
ELIHKSIR Web Server: Evolutionary Links Inferred for Histidine Kinase Sensors Interacting with Response Regulators



Abstract
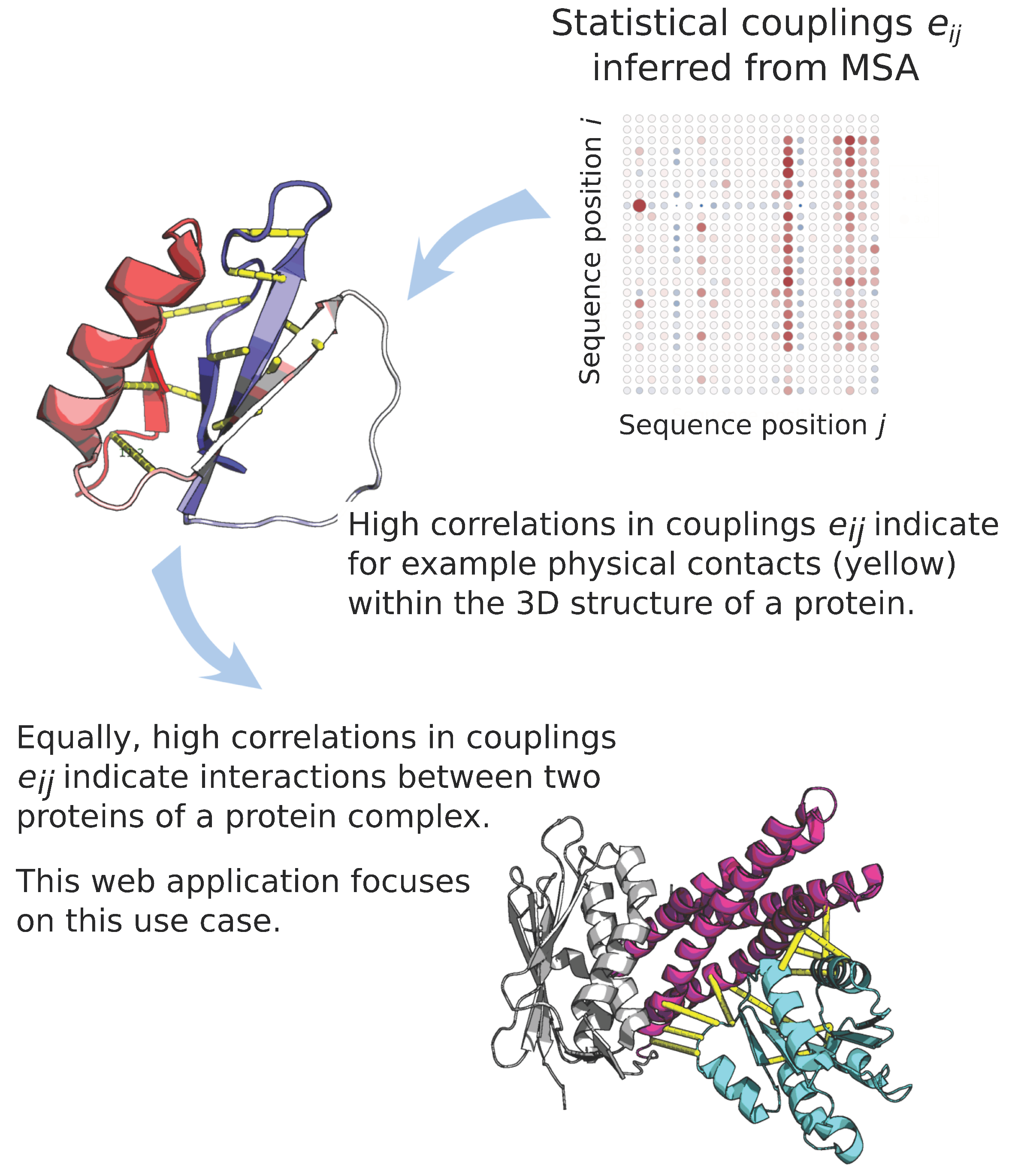
:1. Introduction
2. Results
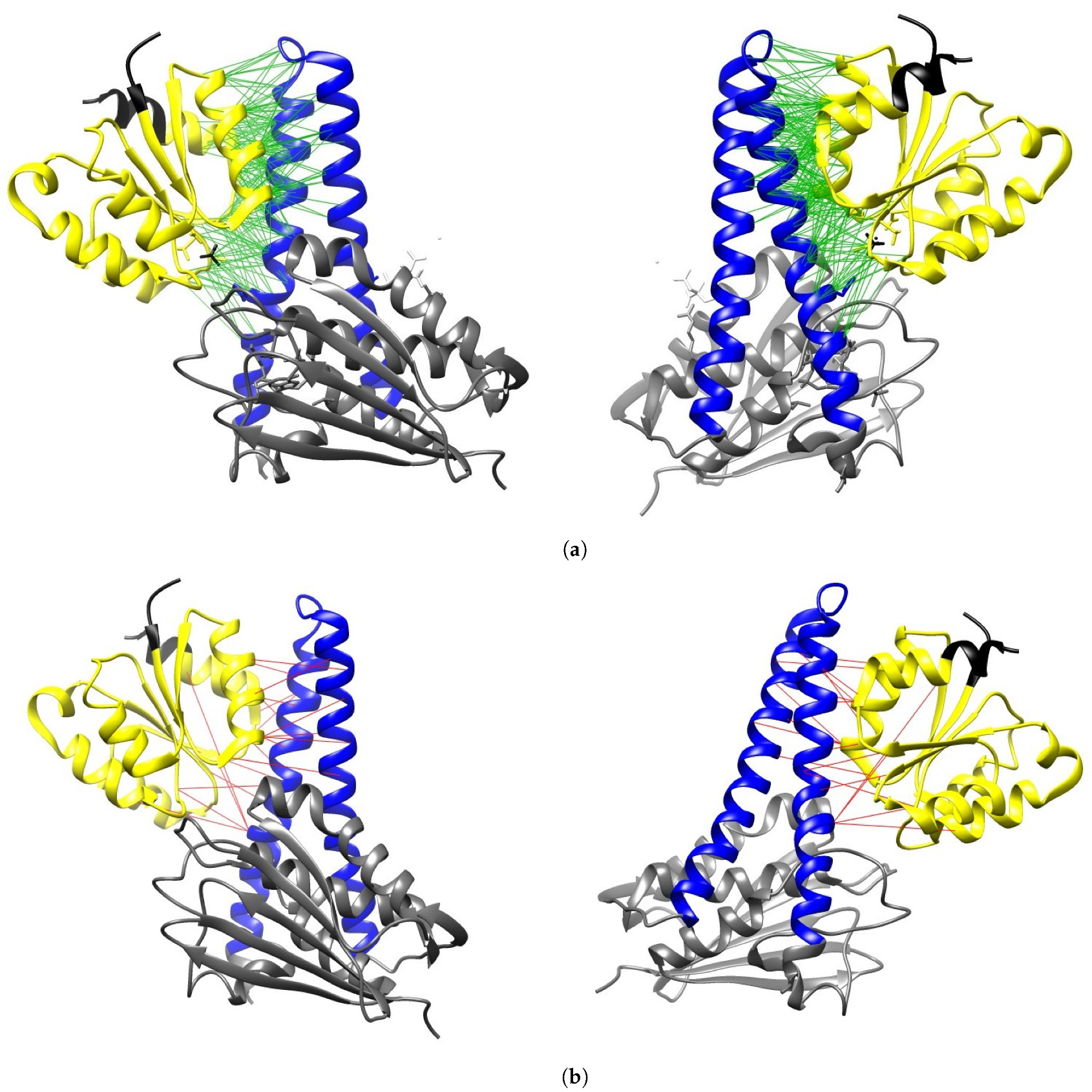
2.1. Validation
2.2. Mutations
2.3. Data Export
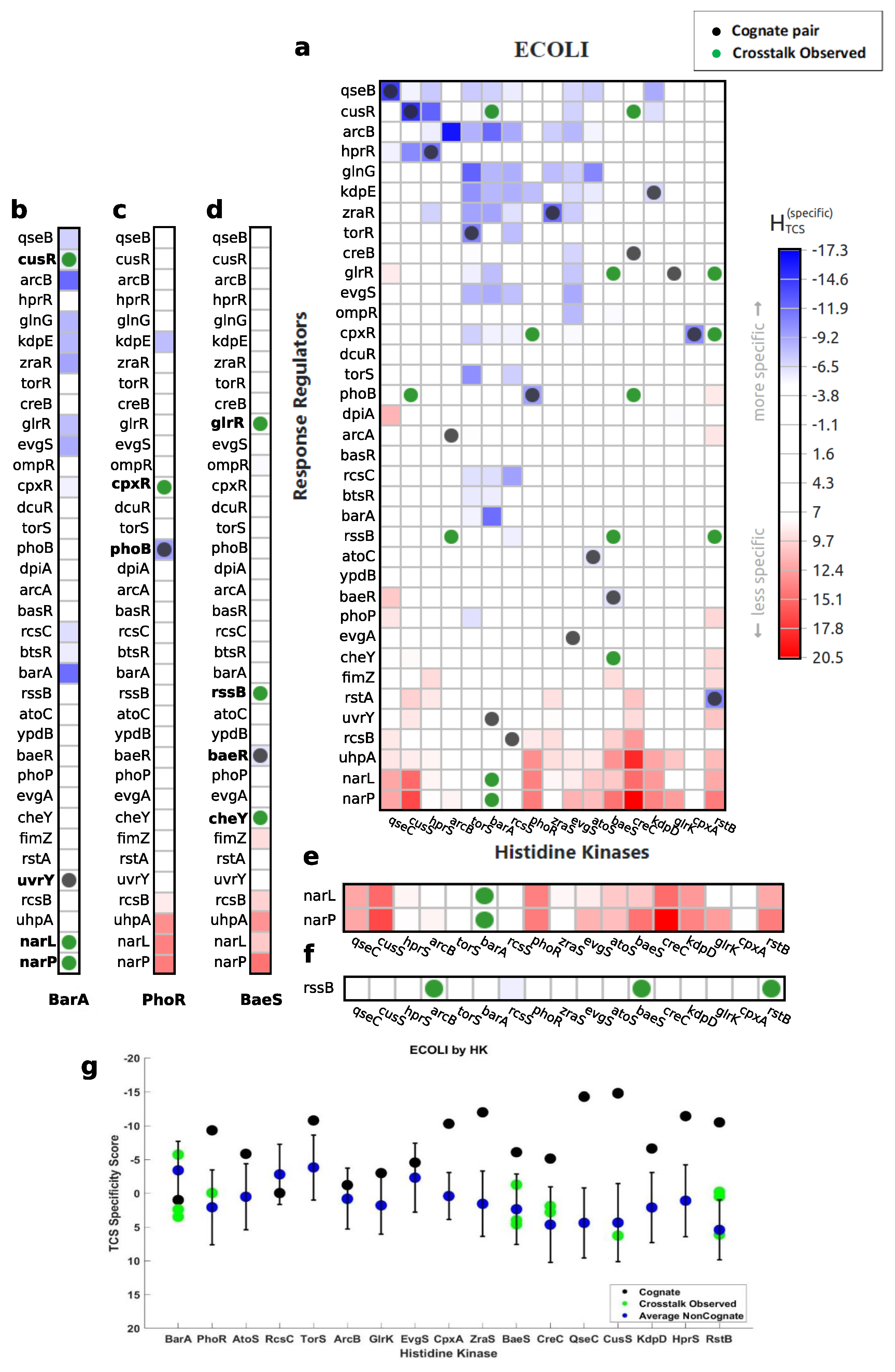
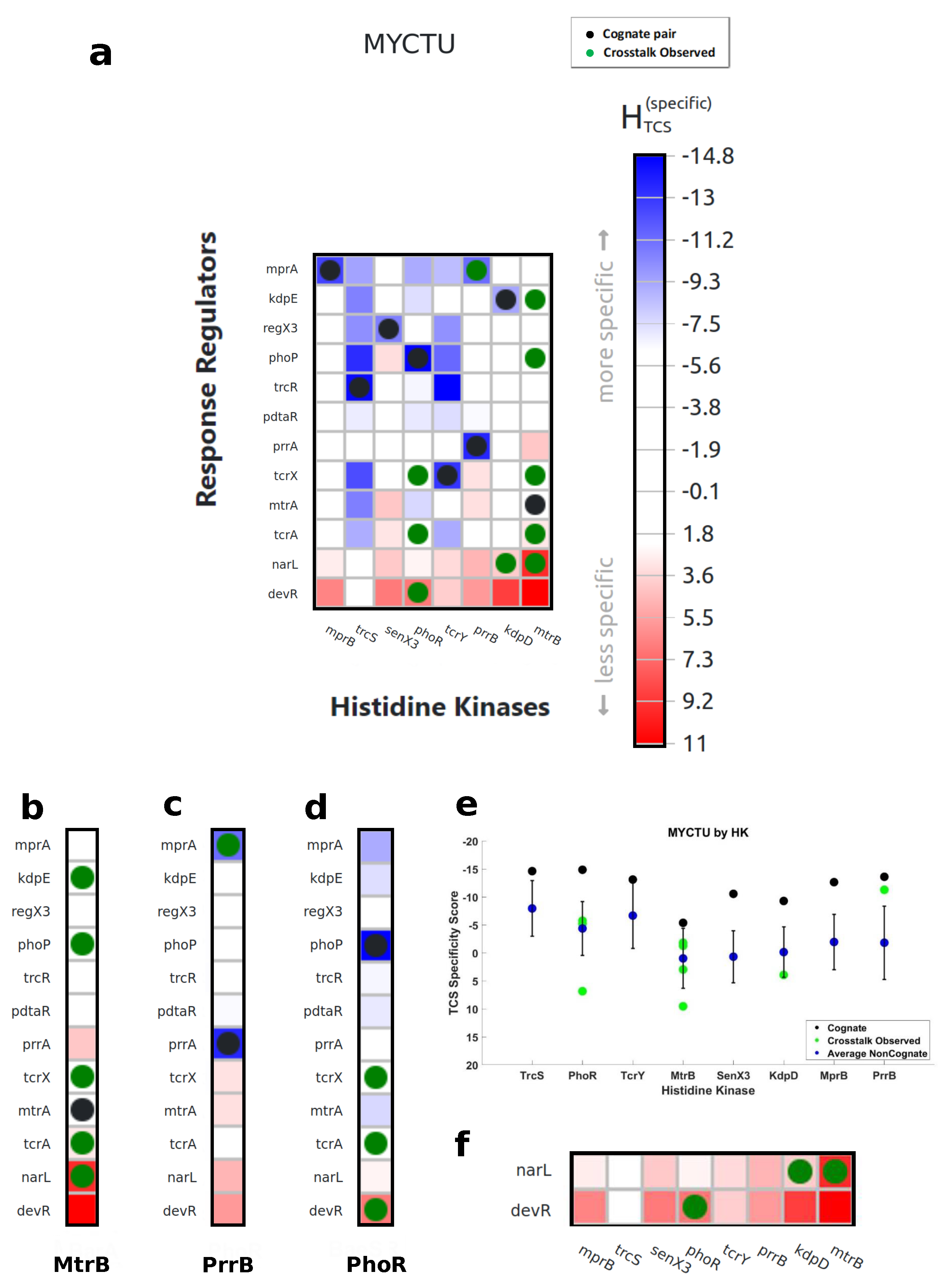
2.4. Negative Selection
3. Discussion
3.1. Characterization of Cognate Specificity
3.2. Exploration of Non-Cognate Interactions
3.3. Revealing Interaction Specificity for Mutation and Variation
4. Materials and Methods
4.1. MSA Construction
4.2. mfDCA Evolutionary Couplings and Hamiltonian Scores
4.3. Software
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ELIHKSIR | Evolutionary Links Inferred for Histidine Kinase Sensors Interacting with Response regulators |
| TCS | Two-Component System |
| DCA | Direct-Coupling Analysis |
| mfDCA | couplings generated by mean-field method as outlined in Morcos, 2011 [1] |
| DI | Direct Information |
| HK | Histidine Kinase, Histidine Kinase family (Pfam:PF00512) [15] |
| RR | Response Regulator, Response Regulator family (Pfam: PF00072) [16] |
| TP | True Positive |
| FN | False Negative |
| PS | Positive Selection |
| NS | Negative Selection |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Endpoint | HTTP Method | URL |
|---|---|---|
| All Organisms | GET | api/list |
| Pairs for heatmap | GET | api/pairs/{ORGANISM_ID::INT} |



| HK | RR | DI | HK | RR | DI | HK | RR | DI | HK | RR | DI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 18 | 77 | 0.102853 | 7 | 147 | 0.00806367 | 14 | 170 | 0.00572271 | 16 | 77 | 0.00460402 |
| 22 | 80 | 0.0722833 | 30 | 83 | 0.00801695 | 19 | 169 | 0.00571496 | 42 | 77 | 0.00460128 |
