice, E.W. Standard Methods for the Examination of Water and Wastewater, 23rd ed.; American Public Health Association: Washington, DC, USA, 2012. [Google Scholar]
- Hach Company. Oxygen Demand, Chemical. 2021. Available online: https://au.hach.com/asset-get.download.jsa?id=7639983816 (accessed on 13 December 2021).
- Hach Company. Nitrogen, Total, Method 10071, doc316.53.01086. 2014. Available online: https://au.hach.com/asset-get.download.jsa?id=763998380410 (accessed on 13 December 2021).
- Hach Company. Nitrate, HR, Method 10020, doc316.53.01068. 2015. Available online: https://au.hach.com/asset-get.download.jsa?id=763998373810 (accessed on 13 December 2021).
- Hach Company. Nitrite, Method 10019, doc316.53.01073. 2015. Available online: https://sea.hach.com/asset-get.download.jsa?id=763998374210 (accessed on 13 December 2021).
- Hach Company. Nitrogen, Ammonia, Method 10031, doc316.53.01079. 2015. Available online: https://au.hach.com/asset-get.download.jsa?id=763998374710 (accessed on 13 December 2021).
- Hach Company. Phosphorus, Total. 2017. Available online: https://au.hach.com/asset-get.download.jsa?id=7639983838 (accessed on 13 December 2021).
- Anaero Technology. Available online: https://www.anaerotech.com/ (accessed on 13 December 2021).
- Qiagen. Dneasy® Powersoil® Pro Kit Handbook; Qiagen: Hilden, Germany, 2023. [Google Scholar]
- Illumina. 16S Metagenomic Sequencing Library Preparation; (15044223 b); Illumina: San Diego, CA, USA, 2013. [Google Scholar]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef]
- Mazzoli, L.; Munz, G.; Lotti, T.; Ramazzotti, M. A novel universal primer pair for prokaryotes with improved performances for anammox containing communities. Sci. Rep. 2020, 10, 15648. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using qiime 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with qiime 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.K.D.; Nierychlo, M.; Andersen, K.S.; Rudkjobing, V.; Knutsson, S.; MiDAS Global Consortium; Albertsen, M.; Nielsen, P.H. Midas 4: A global catalogue of full-length 16S rrna gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. Nat. Commun. 2022, 13, 1908. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.K.D.; Andersen, K.S.; Petersen, A.-K.C.; Rudkjøbing, V.; Nielsen, P.H. MiDAS 5: Global diversity of bacteria and archaea in anaerobic digesters. bioRxiv 2023. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Lahti, L.; Shetty, S. Microbiome R Package. 2012–2019. Available online: https://www.bioconductor.org/packages/release/bioc/html/microbiome.html (accessed on 11 December 2022).
- Oksanen, J.; Simpson, G.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Solymos, P.; Stevens, M.; Szoecs, E.; et al. Vegan: Community Ecology Package. 2022. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 11 December 2022).
- Lin, H.; Eggesbo, M.; Peddada, S.D. Linear and nonlinear correlation estimators unveil undescribed taxa interactions in microbiome data. Nat. Commun. 2022, 13, 4946. [Google Scholar] [CrossRef]
- Eurofins. Trade Waste Testing, 7th ed.; Available online: https://cdnmedia.eurofins.com/apac/media/610106/trade-waste-testing-v7-sept-21.pdf (accessed on 21 February 2024).
- Wang, L.K.; Hung, Y.-T.; Lo, H.H.; Yapijakis, C. Waste Treatment in the Food Processing Industry; Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- El-Abbassi, A.; Hafidi, A.; García-Payo, M.; Khayet, M. Concentration of olive mill wastewater by membrane distillation for polyphenols recovery. Desalination 2009, 245, 670–674. [Google Scholar] [CrossRef]
- Krishnan, S.; Md Din, M.F.; Taib, S.M.; Nasrullah, M.; Sakinah, M.; Wahid, Z.A.; Kamyab, H.; Chelliapan, S.; Rezania, S.; Singh, L. Accelerated two-stage bioprocess for hydrogen and methane production from palm oil mill effluent using continuous stirred tank reactor and microbial electrolysis cell. J. Clean. Prod. 2019, 229, 84–93. [Google Scholar] [CrossRef]
- Braguglia, C.M.; Gallipoli, A.; Gianico, A.; Pagliaccia, P. Anaerobic bioconversion of food waste into energy: A critical review. Bioresour. Technol. 2018, 248, 37–56. [Google Scholar] [CrossRef]
- City West Water. Approved Acceptance Criteria for Discharge to the Sewerage System; City West Water: Melbourne, Australia, 2017. [Google Scholar]
- Zeng, T.; Hu, Q.; Rene, E.R.; Lens, P.N.L. Microbial community and extracellular polymeric substances analysis of anaerobic granular sludge exposed to selenate, cadmium and zinc. Microb. Biotechnol. 2023, 16, 463–473. [Google Scholar] [CrossRef]
- Burdon, F.J.; Bai, Y.; Reyes, M.; Tamminen, M.; Staudacher, P.; Mangold, S.; Singer, H.; Rasanen, K.; Joss, A.; Tiegs, S.D.; et al. Stream microbial communities and ecosystem functioning show complex responses to multiple stressors in wastewater. Glob. Chang. Biol. 2020, 26, 6363–6382. [Google Scholar] [CrossRef]
- Jagaba, A.H.; Kutty, S.R.M.; Isa, M.H.; Ghaleb, A.A.S.; Lawal, I.M.; Usman, A.K.; Birniwa, A.H.; Noor, A.; Abubakar, S.; Umaru, I.; et al. Toxic effects of xenobiotic compounds on the microbial community of activated sludge. ChemBioEng Rev. 2022, 9, 497–535. [Google Scholar] [CrossRef]
