Allostery and Epistasis: Emergent Properties of Anisotropic Networks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
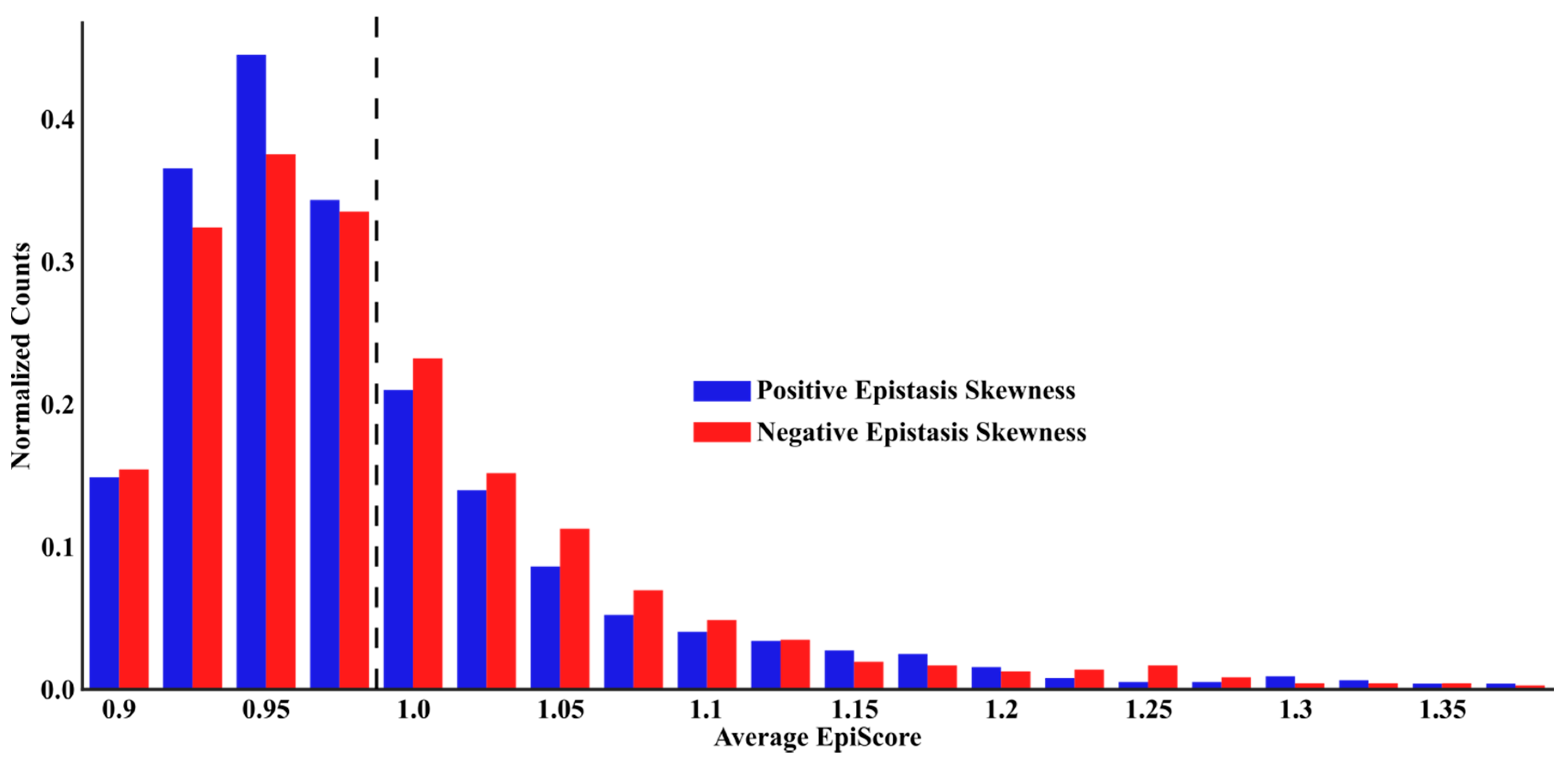
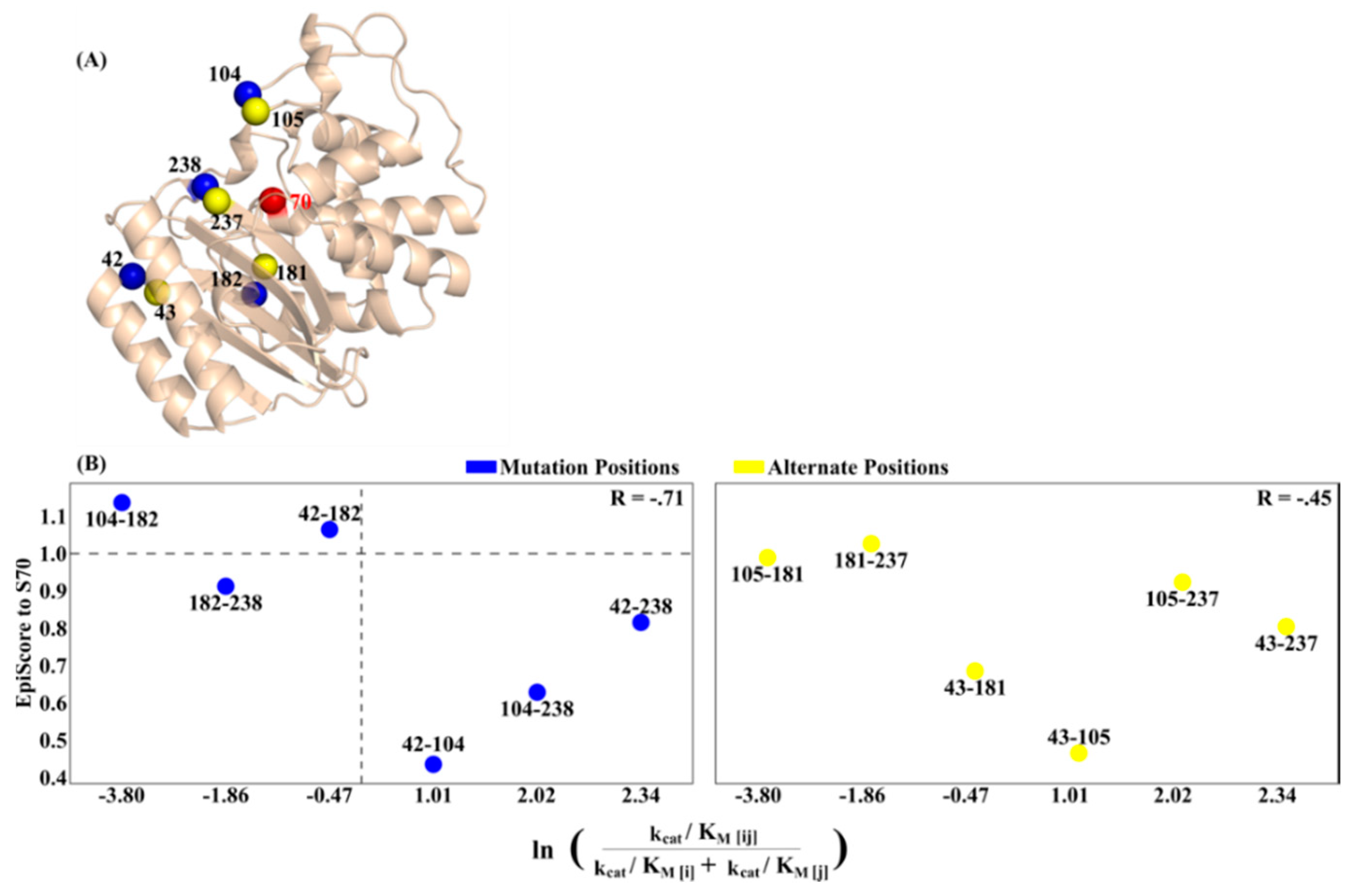
3.1. Epistasis and EpiScore
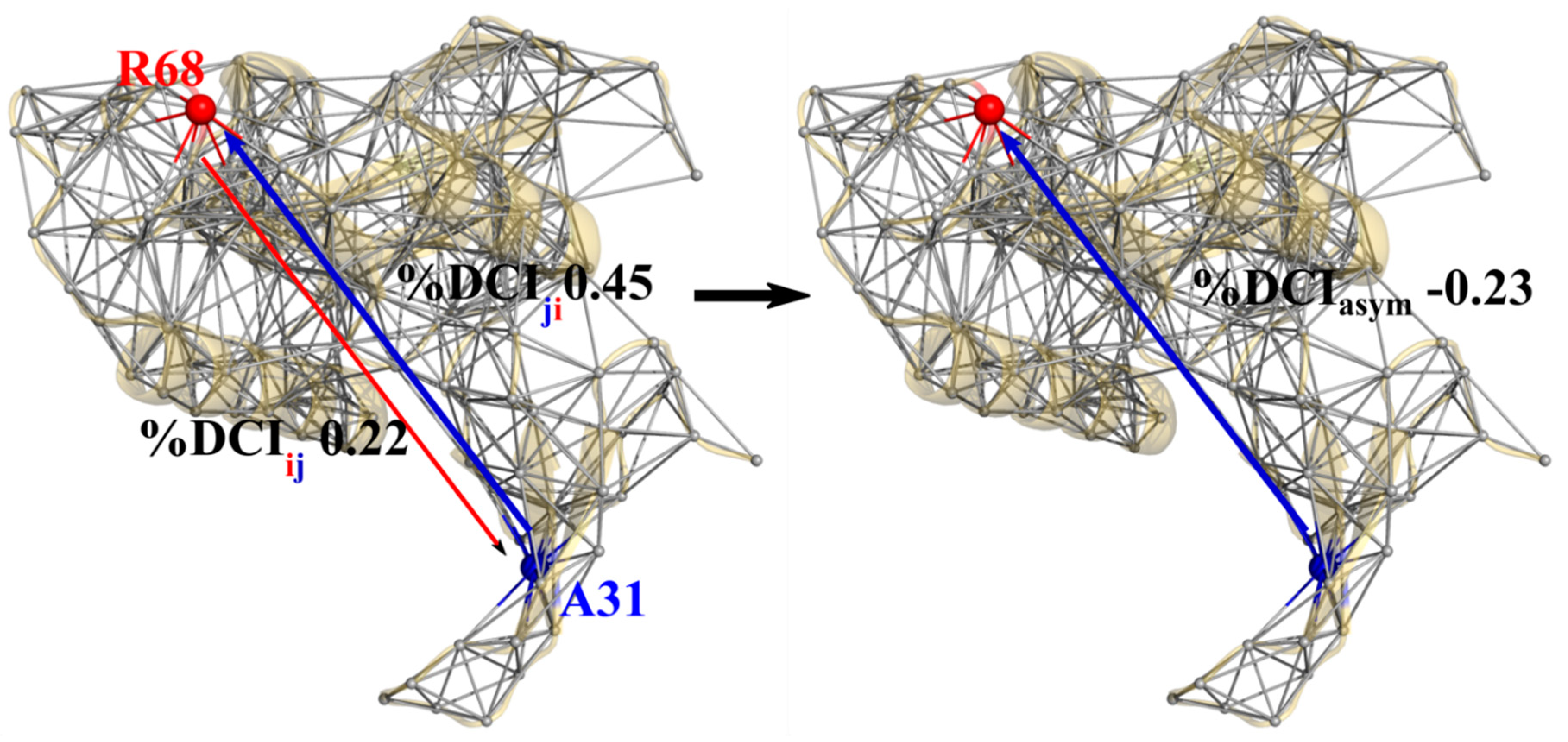
3.2. Asymmetry and Epistasis
3.3. Unidirectional Communication through DCIasym Creates Cause-Effect Relationships in Allosteric Regulations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Subramanian, S.; Kumar, S. Evolutionary anatomies of positions and types of disease-associated and neutral amino acid mutations in the human genome. BMC Genom. 2006, 7, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Dudley, J.T.; Filipski, A.; Liu, L. Phylomedicine: An evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet. 2011, 27, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Swint-Kruse, L. Using evolution to guide protein engineering: The devil is in the details. Biophys. J. 2016, 111, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Starr, T.N.; Thornton, J.W. Epistasis in protein evolution. Protein Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef] [Green Version]
- Domingo, J.; Diss, G.; Lehner, B. Pairwise and higher-order genetic interactions during the evolution of a tRNA. Nature 2018, 558, 117–121. [Google Scholar] [CrossRef]
- Salinas, V.H.; Ranganathan, R. Coevolution-based inference of amino acid interactions underlying protein function. Elife 2018, 7, e34300. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, X.J.; Chen, X.; Bowman, M.E.; Luo, Y.; Noel, J.P.; Ellington, A.D.; Etzkorn, F.A.; Zhang, Y. Structural and kinetic analysis of prolyl-isomerization/phosphorylation cross-talk in the CTD code. ACS Chem. Biol. 2012, 7, 1462–1470. [Google Scholar] [CrossRef] [Green Version]
- Figliuzzi, M.; Jacquier, H.; Schug, A.; Tenaillon, O.; Weigt, M. Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase TEM-1. Mol. Biol. Evol. 2016, 33, 268–280. [Google Scholar] [CrossRef]