| 11 | 167 | 0.0705232 | 33 | 83 | 0.00781679 | 14 | 147 | 0.00570246 | 26 | 176 | 0.004455 |
| 26 | 84 | 0.0515243 | 22 | 171 | 0.00767719 | 22 | 170 | 0.00569941 | 42 | 76 | 0.00444185 |
| 23 | 80 | 0.0492594 | 19 | 79 | 0.00765574 | 16 | 76 | 0.00569788 | 17 | 169 | 0.00442802 |
| 14 | 146 | 0.04276 | 23 | 172 | 0.00763662 | 15 | 76 | 0.00568733 | 27 | 80 | 0.00440244 |
| 46 | 76 | 0.0398581 | 18 | 171 | 0.00759788 | 38 | 80 | 0.0056818 | 15 | 167 | 0.00439432 |
| 19 | 76 | 0.0392644 | 19 | 147 | 0.00753089 | 10 | 147 | 0.00567069 | 19 | 81 | 0.00420284 |
| 25 | 170 | 0.0351779 | 18 | 81 | 0.00752615 | 22 | 172 | 0.00562105 | 38 | 79 | 0.00418686 |
| 25 | 171 | 0.0303173 | 12 | 148 | 0.00734758 | 13 | 147 | 0.00559724 | 41 | 79 | 0.00416339 |
| 11 | 168 | 0.0285807 | 10 | 150 | 0.00732327 | 34 | 87 | 0.00559491 | 15 | 170 | 0.0041264 |
| 15 | 146 | 0.0270048 | 45 | 76 | 0.007169 | 33 | 84 | 0.00556154 | 24 | 173 | 0.00403743 |
| 29 | 87 | 0.0265711 | 22 | 76 | 0.00712089 | 34 | 84 | 0.00552466 | 18 | 146 | 0.00382329 |
| 19 | 77 | 0.0259669 | 21 | 172 | 0.00705718 | 25 | 169 | 0.00545037 | 16 | 147 | 0.00378105 |
| 30 | 87 | 0.0215653 | 30 | 80 | 0.00702085 | 15 | 118 | 0.00544512 | 18 | 172 | 0.00377972 |
| 23 | 76 | 0.0193616 | 15 | 74 | 0.00697282 | 25 | 174 | 0.00543429 | 20 | 168 | 0.00375797 |
| 19 | 80 | 0.0189693 | 18 | 147 | 0.00691656 | 18 | 74 | 0.00541696 | 16 | 169 | 0.00366294 |
| 22 | 77 | 0.0188355 | 45 | 79 | 0.00690392 | 14 | 149 | 0.00540391 | 25 | 81 | 0.00362641 |
| 23 | 79 | 0.0180874 | 22 | 78 | 0.00680398 | 30 | 86 | 0.00538702 | 13 | 169 | 0.00359782 |
| 19 | 74 | 0.0176283 | 19 | 170 | 0.00679327 | 33 | 87 | 0.00538074 | 10 | 148 | 0.00359015 |
| 8 | 147 | 0.0172729 | 23 | 170 | 0.00679065 | 17 | 147 | 0.00537632 | 20 | 77 | 0.00355346 |
| 18 | 169 | 0.0171606 | 18 | 78 | 0.00676363 | 33 | 86 | 0.00534217 | 11 | 146 | 0.00345648 |