- Vanwonterghem, I.; Jensen, P.D.; Dennis, P.G.; Hugenholtz, P.; Rabaey, K.; Tyson, G.W. Deterministic processes guide long-term synchronised population dynamics in replicate anaerobic digesters. ISME J. 2014, 8, 2015–2028. [Google Scholar] [CrossRef]
- Tong, T.; Tong, J.; Xue, K.; Li, Y.; Yu, J.; Wei, Y. Microbial community structure and functional prediction in five full-scale industrial park wastewater treatment plants. Sci. Total Environ. 2023, 904, 166529. [Google Scholar] [CrossRef]
- Rodriguez Lopez, J.; Grande, M.J.; Perez-Pulido, R.; Galvez, A.; Lucas, R. Impact of high-hydrostatic pressure treatments applied singly or in combination with moderate heat on the microbial load, antimicrobial resistance, and bacterial diversity of guacamole. Microorganisms 2020, 8, 909. [Google Scholar] [CrossRef]
- Ferguson, R.M.W.; Coulon, F.; Villa, R. Understanding microbial ecology can help improve biogas production in AD. Sci. Total Environ. 2018, 642, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Waite, D.W.; Vanwonterghem, I.; Rinke, C.; Parks, D.H.; Zhang, Y.; Takai, K.; Sievert, S.M.; Simon, J.; Campbell, B.J.; Hanson, T.E.; et al. Erratum: Addendum: Comparative genomic analysis of the class epsilonproteobacteria and proposed reclassification to epsilonbacteraeota (phyl. nov.). Front. Microbiol. 2018, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, M.; Yang, S.; Gong, H.; Ma, J.; Li, C.; Wang, K. Performance and microbial community evaluation of full-scale two-phase anaerobic digestion of waste activated sludge. Sci. Total Environ. 2022, 814, 152525. [Google Scholar] [CrossRef]
- Boone, D.R.; Castenholz, R.W. The Archaea and the Deeply Branching and Phototrophic Bacteria, 2nd ed.; Springer: New York, NY, USA, 2001; Volume 1. [Google Scholar]
- Brenner, D.J.; Krieg, N.R.; Staley, J.T. The Proteobacteria: Part C: The Alpha-, Beta-, Delta-, and Epsilonproteobacteria, 2nd ed.; Springer: New York, NY, USA, 2005; Volume 2C. [Google Scholar]
- Bonk, F.; Popp, D.; Weinrich, S.; Strauber, H.; Kleinsteuber, S.; Harms, H.; Centler, F. Intermittent fasting for microbes: How discontinuous feeding increases functional stability in anaerobic digestion. Biotechnol. Biofuels 2018, 11, 274. [Google Scholar] [CrossRef]
- Brenner, D.J.; Krieg, N.R.; Staley, J.T. The Proteobacteria: Part B: The Gammaproteobacteria, 2nd ed.; Springer: New York, NY, USA, 2005; Volume 2B. [Google Scholar]
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. Divided into the families Enterobacteriaceae, Ewiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar]
- Boulton, C. Encyclopaedia of Brewing; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chen, H.L.; Zhao, X.Y.; Zhao, G.X.; Huang, H.B.; Li, H.R.; Shi, C.W.; Yang, W.T.; Jiang, Y.L.; Wang, J.Z.; Ye, L.P.; et al. Dissection of the cecal microbial community in chickens after Eimeria tenella infection. Parasit Vectors 2020, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Guo, C.; Gong, Y.; Sun, X.; Wang, W.; Wang, Y.; Yang, H.; Cao, Z.; Li, S. Rumen fermentation, digestive enzyme activity, and bacteria composition between pre-weaning and post-weaning dairy calves. Animals 2021, 11, 2527. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, H.; Liu, S.; Chai, S.; Meng, Q.; Zhou, Z. Dynamic alterations in yak rumen bacteria community and metabolome characteristics in response to feed type. Front. Microbiol. 2019, 10, 1116. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS ONE 2013, 8, e83424. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.W.; Kim, B.Y.; Kim, W.G.; Yoo, K.H.; Yoo, S.H.; Son, J.A.; Weon, H.Y. Paludibacterium yongneupense gen. nov., sp. Nov., isolated from a wetland, yongneup, in korea. Int. J. Syst. Evol. Microbiol. 2008, 58, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rudra, B.; Gupta, R.S. Phylogenomics and molecular signatures support division of the order Neisseriales into emended families Neisseriaceae and Chromobacteriaceae and three new families Aquaspirillaceae fam. nov., Chitinibacteraceae fam. nov., and Leeiaceae fam. nov. Syst. Appl. Microbiol. 2021, 44, 126251. [Google Scholar] [CrossRef] [PubMed]
- Edzwald, J.K. Dissolved air flotation and me. Water Res. 2010, 44, 2077–2106. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.L.; Sueitt, A.P.E.; Dos Santos, P.R.; Leite, L.S.; Daniel, L.A. Removal of protozoan (oo)cysts and bacteria during microalgae harvesting: Outcomes from a lab-scale experiment. Chemosphere 2022, 286, 131767. [Google Scholar] [CrossRef]
- Huber, R.; Kristjansson, J.K.; Stetter, K.O. Pyrobaculum gen. nov., a new genus of neutrophilic, rod-shaped archaebacteria from continental solfataras growing optimally at 100 °C. Arch. Microbiol. 1987, 149, 95–101. [Google Scholar] [CrossRef]
- Nazina, T.N.; Tourova, T.P.; Poltaraus, A.B.; Novikova, E.V.; Grigoryan, A.A.; Ivanova, A.E.; Lysenko, A.M.; Petrunyaka, V.V.; Osipov, G.A.; Belyaev, S.S.; et al. Taxonomic study of aerobic thermophilic bacilli: Descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. From petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermo-catenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermoglucosidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans. Int. J. Syst. Evol. Microbiol. 2001, 51, 433–446. [Google Scholar]
