Upscaling Statistical Patterns from Reduced Storage in Social and Life Science Big Datasets




Abstract
:1. Introduction
2. Materials and Methods
Theoretical Framework
3. Results
3.1. Tests on Rainforest and Human-Activity Data
3.2. Application to Human Single Nucleotide Polymorphism Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TFFD | Type Frequency of Frequencies Distribution |
| TDC | Type-Discovery Curve |
| SNP | Single Nucleotide Polymorphism |
References
- Agarwal, R.; Vasant, D. Big data, data science, and analytics: The opportunity and challenge for is research. Inf. Syst. Res. 2014, 25, 443–448. [Google Scholar] [CrossRef]
- Najafabadi, M.M.; Villanustre, F.; Khoshgoftaar, T.M.; Seliya, N.; Wald, R.; Muharemagic, E. Deep learning applications and challenges in big data analytics. J. Big Data 2015, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Gantz, J.; Reinsel, D. The digital universe in 2020: Big data, bigger digital shadows, and biggest growth in the far east. IDC iView IDC Anal. Future 2012, 2007, 1–16. [Google Scholar]
- Chen, C.L.P.; Zhang, C.Y. Data-intensive applications, challenges, techniques and technologies: A survey on big data. Inf. Sci. 2014, 275, 314–347. [Google Scholar] [CrossRef]
- Dai, L.; Gao, X.; Guo, Y.; Xiao, J.; Zhang, Z. Bioinformatics clouds for big data manipulation. Biol. Direct 2012, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, Y.; Du, X.; Li, C.; Lu, J.; Zhao, S.; Zhou, X. Big data challenge: A data management perspective. Front. Comput. Sci. 2013, 7, 157–164. [Google Scholar] [CrossRef]
- Tsai, C.W.; Lai, C.F.; Chao, H.C.; Vasilakos, A.V. Big data analytics: A survey. J. Big Data 2015, 2, 21. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [Green Version]
- McPherson, J.D. Next-generation gap. Nat. Methods 2009, 6, S2–S5. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.D. On the future of genomic data. Science 2011, 331, 728–729. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.M.; Schmieder, R.; Highnam, G.; Mittelman, D. Big data challenges and opportunities in high-throughput sequencing. Syst. Biomed. 2013, 1, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Tovo, A.; Menzel, P.; Krogh, A.; Lagomarsino, M.C.; Suweis, S. Taxonomic classification method for metagenomics based on core protein families with core-kaiju. Nucleic Acids Res. 2020, 48, e93. [Google Scholar] [CrossRef] [PubMed]
- Colleoni, E.; Rozza, A.; Arvidsson, A. Echo chamber or public sphere? Predicting political orientation and measuring political homophily in twitter using big data. J. Commun. 2014, 64, 317–332. [Google Scholar] [CrossRef]
- Sang, E.T.K.; van den Bosch, A. Dealing with big data: The case of twitter. Dutch J. Appl. Linguist. 2013, 3, 121–134. [Google Scholar]
- Laurila, J.K.; Imad Aad, D.G.P.; Bornet, O.; Do, T.M.T.; Dousse, O.; Eberle, J.; Miettinen, M. The mobile data challenge: Big data for mobile computing research. In Proceedings of the Mobile Data Challenge by Nokia Workshop, in Conjunction with the 10th International Conference on Pervasive Computing, Newcastle, UK, 18–19 June 2012. [Google Scholar]
- Sagiroglu, S.; Sinanc, D. Big data: A review. In Proceedings of the 2013 International Conference on Collaboration Technologies and Systems (CTS), San Diego, CA, USA, 20–24 May 2013; pp. 42–47. [Google Scholar]
- Parsons, M.A.; Godøy, Ø.; LeDrew, E.; De Bruin, E.; Danis, B.; Tomlinson, S.; Carlson, D. A conceptual framework formanaging very diverse data for complex, interdisciplinary science. J. Inf. Sci. 2011, 37, 555–569. [Google Scholar] [CrossRef] [Green Version]