| 29 | 171 | 0.0170736 | 26 | 81 | 0.00675062 | 7 | 149 | 0.00530426 | 21 | 81 | 0.00344168 |
| 16 | 168 | 0.0168674 | 31 | 87 | 0.0067342 | 14 | 169 | 0.00530394 | 42 | 80 | 0.00338203 |
| 15 | 77 | 0.0152404 | 21 | 77 | 0.00670382 | 38 | 83 | 0.00526882 | 28 | 173 | 0.00334077 |
| 25 | 172 | 0.0149784 | 27 | 84 | 0.00667465 | 26 | 82 | 0.00525117 | 22 | 174 | 0.0032904 |
| 39 | 83 | 0.014901 | 22 | 81 | 0.00666247 | 17 | 77 | 0.00524036 | 20 | 170 | 0.00327778 |
| 29 | 172 | 0.0146187 | 46 | 77 | 0.00658625 | 42 | 83 | 0.00521168 | 14 | 168 | 0.0032269 |
| 21 | 170 | 0.014469 | 26 | 79 | 0.00657677 | 34 | 83 | 0.00520958 | 22 | 176 | 0.00319442 |
| 26 | 80 | 0.0141692 | 18 | 168 | 0.00651457 | 34 | 80 | 0.00518935 | 24 | 172 | 0.00310293 |
| 26 | 83 | 0.0139579 | 45 | 80 | 0.00648245 | 20 | 80 | 0.00514101 | 17 | 170 | 0.00307046 |
| 23 | 83 | 0.0130196 | 19 | 172 | 0.00639985 | 46 | 80 | 0.00512939 | 19 | 168 | 0.00298315 |
| 12 | 168 | 0.0128269 | 18 | 76 | 0.00633046 | 18 | 170 | 0.00510377 | 26 | 85 | 0.00290492 |
| 15 | 147 | 0.0123301 | 28 | 172 | 0.00626959 | 23 | 82 | 0.00509604 | 20 | 171 | 0.0027667 |
| 29 | 84 | 0.0121863 | 25 | 175 | 0.00623035 | 25 | 80 | 0.00502442 | 15 | 169 | 0.00275228 |
| 8 | 148 | 0.0119021 | 16 | 74 | 0.00615123 | 49 | 77 | 0.00502117 | 20 | 169 | 0.00269252 |
| 23 | 84 | 0.0118386 | 30 | 88 | 0.00614559 | 45 | 77 | 0.00501361 | 15 | 168 | 0.00266174 |
| 22 | 84 | 0.0113964 | 19 | 75 | 0.00610985 | 24 | 171 | 0.00496918 | 21 | 168 | 0.00254422 |
| 23 | 171 | 0.0108972 | 10 | 149 | 0.00608531 | 17 | 76 | 0.00494564 | 26 | 174 | 0.00238278 |
| 32 | 87 | 0.0108637 | 24 | 80 | 0.00607124 | 14 | 167 | 0.00492586 | 27 | 173 | 0.00238187 |
| 26 | 171 | 0.0107505 | 23 | 169 | 0.00605014 | 15 | 148 | 0.00492344 | 24 | 169 | 0.00237797 |