- Hai, D.; Jiang, H.; Meng, Z.; Qiao, M.; Xu, T.; Song, L.; Huang, X. The impact of high temperature on microbial communities in pork and duck skin. Microorganisms 2023, 11, 2869. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Jones, D.; Rainey, F.A.; Whitman, W.B.; De Vos, P.; Krieg, N.R.; Garrity, G.M.; Schleifer, K.-H. The Firmicutes, 2nd ed.; Springer: New York, NY, USA, 2009; Volume 3. [Google Scholar]
- Maturrano, L.; Valens-Vadell, M.; Rossello-Mora, R.; Anton, J. Salicola marasensis gen. nov., sp. nov., an extremely halophilic bacterium isolated from the Maras solar salterns in Peru. Int. J. Syst. Evol. Microbiol. 2006, 56, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.; DeLong, E.F.; Lory, S.; Stackebrandt, E.; Thompson, F. The Prokaryotes: Other Major Lineages of Bacteria and the Archaea, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Wang, Y.; Cao, L.L.; Tang, S.K.; Lou, K.; Mao, P.H.; Jin, X.; Jiang, C.L.; Xu, L.H.; Li, W.J. Marinococcus luteus sp. nov., a halotolerant bacterium isolated from a salt lake, and emended description of the genus Marinococcus. Int. J. Syst. Evol. Microbiol. 2009, 59, 2875–2879. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Xu, D.; Bi, X.; Ng, H.Y.; Shi, X. Intertidal wetland sediment as a novel inoculation source for developing aerobic granular sludge in membrane bioreactor treating high-salinity antibiotic manufacturing wastewater. Bioresour. Technol. 2020, 314, 123715. [Google Scholar] [CrossRef]
- Yoon, J.H.; Lee, K.C.; Weiss, N.; Kang, K.H.; Park, Y.H. Jeotgalicoccus halotolerans gen. nov., sp. nov. and Jeotgalicoccus psychrophilus sp. nov., isolated from the traditional Korean fermented seafood jeotgal. Int. J. Syst. Evol. Microbiol. 2003, 53, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Whitman, W.B.; Goodfellow, M.; Kämpfer, P.; Busse, H.-J.; Trujillo, M.E.; Ludwig, W.; Suzuki, K.-I. The Actinobacteria, 2nd ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Liaw, H.J.; Mah, R.A. Isolation and characterization of haloanaerobacter Chitinovorans gen. nov., sp. nov., a halophilic, anaerobic, chitinolytic bacterium from a solar saltern. Appl. Environ. Microbiol. 1992, 58, 260–266. [Google Scholar] [CrossRef]
- Korshunova, T.; Kuzina, E.; Mukhamatdyarova, S.; Sharipova, Y.; Iskuzhina, M. Promising strains of hydrocarbon-oxidizing pseudomonads with herbicide resistance and plant growth-stimulating properties for bioremediation of oil-contaminated agricultural soils. Agriculture 2023, 13, 1111. [Google Scholar] [CrossRef]
- Peng, X.; Pan, X.; Wang, X.; Li, D.; Huang, P.; Qiu, G.; Shan, K.; Chu, X. Accelerated removal of high concentration p-chloronitrobenzene using bioelectrocatalysis process and its microbial communities analysis. Bioresour. Technol. 2018, 249, 844–850. [Google Scholar] [CrossRef]
- Yuan, K.; Li, S.; Zhong, F. Treatment of coking wastewater in biofilm-based bioaugmentation process: Biofilm formation and microbial community analysis. J. Hazard. Mater. 2020, 400, 123117. [Google Scholar] [CrossRef]
- Luijten, M.L.; Weelink, S.A.; Godschalk, B.; Langenhoff, A.A.; van Eekert, M.H.; Schraa, G.; Stams, A.J. Anaerobic reduction and oxidation of quinone moieties and the reduction of oxidized metals by halorespiring and related organisms. FEMS Microbiol. Ecol. 2004, 49, 145–150. [Google Scholar] [CrossRef]
- Hubert, C.; Voordouw, G. Oil field souring control by nitrate-reducing Sulfurospirillum spp. that outcompete sulfate-reducing bacteria for organic electron donors. Appl. Environ. Microbiol. 2007, 73, 2644–2652. [Google Scholar] [CrossRef] [PubMed]
- Eisenmann, E.; Beuerle, J.; Sulger, K.; Kroneck, P.M.H.; Schumacher, W. Lithotrophic growth of Sulfurospirillum deleyianum with sulfide as electron donor coupled to respiratory reduction of nitrate to ammonia. Arch. Microbiol. 1995, 164, 180–185. [Google Scholar] [CrossRef]
- Petriglieri, F.; Singleton, C.M.; Kondrotaite, Z.; Dueholm, M.K.D.; McDaniel, E.A.; McMahon, K.D.; Nielsen, P.H.; McGrath, J. Reevaluation of the phylogenetic diversity and global distribution of the genus “Candidatus accumulibacter”. mSystems 2022, 7, e00016-22. [Google Scholar] [CrossRef] [PubMed]
- Malfliet, S.; Juste, A.; Crauwels, S.; Willems, K.; De Cooman, L.; Lievens, B.; Aerts, G. Assessing the xylanolytic bacterial diversity during the malting process. Food Microbiol. 2013, 36, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Dilek, F.B.; Anderson, G.K.; Bloor, J. Investigation into the microbiology of a high rate jet-loop activated sludge reactor treating brewery wastewater. Water Sci. Technol. 1996, 34, 107–112. [Google Scholar] [CrossRef]