- Wang, Z.O.; Pollock, D.D. Context dependence and coevolution among amino acid residues in proteins. Methods Enzymol. 2005, 395, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Payne, J.L.; Wagner, A. The causes of evolvability and their evolution. Nat. Rev. Genet. 2019, 20, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Larrimore, K.E.; Kazan, I.C.; Kannan, L.; Kendle, R.P.; Jamal, T.; Barcus, M.; Bolia, A.; Brimijoin, S.; Zhan, C.-G.; Ozkan, S.B.; et al. Plant-expressed cocaine hydrolase variants of butyrylcholinesterase exhibit altered allosteric effects of cholinesterase activity and increased inhibitor sensitivity. Sci. Rep. 2017, 7, 10419. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Glembo, T.J.; Ozkan, S.B. The role of conformational dynamics and allostery in the disease development of human ferritin. Biophys. J. 2015, 109, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Modi, T.; Huihui, J.; Ghosh, K.; Ozkan, S.B. Ancient thioredoxins evolved to modern-day stability–function requirement by altering native state ensemble. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuriki, N.; Tawfik, D.S. Protein dynamism and evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Bhabha, G.; Ekiert, D.C.; Jennewein, M.; Zmasek, C.M.; Tuttle, L.M.; Kroon, G.; Dyson, H.J.; Godzik, A.; Wilson, I.A.; Wright, P.E. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Meinhardt, S.; Manley, M.W.; Parente, D.J.; Swint-Kruse, L. Rheostats and toggle switches for modulating protein function. PLoS ONE 2013, 8, e83502. [Google Scholar] [CrossRef] [Green Version]
- Saavedra, H.G.; Wrabl, J.O.; Anderson, J.A.; Li, J.; Hilser, V.J. Dynamic allostery can drive cold adaptation in enzymes. Nature 2018, 558, 324–328. [Google Scholar] [CrossRef] [PubMed]
- McLeish, T.C.B.; Rodgers, T.L.; Wilson, M.R. Allostery without conformation change: Modelling protein dynamics at multiple scales. Phys. Biol. 2013, 10, 56004. [Google Scholar] [CrossRef] [Green Version]
- Gobeil, S.M.C.; Ebert, M.C.C.J.C.; Park, J.; Gagné, D.; Doucet, N.; Berghuis, A.M.; Pleiss, J.; Pelletier, J.N. The structural dynamics of engineered β-lactamases vary broadly on three timescales yet sustain native function. Sci. Rep. 2019, 9, 6656. [Google Scholar] [CrossRef]
- Rollins, N.J.; Brock, K.P.; Poelwijk, F.J.; Stiffler, M.A.; Gauthier, N.P.; Sander, C.; Marks, D.S. Inferring protein 3D structure from deep mutation scans. Nat. Genet. 2019, 51, 1170–1176. [Google Scholar] [CrossRef]