| 25 | 84 | 0.0104978 | 33 | 88 | 0.00604033 | 23 | 173 | 0.00489615 | 20 | 172 | 0.00233119 |
| 14 | 148 | 0.0104055 | 7 | 150 | 0.00599047 | 24 | 170 | 0.00488941 | 18 | 145 | 0.00222389 |
| 30 | 84 | 0.0102802 | 48 | 76 | 0.00596518 | 44 | 76 | 0.00484498 | 13 | 168 | 0.00190674 |
| 26 | 87 | 0.0102777 | 21 | 80 | 0.0059512 | 21 | 173 | 0.00483877 | 10 | 169 | 0.00186603 |
| 23 | 78 | 0.01007 | 25 | 173 | 0.00592837 | 29 | 173 | 0.00481891 | 18 | 167 | 0.00162171 |
| 23 | 77 | 0.00997545 | 28 | 171 | 0.00591566 | 10 | 168 | 0.00480323 | 12 | 169 | 0.00147153 |
| 22 | 169 | 0.00996165 | 42 | 79 | 0.00586154 | 17 | 171 | 0.00478502 | 15 | 145 | 0.00135628 |
| 43 | 80 | 0.00972282 | 22 | 173 | 0.00585409 | 22 | 168 | 0.00478308 | 11 | 149 | 0.00105631 |
| 22 | 83 | 0.00952424 | 26 | 172 | 0.0058532 | 12 | 147 | 0.0047804 | 11 | 169 | 0.000969333 |
| 18 | 80 | 0.0093605 | 49 | 76 | 0.005838 | 26 | 173 | 0.00476983 | 11 | 150 | 0.000862052 |
| 7 | 148 | 0.00897782 | 26 | 175 | 0.00581626 | 22 | 82 | 0.00476307 | 11 | 148 | 0.000856593 |
| 29 | 83 | 0.00882006 | 30 | 172 | 0.00577961 | 45 | 75 | 0.00475623 | 11 | 118 | 0.000790599 |
| 21 | 171 | 0.00865425 | 22 | 79 | 0.00577265 | 39 | 80 | 0.00473036 | 11 | 147 | 0.000413171 |
| 26 | 170 | 0.00854487 | 15 | 73 | 0.00576882 | 27 | 172 | 0.00472585 | |||
| 19 | 171 | 0.00831504 | 41 | 80 | 0.00576018 | 41 | 76 | 0.00472158 | |||
| 17 | 168 | 0.008266 | 30 | 173 | 0.00574398 | 27 | 83 | 0.0046522 | |||
| 19 | 78 | 0.00818936 | 23 | 81 | 0.00573789 | 20 | 76 | 0.00465154 | |||
| 21 | 169 | 0.0080992 | 22 | 175 | 0.00573107 | 16 | 77 | 0.00460402 |
References