- Geissler, A.J.; Behr, J.; von Kamp, K.; Vogel, R.F. Metabolic strategies of beer spoilage lactic acid bacteria in beer. Int. J. Food Microbiol. 2016, 216, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Cocuzza, S.; Biendl, M.; Peifer, F.; Hans, S.; Methner, Y.; Pehl, F.; Back, W.; Jacob, F.; Hutzler, M. The impact of different hop compounds on the growth of selected beer spoilage bacteria in beer. J. Inst. Brew. 2020, 126, 354–361. [Google Scholar]
- Ueki, A.; Akasaka, H.; Satoh, A.; Suzuki, D.; Ueki, K. Prevotella paludivivens sp. nov., a novel strictly anaerobic, gram-negative, hemicellulose-decomposing bacterium isolated from plant residue and rice roots in irrigated rice-field soil. Int. J. Syst. Evol. Microbiol. 2007, 57, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Ueki, A.; Akasaka, H.; Suzuki, D.; Hattori, S.; Ueki, K. Xylanibacter oryzae gen. nov., sp. nov., a novel strictly anaerobic, gram-negative, xylanolytic bacterium isolated from rice-plant residue in flooded rice-field soil in japan. Int. J. Syst. Evol. Microbiol. 2006, 56, 2215–2221. [Google Scholar] [CrossRef]
- Keyser, M.; Britz, T.J.; Witthuhn, R.C. Fingerprinting and identification of bacteria present in UASB granules used to treat winery, brewery, distillery or peach-lye canning wastewater. S. Afr. J. Enol. Vitic. 2007, 28, 69–79. [Google Scholar] [CrossRef]
- Wright, M.S.; Haft, D.H.; Harkins, D.M.; Perez, F.; Hujer, K.M.; Bajaksouzian, S.; Benard, M.F.; Jacobs, M.R.; Bonomo, R.A.; Adams, M.D. New insights into dissemination and variation of the health care-associated pathogen Acinetobacter baumannii from genomic analysis. mBio 2014, 5, e00963-13. [Google Scholar] [CrossRef] [PubMed]
- Arredondo-Alonso, S.; Top, J.; McNally, A.; Puranen, S.; Pesonen, M.; Pensar, J.; Marttinen, P.; Braat, J.C.; Rogers, M.R.C.; van Schaik, W.; et al. Plasmids shaped the recent emergence of the major nosocomial pathogen Enterococcus faecium. mBio 2020, 11, e03284-19. [Google Scholar] [CrossRef] [PubMed]
- Antoun, M.; Hattab, Y.; Akhrass, F.A.; Hamilton, L.D. Uncommon pathogen, Lactobacillus, causing infective endocarditis: Case report and review. Case Rep. Infect. Dis. 2020, 2020, 8833948. [Google Scholar] [CrossRef] [PubMed]
- Chery, J.; Dvoskin, D.; Morato, F.P.; Fahoum, B. Lactobacillus fermentum, a pathogen in documented cholecystitis. Int. J. Surg. Case Rep. 2013, 4, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Kita, A.; Miura, T.; Okamura, Y.; Aki, T.; Matsumura, Y.; Tajima, T.; Kato, J.; Nakashimada, Y. Dysgonomonas alginatilytica sp. nov., an alginate-degrading bacterium isolated from a microbial consortium. Int. J. Syst. Evol. Microbiol. 2015, 65, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Efsa Panel on Additives and Products or Substances Used in Animal Feed; Rychen, G.; Aquilina, G.; Azimonti, G.; Bampidis, V.; Bastos, M.L.; Bories, G.; Chesson, A.; Cocconcelli, P.S.; Flachowsky, G.; et al. Safety and efficacy of sodium and potassium alginate for pets, other non food-producing animals and fish. EFSA J. 2017, 15, e04945. [Google Scholar] [PubMed]
- Hay, I.D.; Ur Rehman, Z.; Ghafoor, A.; Rehm, B.H.A. Bacterial biosynthesis of alginates. J. Chem. Technol. Biotechnol. 2010, 85, 752–759. [Google Scholar] [CrossRef]
- Bouchez, T.; Patureau, D.; Delgenes, J.P.; Moletta, R. Successful bacterial incorporation into activated sludge flocs using alginate. Bioresour. Technol. 2009, 100, 1031–1032. [Google Scholar] [CrossRef]
- Kim, B.C.; Seung Jeon, B.; Kim, S.; Kim, H.; Um, Y.; Sang, B.I. Caproiciproducens galactitolivorans gen. nov., sp. nov., a bacterium capable of producing caproic acid from galactitol, isolated from a wastewater treatment plant. Int. J. Syst. Evol. Microbiol. 2015, 65, 4902–4908. [Google Scholar] [CrossRef]
- Tang, J.; Dai, K.; Wang, Q.T.; Zheng, S.J.; Hong, S.D.; Jianxiong Zeng, R.; Zhang, F. Caproate production from xylose via the fatty acid biosynthesis pathway by genus Caproiciproducens dominated mixed culture fermentation. Bioresour. Technol. 2022, 351, 126978. [Google Scholar] [CrossRef]
- Nishiyama, T.; Ueki, A.; Kaku, N.; Watanabe, K.; Ueki, K. Bacteroides graminisolvens sp. nov., a xylanolytic anaerobe isolated from a methanogenic reactor treating cattle waste. Int. J. Syst. Evol. Microbiol. 2009, 59, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, A.M.; Schmidt, T.; Lv, Z.; Liebetrau, J.; Richnow, H.H.; Kleinsteuber, S.; Nikolausz, M. Reduction of the hydraulic retention time at constant high organic loading rate to reach the microbial limits of anaerobic digestion in various reactor systems. Bioresour. Technol. 2016, 217, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Pagnier, I.; Raoult, D.; La Scola, B. Isolation and characterization of Reyranella massiliensis gen. nov., sp. nov. from freshwater samples by using an amoeba co-culture procedure. Int. J. Syst. Evol. Microbiol. 2011, 61, 2151–2154. [Google Scholar] [CrossRef] [PubMed]
- van der Kooij, D.; Veenendaal, H.R.; Italiaander, R.; van der Mark, E.J.; Dignum, M. Primary colonizing Betaproteobacteriales play a key role in the growth of Legionella pneumophila in biofilms on surfaces exposed to drinking water treated by slow sand filtration. Appl. Environ. Microbiol. 2018, 84, e01732-18. [Google Scholar] [CrossRef] [PubMed]