- ur Rehman, M.H.; Liew, C.S.; Abbas, A.; Jayaraman, P.P.; Wah, T.Y.; Khan, S.U. Big data reduction methods: A survey. DSE 2016, 1, 265–284. [Google Scholar] [CrossRef] [Green Version]
- Good, I.J.; Toulmin, G.H. The number of new species, and the increase in population coverage, when a sample is increased. Biometrika 1956, 43, 45–63. [Google Scholar] [CrossRef]
- Harte, J.; Smith, A.B.; Storch, D. Biodiversity scales from plots to biomes with a universal species–area curve. Ecol. Lett. 2009, 12, 789–797. [Google Scholar] [CrossRef]
- Chao, A.; Chiu, C.-H. Species richness: Estimation and comparison. In Wiley StatsRef: Statistics Reference Online; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 1–26. [Google Scholar]
- Slik, J.W.F.; Arroyo-Rodríguez, V.; Aiba, S.I.; Alvarez-Loayza, P.; Alves, F.A.; Ashton, P.; Balvanera, P.; Bastian, M.L.; Bellingham, P.J.; van den Berg, E.; et al. An estimate of the number of tropical tree species. Proc. Natl. Acad. Sci. USA 2015, 112, 7472–7477. [Google Scholar] [CrossRef] [Green Version]
- Orlitsky, A.; Suresh, A.T.; Wu, Y. Optimal prediction of the number of unseen species. Proc. Natl. Acad. Sci. USA 2016, 113, 13283–13288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovo, A.; Suweis, S.; Formentin, M.; Favretti, M.; Volkov, I.; Banavar, J.R.; Azaele, S.; Maritan, A. Upscaling species richness and abundances in tropical forests. Sci. Adv. 2017, 3, e1701438. [Google Scholar] [CrossRef] [Green Version]
- Tovo, A.; Formentin, M.; Suweis, S.; Stivanello, S.; Azaele, S.; Maritan, A. Inferring macro-ecological patterns from local presence/absence data. Oikos 2019, 128, 1641–1652. [Google Scholar] [CrossRef]
- Formentin, M.; Lovison, A.; Maritan, A.; Zanzotto, G. Hidden scaling patterns and universality in written communication. Phys. Rev. E 2014, 90, 012817. [Google Scholar] [CrossRef] [Green Version]
- Monechi, B.; Ruiz-Serrano, A.; Tria, F.; Loreto, V. Waves of novelties in the expansion into the adjacent possible. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Birney, E.; Soranzo, N. Human genomics: The end of the start for population sequencing. Nature 2015, 526, 52–53. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.Y.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayood, K. Introduction to Data Compression, 5th ed.; Morgan Kaufmann: Cambridge, MA, USA, 2017. [Google Scholar]
- Chao, A.; Wang, Y.T.; Jost, L. Entropy and the species accumulation curve: A novel entropy estimator via discovery rates of new species. Methods Ecol. Evol. 2013, 4, 1091–1100. [Google Scholar] [CrossRef]
- Nabout, J.C.; da Silva Rocha, B.; Carneiro, F.M.; Sant’Anna, C.L. How many species of cyanobacteria are there? Using a discovery curve to predict the species number. Biodivers. Conserv. 2013, 22, 2907–2918. [Google Scholar] [CrossRef]
- Tovo, A.; Stivanello, S.; Maritan, A.; Suweis, S.; Favaro, S.; Formentin, M. Upscaling human activity data: An ecological perspective. arXiv 2019, arXiv:1912.03023. [Google Scholar]
- Volkov, I.; Banavar, J.R.; Hubbell, S.P.; Maritan, A. Patterns of relative species abundance in rainforests and coral reefs. Nature 2007, 450, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Azaele, S.; Suweis, S.; Grilli, J.; Volkov, I.; Banavar, J.R.; Maritan, A. Statistical mechanics of ecological systems: Neutral theory and beyond. Rev. Mod. Phys. 2016, 88, 035003. [Google Scholar] [CrossRef]