- Morcos, F.; Pagnani, A.; Lunt, B.; Bertolino, A.; Marks, D.S.; Sander, C.; Zecchina, R.; Onuchic, J.N.; Hwa, T.; Weigt, M. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. USA 2011, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.R.; Morcos, F.; Levine, H.; Onuchic, J.N. Toward rationally redesigning bacterial two-component signaling systems using coevolutionary information. Proc. Natl. Acad. Sci. USA 2014, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, J.S.; Cheng, R.R.; Paddock, M.L.; Sancar, C.; Morcos, F.; Golden, S.S. A combined computational and genetic approach uncovers network interactions of the cyanobacterial circadian clock. J. Bacteriol. 2016, 198, 2439–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.R.; Nordesjö, O.; Hayes, R.L.; Levine, H.; Flores, S.C.; Onuchic, J.N.; Morcos, F. Connecting the sequence-space of bacterial signaling proteins to phenotypes using coevolutionary landscapes. Mol. Biol. Evol. 2016, 33, 3054–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.R.; Haglund, E.; Tiee, N.S.; Morcos, F.; Levine, H.; Adams, J.A.; Jennings, P.A.; Onuchic, J.N. Designing bacterial signaling interactions with coevolutionary landscapes. PLoS ONE 2018, 13, e0201734. [Google Scholar] [CrossRef]
- Morcos, F.; Hwa, T.; Onuchic, J.N.; Weigt, M. Direct Coupling Analysis for Protein Contact Prediction. In Protein Structure Prediction; Springer: New York, NY, USA, 2014; pp. 55–70. [Google Scholar] [CrossRef]
- Muscat, M.; Croce, G.; Sarti, E.; Weigt, M. FilterDCA: Interpretable supervised contact prediction using inter-domain coevolution. bioRxiv 2019. Available online: https://www.biorxiv.org/content/early/2019/12/24/2019.12.24.887877.full.pdf (accessed on 11 December 2020).
- Szurmant, H.; Weigt, M. Inter-residue, inter-protein and inter-family coevolution: Bridging the scales. Curr. Opin. Struct. Biol. 2018, 50, 26–32. [Google Scholar] [CrossRef]