- Faber, M.S.; Wrenbeck, E.E.; Azouz, L.R.; Steiner, P.J.; Whitehead, T.A. Impact of In Vivo Protein Folding Probability on Local Fitness Landscapes. Mol. Biol. Evol. 2019, 36, 2764–2777. [Google Scholar] [CrossRef] [PubMed]
- Lozovsky, E.R.; Chookajorn, T.; Brown, K.M.; Imwong, M.; Shaw, P.J.; Kamchonwongpaisan, S.; Neafsey, D.E.; Weinreich, D.M.; Hartl, D.L. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc. Natl. Acad. Sci. USA 2009, 106, 12025–12030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knies, J.L.; Cai, F.; Weinreich, D.M. Enzyme efficiency but not thermostability drives cefotaxime resistance evolution in TEM-1 β-lactamase. Mol. Biol. Evol. 2017, 34, 1040–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.A.; Wu, N.C.; Sun, R. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr. Biol. 2014, 24, 2643–2651. [Google Scholar] [CrossRef] [Green Version]
- Varona, L.; Legarra, A.; Toro, M.A.; Vitezica, Z.G. Non-additive effects in genomic selection. Front. Genet. 2018, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Tsai, C.-J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.-J.; Liu, J. Principles of allosteric interactions in cell signaling. J. Am. Chem. Soc. 2014, 136, 17692–17701. [Google Scholar] [CrossRef] [Green Version]
- Popovych, N.; Sun, S.; Ebright, R.H.; Kalodimos, C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006, 13, 831–838. [Google Scholar] [CrossRef]
- Tsai, C.-J.; del Sol, A.; Nussinov, R. Allostery: Absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2008, 378, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Doruker, P.; Kaynak, B.; Zhang, S.; Krieger, J.; Li, H.; Bahar, I. Intrinsic dynamics is evolutionarily optimized to enable allosteric behavior. Curr. Opin. Struct. Biol. 2019, 62, 14–21. [Google Scholar] [CrossRef]
- Loutchko, D.; Flechsig, H. Allosteric communication in molecular machines via information exchange: What can be learned from dynamical modeling. Biophys. Rev. 2020, 12, 443–452. [Google Scholar] [CrossRef]
- Dutta, S.; Eckmann, J.-P.; Libchaber, A.; Tlusty, T. Green function of correlated genes in a minimal mechanical model of protein evolution. Proc. Natl. Acad. Sci. USA 2018, 115, E4559–E4568. [Google Scholar] [CrossRef] [Green Version]