- Frayne, C. (Ed.) Chemical treatments and programs for cooling water. In Cooling Water Treatment—Principles and Practice; Chemical Publishing Company: Revere, MA, USA, 1999. [Google Scholar]
- de Carvalho, C.C.; da Fonseca, M.M. The remarkable Rhodococcus erythropolis. Appl. Microbiol. Biotechnol. 2005, 67, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Ryu, S.H.; Hwang, H.W.; Kim, Y.J.; Park, M.; Lee, J.R.; Lee, S.S.; Jeon, C.O. Pseudoxanthomonas sacheonensis sp. nov., isolated from btex-contaminated soil in Korea, transfer of Stenotrophomonas dokdonensis Yoon et al. 2006 to the genus Pseudoxanthomonas as Pseudoxanthomonas dokdonensis comb. nov. and emended description of the genus Pseudoxanthomonas. Int. J. Syst. Evol. Microbiol. 2008, 58, 2235–2240. [Google Scholar] [PubMed]
- Bedics, A.; Tancsics, A.; Banerjee, S.; Toth, E.; Harkai, P.; Gottschall, G.G.; Boka, K.; Kriszt, B. Acidovorax benzenivorans sp. nov., a novel aromatic hydrocarbon-degrading bacterium isolated from a xylene-degrading enrichment culture. Int. J. Syst. Evol. Microbiol. 2024, 74, 006219. [Google Scholar] [CrossRef]
- Corteselli, E.M.; Aitken, M.D.; Singleton, D.R. Description of Immundisolibacter cernigliae gen. nov., sp. nov., a high-molecular-weight polycyclic aromatic hydrocarbon-degrading bacterium within the class Gammaproteobacteria, and proposal of Immundisolibacterales ord. nov. and Immundisolibacteraceae fam. nov. Int. J. Syst. Evol. Microbiol. 2017, 67, 925–931. [Google Scholar] [PubMed]
- Schleheck, D.; Tindall, B.J.; Rossello-Mora, R.; Cook, A.M. Parvibaculum lavamentivorans gen. nov., sp. nov., a novel heterotroph that initiates catabolism of linear alkylbenzenesulfonate. Int. J. Syst. Evol. Microbiol. 2004, 54, 1489–1497. [Google Scholar] [CrossRef]
- Sperfeld, M.; Diekert, G.; Studenik, S. Anaerobic aromatic compound degradation in Sulfuritalea hydrogenivorans sk43h. FEMS Microbiol. Ecol. 2019, 95, fiy199. [Google Scholar] [CrossRef]
- Park, H.; Shah, S.S.A.; Korshin, G.; Angelidaki, I.; Choo, K.-H. The impact of sunlight on fouling behaviors and microbial communities in membrane bioreactors. J. Membr. Sci. 2023, 672, 121443. [Google Scholar] [CrossRef]
- Petriglieri, F.; Singleton, C.; Peces, M.; Petersen, J.F.; Nierychlo, M.; Nielsen, P.H. “Candidatus dechloromonas phosphoritropha” and “Ca. D. Phosphorivorans”, novel polyphosphate accumulating organisms abundant in wastewater treatment systems. ISME J. 2021, 15, 3605–3614. [Google Scholar] [CrossRef] [PubMed]
- Hiraishi, A.; Shin, Y.K.; Sugiyama, J. Brachymonas denitrificans gen. nov., sp. nov., an aerobic chemoorganotrophic bacterium which contains rhodoquinones, and evolutionary relationships of rhodoquinone producers to bacterial species with various quinone classes. J. Gen. Appl. Microbiol. 1995, 41, 99–117. [Google Scholar] [CrossRef]
- Halpern, M.; Shaked, T.; Schumann, P. Brachymonas chironomi sp. nov., isolated from a chironomid egg mass, and emended description of the genus Brachymonas. Int. J. Syst. Evol. Microbiol. 2009, 59, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Volmer, J.G.; Soo, R.M.; Evans, P.N.; Hoedt, E.C.; Astorga Alsina, A.L.; Woodcroft, B.J.; Tyson, G.W.; Hugenholtz, P.; Morrison, M. Isolation and characterisation of novel Methanocorpusculum species indicates the genus is ancestrally host-associated. BMC Biol. 2023, 21, 59. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Tamaki, H.; Kyrpides, N.; Woyke, T.; Goodwin, L.; Imachi, H.; Brauer, S.; Yavitt, J.B.; Liu, W.T.; Zinder, S.; et al. Genomic composition and dynamics among Methanomicrobiales predict adaptation to contrasting environments. ISME J. 2017, 11, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Ueki, A.; Ohtaki, Y.; Kaku, N.; Watanabe, K.; Ueki, K. Anaerocella delicata gen. nov., sp. nov., a strictly anaerobic bacterium in the phylum Bacteroidetes isolated from a methanogenic reactor of cattle farms. J. Gen. Appl. Microbiol. 2012, 58, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Jinno, C.; Wong, B.; Klunemann, M.; Htoo, J.; Li, X.; Liu, Y. Effects of supplementation of Bacillus amyloliquefaciens on performance, systemic immunity, and intestinal microbiota of weaned pigs experimentally infected with a pathogenic enterotoxigenic E. coli f18. Front. Microbiol. 2023, 14, 1101457. [Google Scholar] [CrossRef]
- Ramees, T.P.; Dhama, K.; Karthik, K.; Rathore, R.S.; Kumar, A.; Saminathan, M.; Tiwari, R.; Malik, Y.S.; Singh, R.K. Arcobacter: An emerging food-borne zoonotic pathogen, its public health concerns and advances in diagnosis and control—A comprehensive review. Vet. Q. 2017, 37, 136–161. [Google Scholar] [CrossRef]
- Pikuta, E.V.; Lyu, Z.; Hoover, R.B.; Liu, Y.; Patel, N.B.; Busse, H.J.; Lawson, P.A. Williamwhitmania taraxaci gen. nov., sp. nov., a proteolytic anaerobe with a novel type of cytology from Lake Untersee in Antarctica, description of Williamwhitmaniaceae fam. nov., and emendation of the order Bacteroidales krieg 2012. Int. J. Syst. Evol. Microbiol. 2017, 67, 4132–4145. [Google Scholar] [CrossRef]