- Georgii, H.O. Stochastics: Introduction to Probability and Statistics; Walter de Gruyter: Berlin, Germany, 2012; ISBN 978-311-029-360-9. [Google Scholar]
- Rimoin, D.L.; Connor, J.M.; Pyeritz, R.E.; Korf, B.K. Emery and Rimoin’s Principles and Practice of Medical Genetics; Churcill Livingstone Elsevier: London, UK, 2007; ISBN 978-012-812-537-3. [Google Scholar]
- Zhang, K.; Qin, Z.S.; Liu, J.S.; Chen, T.; Waterman, M.S.; Sun, F. Haplotype block partitioning and tag SNP selection using genotype data and their applications to association studies. Genome Res. 2004, 14, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Ionita-Laza, I.; Lange, C.; Laird, N.M. Estimating the number of unseen variants in the human genome. Proc. Natl. Acad. Sci. USA 2009, 106, 5008–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erichsen, H.; Chanock, S. SNPs in cancer research and treatment. Br. J. Cancer 2004, 90, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Martinez, P.; Kimberley, C.; BirkBak, N.J.; Marquard, A.; Szallasi, Z.; Graham, T.A. Quantification of within-sample genetic heterogeneity from SNP-array data. Sci. Rep. 2017, 7, 3248. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, J.Z. A general framework for analyzing tumor subclonality using SNP array and DNA sequencing data. Genome Biol. 2014, 15, 473. [Google Scholar] [CrossRef]



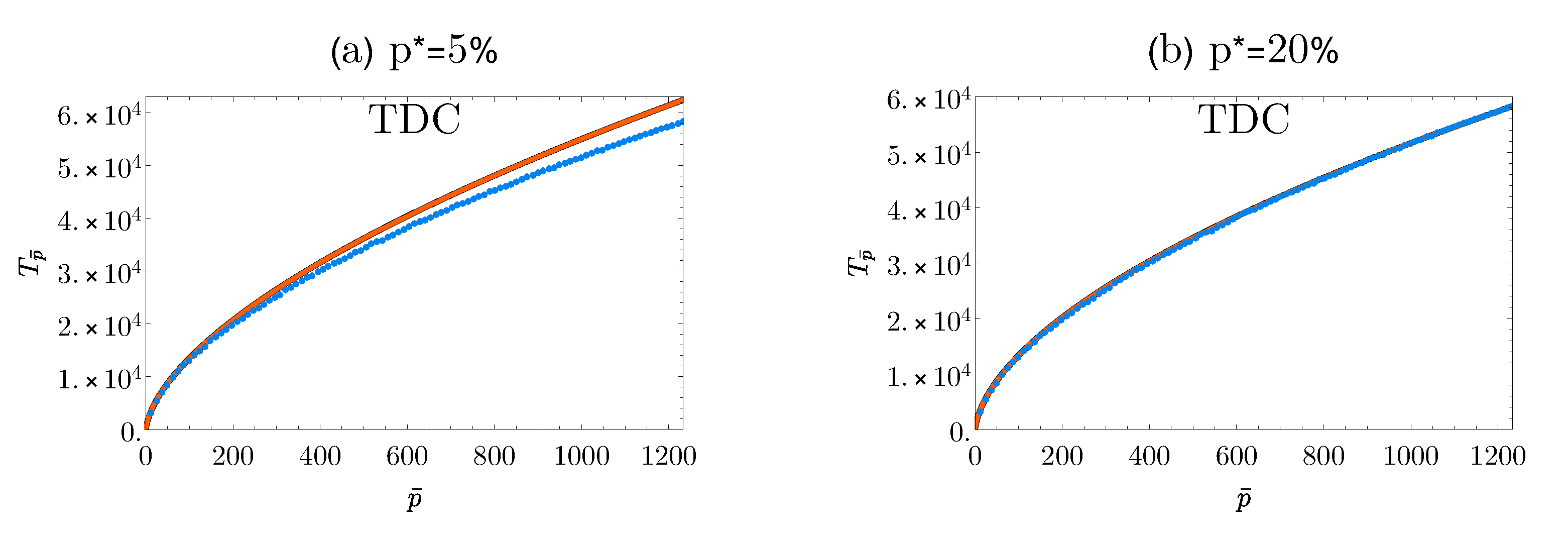

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | T | N | Relative Error | |||
|---|---|---|---|---|---|---|
| BCI | 301 | 222,602 | 0.073 ± 0.002 | |||
| Pasoh | 927 | 310,520 | ||||
| Emails | 752,299 | 6,914,872 | 741,036 ± 3646 | |||
| 6,972,453 | 34,696,973 | 729,288 ± 8357 | ||||
| Wikipedia | 673,872 | 29,606,116 | 720,432 ± 3295 | |||
| Gutenberg | 554,193 | 126,289,661 | 532,555 ± 2110 |
| Dataset | Estimator | Relative Error | ||
|---|---|---|---|---|
| BCI | Equation (6) | 0.073 | 0.9998 | −7.1% |
| Ref. [26] | 0.20 | 0.9991 | 1.8% | |
| Pasoh | Equation (6) | 0.20 | 0.9992 | 2.9% |
| Ref. [26] | 0.24 | 0.9998 | 1.8% | |
| Emails | Equation (6) | −0.795 | ∼1 | 1.5% |
| Ref. [35] | −0.798 | ∼1 | 0.11% | |
| Equation (6) | −0.824 | ∼1 | −4.6% | |
| Ref. [35] | −0.828 | ∼1 | 3.33% | |
| Wikipedia | Equation (6) | −0.543 | ∼ 1 | −6.9% |
| Ref. [35] | −0.544 | ∼1 | 6.11% | |
| Gutenberg | Equation (6) | −0.426 | ∼1 | 3.9% |
| Ref. [35] | −0.420 | ∼1 | −2.3% |
| Fraction | T | N | Relative Error | |||
|---|---|---|---|---|---|---|
| 58,671 | 839,459 | |||||
| 58,671 | 839,459 |
| Relative Error | ||
|---|---|---|
| 62,414 ± 816 | ||
| 60,471 ± 539 | ||
| 58,297 ± 370 | ||
| 58,070 ± 197 | ||
| 58,371 ± 143 | ||
| 58,504 ± 111 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garlaschi, S.; Fochesato, A.; Tovo, A. Upscaling Statistical Patterns from Reduced Storage in Social and Life Science Big Datasets. Entropy 2020, 22, 1084. https://doi.org/10.3390/e22101084
Garlaschi S, Fochesato A, Tovo A. Upscaling Statistical Patterns from Reduced Storage in Social and Life Science Big Datasets. Entropy. 2020; 22(10):1084. https://doi.org/10.3390/e22101084
Chicago/Turabian StyleGarlaschi, Stefano, Anna Fochesato, and Anna Tovo. 2020. "Upscaling Statistical Patterns from Reduced Storage in Social and Life Science Big Datasets" Entropy 22, no. 10: 1084. https://doi.org/10.3390/e22101084
APA StyleGarlaschi, S., Fochesato, A., & Tovo, A. (2020). Upscaling Statistical Patterns from Reduced Storage in Social and Life Science Big Datasets. Entropy, 22(10), 1084. https://doi.org/10.3390/e22101084