- Flechsig, H.; Togashi, Y. Designed Elastic Networks: Models of Complex Protein Machinery. Int. J. Mol. Sci. 2018, 19, 3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandaru, P.; Shah, N.H.; Bhattacharyya, M.; Barton, J.P.; Kondo, Y.; Cofsky, J.C.; Gee, C.L.; Chakraborty, A.K.; Kortemme, T.; Ranganathan, R.; et al. Deconstruction of the Ras switching cycle through saturation mutagenesis. Elife 2017, 6, e27810. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef] [Green Version]
- Melamed, D.; Young, D.L.; Gamble, C.E.; Miller, C.R.; Fields, S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA 2013, 19, 1537–1551. [Google Scholar] [CrossRef] [Green Version]
- Gerek, Z.N.; Ozkan, S.B. Change in allosteric network affects binding affinities of PDZ domains: Analysis through perturbation response scanning. PLoS Comput. Biol. 2011, 7, e1002154. [Google Scholar] [CrossRef] [PubMed]
- Nevin Gerek, Z.; Kumar, S.; Banu Ozkan, S. Structural dynamics flexibility informs function and evolution at a proteome scale. Evol. Appl. 2013, 6, 423–433. [Google Scholar] [CrossRef]
- Schrodinger L.L.C. The PyMOL Molecular Graphics System. Available online: https://pymol.org/2/ (accessed on 10 June 2020).
- Jelsch, C.; Mourey, L.; Masson, J.M.; Samama, J.P. Crystal structure of Escherichia coli TEM1 beta-lactamase at 1.8 A resolution. Proteins 1993, 16, 364–383. [Google Scholar] [CrossRef] [PubMed]
- Frericks Schmidt, H.L.; Sperling, L.J.; Gao, Y.G.; Wylie, B.J.; Boettcher, J.M.; Wilson, S.R.; Rienstra, C.M. Crystal polymorphism of protein GB1 examined by solid-state NMR and X-ray diffraction. J. Phys. Chem. B 2007, 111, 14362–14369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.T.; Hanoian, P.; French, J.B.; Pringle, T.H.; Hammes-Schiffer, S.; Benkovic, S.J. Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. USA 2013, 110, 10159–10164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cody, V.; Schwalbe, C.H. Structural characteristics of antifolate dihydrofolate reductase enzyme interactions. Crystallogr. Rev. 2006, 12, 301–333. [Google Scholar] [CrossRef]