- Singh, S.; Keating, C.; Ijaz, U.Z.; Hassard, F. Molecular insights informing factors affecting low temperature anaerobic applications: Diversity, collated core microbiomes and complexity stability relationships in LCFA-fed systems. Sci. Total Environ. 2023, 874, 162420. [Google Scholar] [CrossRef] [PubMed]
- Engle, M.; Li, Y.; Rainey, F.; Deblois, S.; Mai, V.; Reichert, A.; Mayer, F.; Messner, P.; Wiegel, J. Thermobrachium celere gen. nov., sp. nov., a rapidly growing thermophilic, alkalitolerant, and proteolytic obligate anaerobe. Int. J. Syst. Evol. Microbiol. 1996, 46, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanović, N.; Fagorzi, C.; Mengoni, A.; Lassalle, F.; Dicenzo, G.C. Taxonomy of Rhizobiaceae revisited: Proposal of a new framework for genus delimitation. Int. J. Syst. Evol. Microbiol. 2022, 72, 005243. [Google Scholar] [CrossRef] [PubMed]
- Riisgaard-Jensen, M.; Dottorini, G.; Nierychlo, M.; Nielsen, P.H. Primary settling changes the microbial community of influent wastewater to wastewater treatment plants. Water Res. 2023, 244, 120495. [Google Scholar] [CrossRef] [PubMed]
- Grabovich, M.; Gavrish, E.; Kuever, J.; Lysenko, A.M.; Podkopaeva, D.; Dubinina, G. Proposal of Giesbergeria voronezhensis gen. nov., sp. nov. and G. kuznetsovii sp. nov. and reclassification of [Aquaspirillum] anulus, [A.] sinuosum and [A.] giesbergeri as Giesbergeria anulus comb. nov., G. sinuosa comb. nov. and G. giesbergeri comb. nov., and [Aquaspirillum] metamorphum and [A.] psychrophilum as Simplicispira metamorpha gen. nov., comb. nov. and S. psychrophila comb. nov. Int. J. Syst. Evol. Microbiol. 2006, 56, 569–576. [Google Scholar] [PubMed]
- Sun, L.; Toyonaga, M.; Ohashi, A.; Tourlousse, D.M.; Matsuura, N.; Meng, X.Y.; Tamaki, H.; Hanada, S.; Cruz, R.; Yamaguchi, T.; et al. Lentimicrobium saccharophilum gen. nov., sp. nov., a strictly anaerobic bacterium representing a new family in the phylum Bacteroidetes, and proposal of Lentimicrobiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 2635–2642. [Google Scholar] [CrossRef]
- Garcia, R.O.; Reichenbach, H.; Ring, M.W.; Muller, R. Phaselicystis flava gen. nov., sp. nov., an arachidonic acid-containing soil myxobacterium, and the description of Phaselicystidaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 1524–1530. [Google Scholar] [CrossRef]
Figure 1.
Diversity measurements of microbial communities sequenced from wastewater samples. Colour of box-plot denotes whether sample was measured before or after on-site treatment; colour and shape of dots depict level of methane yield in BMP tests.
Figure 1.
Diversity measurements of microbial communities sequenced from wastewater samples. Colour of box-plot denotes whether sample was measured before or after on-site treatment; colour and shape of dots depict level of methane yield in BMP tests.

Figure 2.
PCA of microbial communities in wastewater samples. Circles represent untreated streams, squares represent wastewater ready for discharge to sewers (treated), and colours denote sites. 5C represents the source-separated waste from Site 5. The letter at each point denotes a high, medium, or low (H, M, or L, respectively) gas yield in BMP tests. Groups shown by ellipses; 12.6% and 11.5% of the variance between samples are mapped by the x- and y-axes, respectively.
Figure 2.
PCA of microbial communities in wastewater samples. Circles represent untreated streams, squares represent wastewater ready for discharge to sewers (treated), and colours denote sites. 5C represents the source-separated waste from Site 5. The letter at each point denotes a high, medium, or low (H, M, or L, respectively) gas yield in BMP tests. Groups shown by ellipses; 12.6% and 11.5% of the variance between samples are mapped by the x- and y-axes, respectively.

Figure 3.
Genera with statistically significant (p < 0.05) differences in relative abundance in untreated samples relative to treated samples. Samples limited to sites with both treated and untreated samples available. Y-axis shows log-fold change in relative abundance in untreated samples vs. treated samples. Positive values, in brown, denote genera more abundant in untreated samples; negative values, in blue, denote genera more abundant in treated samples; n = 12.
Figure 3.
Genera with statistically significant (p < 0.05) differences in relative abundance in untreated samples relative to treated samples. Samples limited to sites with both treated and untreated samples available. Y-axis shows log-fold change in relative abundance in untreated samples vs. treated samples. Positive values, in brown, denote genera more abundant in untreated samples; negative values, in blue, denote genera more abundant in treated samples; n = 12.

Figure 4.
Genera with statistically significant (p < 0.05) differences in relative abundance. Y-axis shows log-fold change in relative abundance in high-yielding samples relative to low-yielding samples. Positive values, in green, denote genera more abundant in untreated samples; negative values, in red, denote genera more abundant in low-yielding samples; n = 16.
Figure 4.