- Jiang, X.L.; Martinez-Ledesma, E.; Morcos, F. Revealing protein networks and gene-drug connectivity in cancer from direct information. Sci. Rep. 2017, 7, 3739. [Google Scholar] [CrossRef] [Green Version]
- Jacquin, H.; Gilson, A.; Shakhnovich, E.; Cocco, S.; Monasson, R. Benchmarking Inverse Statistical Approaches for Protein Structure and Design with Exactly Solvable Models. PLoS Comput. Biol. 2016, 12, e1004889. [Google Scholar] [CrossRef]
- Levy, R.M.; Haldane, A.; Flynn, W.F. Potts Hamiltonian models of protein co-variation, free energy landscapes, and evolutionary fitness. Curr. Opin. Struct. Biol. 2017, 43, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Figliuzzi, M.; Jacquier, H.; Schug, A.; Tenaillon, O.; Weigt, M. Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase tem-1. Mol. Biol. Evol. 2016, 33, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Ekeberg, M.; Hartonen, T.; Aurell, E. Fast pseudolikelihood maximization for direct-coupling analysis of protein structure from many homologous. J. Comput. Phys. 2014, 276, 341–356. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. Available online: https://academic.oup.com/nar/article-pdf/47/D1/D427/27436497/gky995.pdf (accessed on 11 December 2020). [CrossRef] [PubMed]
- Pfam. Family: HisKA (PF00512)—His Kinase A (Phospho-Acceptor) Domain; Pfam: Hinxton, UK, 2020. [Google Scholar]
- Pfam. Family: Response_reg (PF00072) Response Regulator Receiver Domain; Pfam: Hinxton, UK, 2020. [Google Scholar]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Schug, A.; Weigt, M.; Onuchic, J.N.; Hwa, T.; Szurmant, H. High-resolution protein complexes from integrating genomic information with molecular simulation. Proc. Natl. Acad. Sci. USA 2009, 106, 22124–22129. [Google Scholar] [CrossRef] [Green Version]