- Mhashal, A.R.; Vardi-Kilshtain, A.; Kohen, A.; Major, D.T. The role of the Met20 loop in the hydride transfer in Escherichia coli dihydrofolate reductase. J. Biol. Chem. 2017, 292, 14229–14239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef]
- Behiry, E.M.; Evans, R.M.; Guo, J.; Loveridge, E.J.; Allemann, R.K. Loop interactions during catalysis by dihydrofolate reductase from Moritella profunda. Biochemistry 2014, 53, 4769–4774. [Google Scholar] [CrossRef] [Green Version]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosomal dihydrofolate reductase reveals natural antifolate resistance. ACS Chem. Biol. 2011, 6, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Salverda, M.L.M.; de Visser, J.A.G.M.; Barlow, M. Natural evolution of TEM-1 β-lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiol. Rev. 2010, 34, 1015–1036. [Google Scholar] [CrossRef] [Green Version]
- Ingles-Prieto, A.; Ibarra-Molero, B.; Delgado-Delgado, A.; Perez-Jimenez, R.; Fernandez, J.M.; Gaucher, E.A.; Sanchez-Ruiz, J.M.; Gavira, J.A. Conservation of protein structure over four billion years. Structure 2013, 21, 1690–1697. [Google Scholar] [CrossRef] [Green Version]
- Weinreich, D.M.; Delaney, N.F.; Depristo, M.A.; Hartl, D.L. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science 2006, 312, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Risso, V.A.; Gavira, J.A.; Sanchez-Ruiz, J.M.; Ozkan, S.B. Evolution of conformational dynamics determines the conversion of a promiscuous generalist into a specialist enzyme. Mol. Biol. Evol. 2015, 32, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Modi, T.; Ozkan, B. Allostery modulates resistance driver mutations in TEM-1. Biophys. J. 2019, 116, 342a. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Daum, S.; Wildemann, D.; Zhou, X.Z.; Verdecia, M.A.; Bowman, M.E.; Lücke, C.; Hunter, T.; Lu, K.-P.; Fischer, G.; et al. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem. Biol. 2007, 2, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdecia, M.A.; Bowman, M.E.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 2000, 7, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Balastik, M.; Lim, J.; Pastorino, L.; Lu, K.P. Pin1 in Alzheimer’s disease: Multiple substrates, one regulatory mechanism? Biochim. Biophys. Acta-Mol. Bas. Dis. 2007, 1772, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.P. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem. Sci. 2004, 29, 200–209. [Google Scholar] [CrossRef]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef]