Genera with statistically significant (p < 0.05) differences in relative abundance. Y-axis shows log-fold change in relative abundance in high-yielding samples relative to low-yielding samples. Positive values, in green, denote genera more abundant in untreated samples; negative values, in red, denote genera more abundant in low-yielding samples; n = 16.

Figure 5.
Abundance plot at phylum level, showing sites, treatment status, and level of gas yield from BMP tests. Proteobacteria were the most highly abundant across the samples. Phylum abundance had no significant difference between treatment levels.
Figure 5.
Abundance plot at phylum level, showing sites, treatment status, and level of gas yield from BMP tests. Proteobacteria were the most highly abundant across the samples. Phylum abundance had no significant difference between treatment levels.

Figure 6.
Canonical correspondence analysis (CCA) plots of abundances (Bray–Curtis dissimilarities) of all the samples, showing the 10 most common phyla (a) and the influence of the measured chemical data on their abundance (b). Plot b axes are at a smaller scale for clarity. CCA1 represents 31.3% of the inertia; CCA2 represents 19.9%.
Figure 6.
Canonical correspondence analysis (CCA) plots of abundances (Bray–Curtis dissimilarities) of all the samples, showing the 10 most common phyla (a) and the influence of the measured chemical data on their abundance (b). Plot b axes are at a smaller scale for clarity. CCA1 represents 31.3% of the inertia; CCA2 represents 19.9%.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
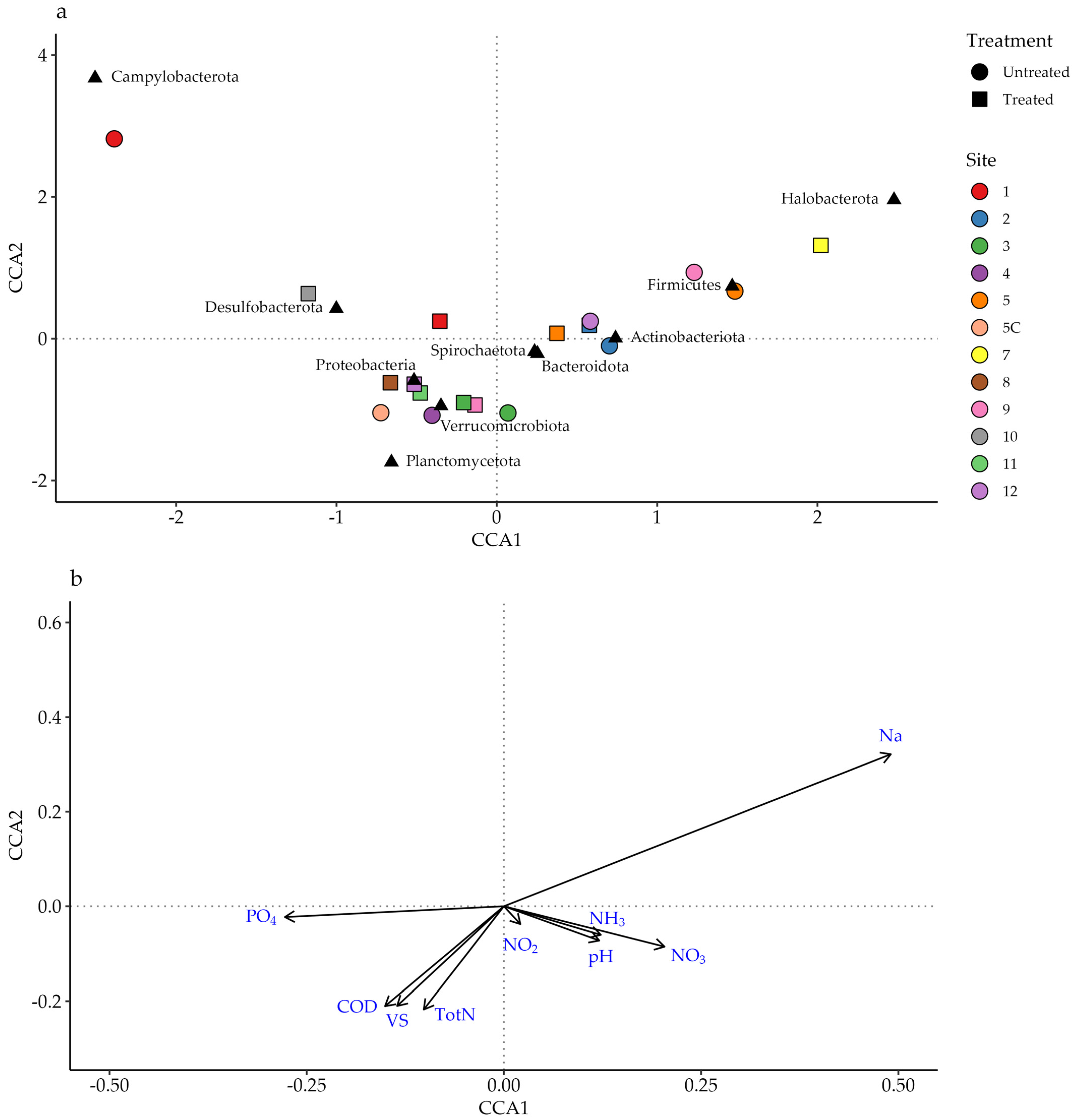{kind=link}
{kind=link}
Table 1.
Measured characteristics of wastewater samples, including industries from which they were taken, whether before or after on-site treatment, chemical data, BMP yield, and microbial diversity.
Table 1.