- Szurmant, H.; Hoch, J.A. Interaction fidelity in two-component signaling. Curr. Opin. Microbiol. 2010, 13, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Capra, E.J.; Laub, M.T. The Evolution of Two-Component. Annu. Rev. Microbiol. 2012, 66, 325–347. [Google Scholar] [CrossRef] [Green Version]
- Heath, J.D.; Charles, T.C.; Nester, E.W. Ti Plasmid and Chromosomally Encoded Two-Component Systems Important in Plant Cell Transformation by Agrobacterium Species. In Two—Component Signal Transduction; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1995; Chapter 23; pp. 367–385. [Google Scholar]
- Stewart, R.C.; Jahreis, K.; Parkinson, J.S. Rapid phosphotransfer to CheY from a CheA protein lacking the CheY-binding domain. Biochemistry 2000, 39, 13157–13165. [Google Scholar] [CrossRef]
- Yamamoto, K.; Hirao, K.; Oshima, T.; Aiba, H.; Utsumi, R.; Ishihama, A. Functional characterization in vitro of all two-component signal transduction systems from Escherichia coli. J. Biol. Chem. 2005, 280, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, R.; Pandey, A.; Rajankar, M.P.; Dixit, N.M.; Saini, D.K. The two-component signalling networks of Mycobacterium tuberculosis display extensive cross-talk in vitro. Biochem. J. 2015, 469, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Becker, G.; Klauck, E.; Hengge-Aronis, R. Regulation of RpoS proteolysis in Escherichia coli: The response regulator RssB is a recognition factor that interacts with the turnover element in RpoS. Proc. Natl. Acad. Sci. USA 1999, 96, 6439–6444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauck, E.; Lingnau, M.; Hengge-Aronis, R. Role of the response regulator RssB in σS recognition and initiation of σS proteolysis in Escherichia coli. Mol. Microbiol. 2001, 40, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2009, 38, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.H.; Whitworth, D.E. The genetic organisation of prokaryotic two-component system signalling pathways. BMC Genom. 2010, 11, 720. [Google Scholar] [CrossRef] [Green Version]