- Guo, J.; Pang, X.; Zhou, H.-X. Two pathways mediate inter-domain allosteric regulation in Pin1. Structure 2014, 23, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.W. Investigating dynamic interdomain allostery in Pin1. Biophys. Rev. 2015, 7, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, H.; Devasahayam, G.; Gemmill, T.; Chaturvedi, V.; Hanes, S.D.; van Roey, P. The structure of the Candida albicans Ess1 prolyl isomerase reveals a well-ordered linker that restricts domain mobility. Biochemistry 2005, 44, 6180–6189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namanja, A.T.; Wang, X.J.; Xu, B.; Mercedes-Camacho, A.Y.; Wilson, K.A.; Etzkorn, F.A.; Peng, J.W. Stereospecific gating of functional motions in Pin1. Proc. Natl. Acad. Sci. USA 2011, 108, 12289–12294. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.W.; Wilson, B.D.; Namanja, A.T. Mapping the dynamics of ligand reorganization via 13CH3 and 13CH2 relaxation dispersion at natural abundance. J. Biomol. NMR 2009, 45, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Campitelli, P.; Guo, J.; Zhou, H.-X.; Ozkan, S.B. Hinge-shift mechanism modulates allosteric regulations in human Pin1. J. Phys. Chem. B 2018, 122, 5623–5629. [Google Scholar] [CrossRef]
- Campitelli, P.; Modi, T.; Kumar, S.; Ozkan, S.B. The role of conformational dynamics and allostery in modulating protein evolution. Annu. Rev. Biophys. 2020, 49, 267–288. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campitelli, P.; Ozkan, S.B. Allostery and Epistasis: Emergent Properties of Anisotropic Networks. Entropy 2020, 22, 667. https://doi.org/10.3390/e22060667
Campitelli P, Ozkan SB. Allostery and Epistasis: Emergent Properties of Anisotropic Networks. Entropy. 2020; 22(6):667. https://doi.org/10.3390/e22060667
Chicago/Turabian StyleCampitelli, Paul, and S. Banu Ozkan. 2020. "Allostery and Epistasis: Emergent Properties of Anisotropic Networks" Entropy 22, no. 6: 667. https://doi.org/10.3390/e22060667