Measured characteristics of wastewater samples, including industries from which they were taken, whether before or after on-site treatment, chemical data, BMP yield, and microbial diversity.
| Sample Name | Site | Treatment Level | Industry | COD (mg/L) | VS (mg/L) | PO4 (mg/L) | pH | NO2 (mg/L) | NO3 (mg/L) | NH3 (mg/L) | TotN (mg/L) | Na (mg/L) | Gas Yield | Observed Taxa | Shannon Diversity | Pielou’s Evenness |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1U | 1 | Untreated | Edible Fats | 44,900 | 7430 | 272 | 6.02 | 0.7 | 5 | 0 | 11 | 2.1 | Low | 37 | 2.182 | 0.604 |
| 1T | 1 | Treated | Edible Fats | 1086 | 614 | 170 | 6.33 | 0 | 10 | 0 | 8 | 2.1 | Low | 14 | 2.393 | 0.907 |
| 2U | 2 | Untreated | Brewery | 13,800 | 3778 | 3.47 | 3.89 | 0.17 | 2.8 | 0 | 93 | 0.9 | Low | 37 | 2.896 | 0.802 |
| 2T | 2 | Treated | Brewery | 5530 | 1864 | 63 | 4.02 | 0.4 | 7 | 0 | 49 | 0.9 | Medium | 46 | 3.243 | 0.847 |
| 3U | 3 | Untreated | Logistics | 9710 | 5556 | 72 | 12.29 | 0.8 | 9 | 0 | 83 | 1.5 | High | 45 | 3.247 | 0.853 |
| 3T | 3 | Treated | Logistics | 2880 | 874 | 53 | 6.72 | 0.6 | 7 | 20 | 65 | 1.7 | Low | 58 | 3.164 | 0.779 |
| 4U | 4 | Untreated | Chemicals | 20 | 84 | 10 | 6.89 | 0.02 | 8 | 0 | 4 | 0.2 | Low | 60 | 3.57 | 0.872 |
| 5U | 5 | Untreated | Foods | 22,200 | 6982 | 263 | 5.39 | 0 | 12 | 20 | 239 | 1.8 | High | 44 | 3.107 | 0.821 |
| 5CU * | 5 | Untreated * | Foods | 1,466,300 | 279,271 | 1090 | 4.86 | 0.62 | 8 | 210 | 7500 | 3.7 | High | 19 | 1.984 | 0.674 |
| 5T | 5 | Treated | Foods | 9210 | 3332 | 170 | 6.32 | 0.5 | 9 | 0 | 103 | 1.5 | High | 91 | 3.24 | 0.718 |
| 7T | 7 | Treated | Animal Products | >100,000 | 19,460 | 3.06 | 6.77 | 2.3 | 8 | 100 | 870 | 310 | Low | 50 | 2.571 | 0.657 |
| 8T | 8 | Treated | Chemicals | 10,000 | 1326 | 10 | 6.50 | 0.15 | 11 | 0 | 0 | 0.4 | Low | 24 | 2.521 | 0.793 |
| 9U | 9 | Untreated | Logistics | 17,700 | 9992 | 3.34 | 12.16 | 0 | 6.2 | 0 | 19 | 2.4 | High | 64 | 3.577 | 0.86 |
| 9T | 9 | Treated | Logistics | 3650 | 1173 | 2.46 | 5.63 | 0.04 | 1.4 | 0 | 50 | 1.2 | Medium | 52 | 3.041 | 0.77 |
| 10T | 10 | Treated | Chemicals | 2560 | 615 | 235 | 6.21 | 0.09 | 1 | 0 | 8 | 0.6 | Low | 47 | 2.981 | 0.774 |
| 11T | 11 | Treated | Chemicals | 140 | 443 | 1.36 | 7.91 | 4.9 | 10.3 | 0 | 9 | 0.6 | Low | 153 | 4.465 | 0.888 |
| 12U | 12 | Untreated | Animal Products | 2920 | 1109 | 95 | 6.72 | 0.03 | 2 | 270 | 254 | 1.4 | High | 128 | 4.175 | 0.86 |
| 12T | 12 | Treated | Animal Products | 2000 | 211 | 16 | 6.96 | 0.04 | 0.7 | 120 | 96 | 1 | Low | 177 | 4.788 | 0.925 |
* Sample 5CU is untreated source-separated wastewater from a single cooking process at Site 5, all the other untreated samples are the final combined effluents sampled immediately before treatment. COD: chemical oxygen demand; VS: volatile solids; PO4: phosphate; NO2: nitrite; NO3: nitrate; NH3: ammonia; TotN: total nitrogen; Na: sodium. Adapted with permission from [16]. 2023, Elsevier.
Table 2.
Treatments undertaken at each site. Treatment processes are not in order. Treatments are general processes and are not performed identically at each site.
Table 2.
Treatments undertaken at each site. Treatment processes are not in order. Treatments are general processes and are not performed identically at each site.
| Site | Industry | Straining | Settling | Mixing | Oil Interception | Neutralisation | Cooling | Screening | Ferric Sulphate | Polymer | DAF * | Sludge Removal | Sulphide Control | Aeration | Separation | pH Adjustment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Edible Fats | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ |
| 2 | Foods | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 3 | Logistics | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 4 | Chemicals | ✗ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 5 | Foods | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 7 | Animal Products | ✓ | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 8 | Chemicals | ✗ | ✗ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ |
| 9 | Logistics | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| 10 | Chemicals | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ |
| 11 | Chemicals | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ |
| 12 | Animal Products | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ |
* Dissolved-air flotation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Elliott, J.A.K.; Krohn, C.; Ball, A.S. Diversity of Microbial Communities in Trade Wastes—Implications for Treatments and Operations. Appl. Microbiol. 2024, 4, 682-703. https://doi.org/10.3390/applmicrobiol4020047
AMA Style
Elliott JAK, Krohn C, Ball AS. Diversity of Microbial Communities in Trade Wastes—Implications for Treatments and Operations. Applied Microbiology. 2024; 4(2):682-703. https://doi.org/10.3390/applmicrobiol4020047
Chicago/Turabian StyleElliott, Jake A. K., Christian Krohn, and Andrew S. Ball. 2024. "Diversity of Microbial Communities in Trade Wastes—Implications for Treatments and Operations" Applied Microbiology 4, no. 2: 682-703. https://doi.org/10.3390/applmicrobiol4020047