- Facebook Inc. React—A JavaScript Library for Building User Interfaces; Facebook Inc.: Menlo Park, CA, USA, 2020. [Google Scholar]
- Fielding, R.T.; Taylor, R.N. Architectural Styles and the Design of Network-Based Software Architectures. Ph.D. Thesis, University of California, Irvine, CA, USA, 2000. [Google Scholar]




| Total Organisms | 6676 |
| Bacteria | 6412 |
| Archaea | 65 |
| Eukaryotes | 188 |
| Unknown Organisms/Metagenomes | 11 |
| Total Interactions Evaluated | 6,272,607 |
| Number of HKs | 111,032 |
| Number of RRs | 225,616 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinner, C.; Ziegler, C.; Jung, Y.H.; Jiang, X.; Morcos, F. ELIHKSIR Web Server: Evolutionary Links Inferred for Histidine Kinase Sensors Interacting with Response Regulators. Entropy 2021, 23, 170. https://doi.org/10.3390/e23020170
Sinner C, Ziegler C, Jung YH, Jiang X, Morcos F. ELIHKSIR Web Server: Evolutionary Links Inferred for Histidine Kinase Sensors Interacting with Response Regulators. Entropy. 2021; 23(2):170. https://doi.org/10.3390/e23020170
Chicago/Turabian StyleSinner, Claude, Cheyenne Ziegler, Yun Ho Jung, Xianli Jiang, and Faruck Morcos. 2021. "ELIHKSIR Web Server: Evolutionary Links Inferred for Histidine Kinase Sensors Interacting with Response Regulators" Entropy 23, no. 2: 170. https://doi.org/10.3390/e23020170
APA StyleSinner, C., Ziegler, C., Jung, Y. H., Jiang, X., & Morcos, F. (2021). ELIHKSIR Web Server: Evolutionary Links Inferred for Histidine Kinase Sensors Interacting with Response Regulators. Entropy, 23(2), 170. https://doi.org/10.3390/e23020170