
Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers


Abstract
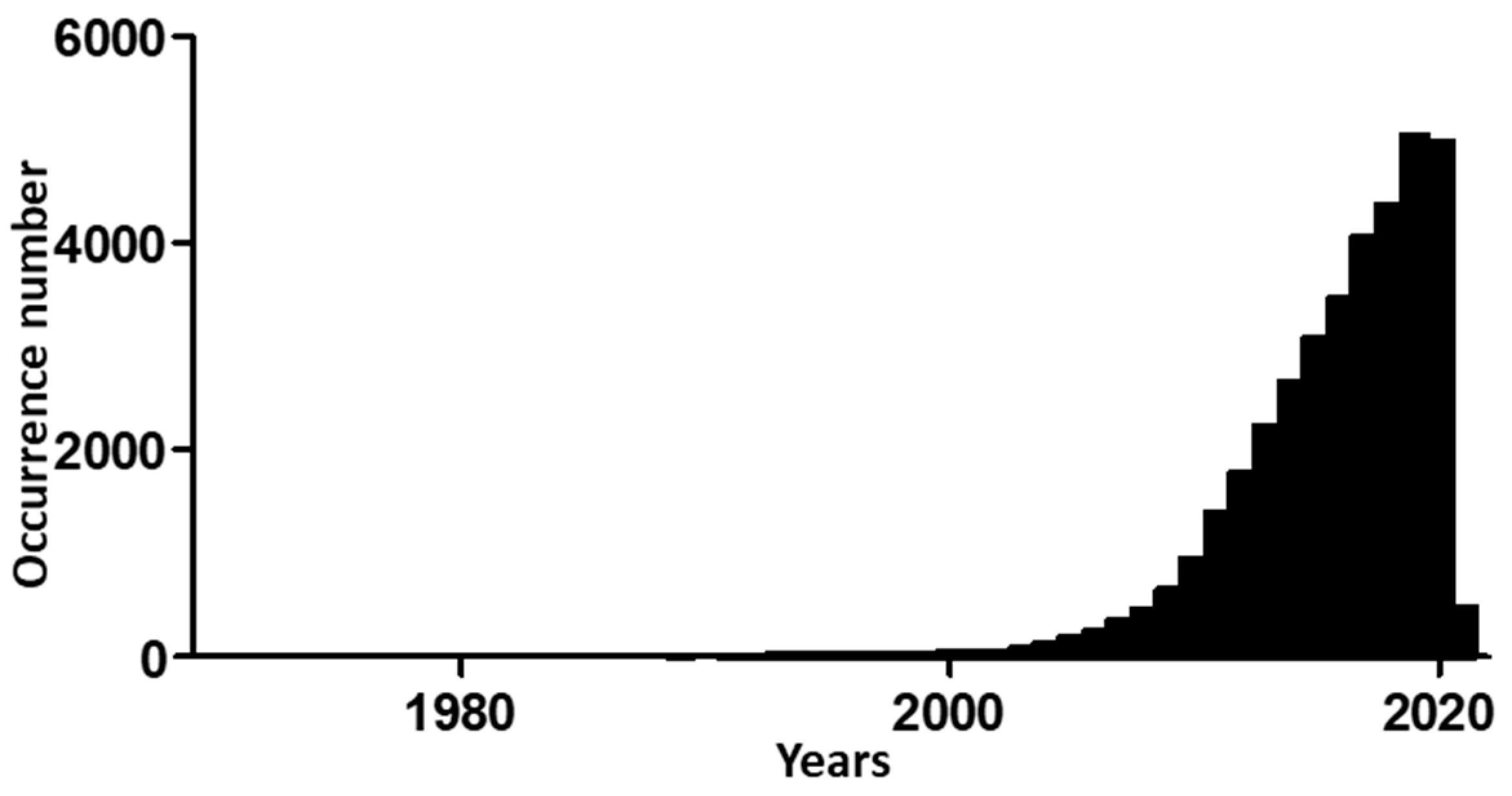
:1. Introduction
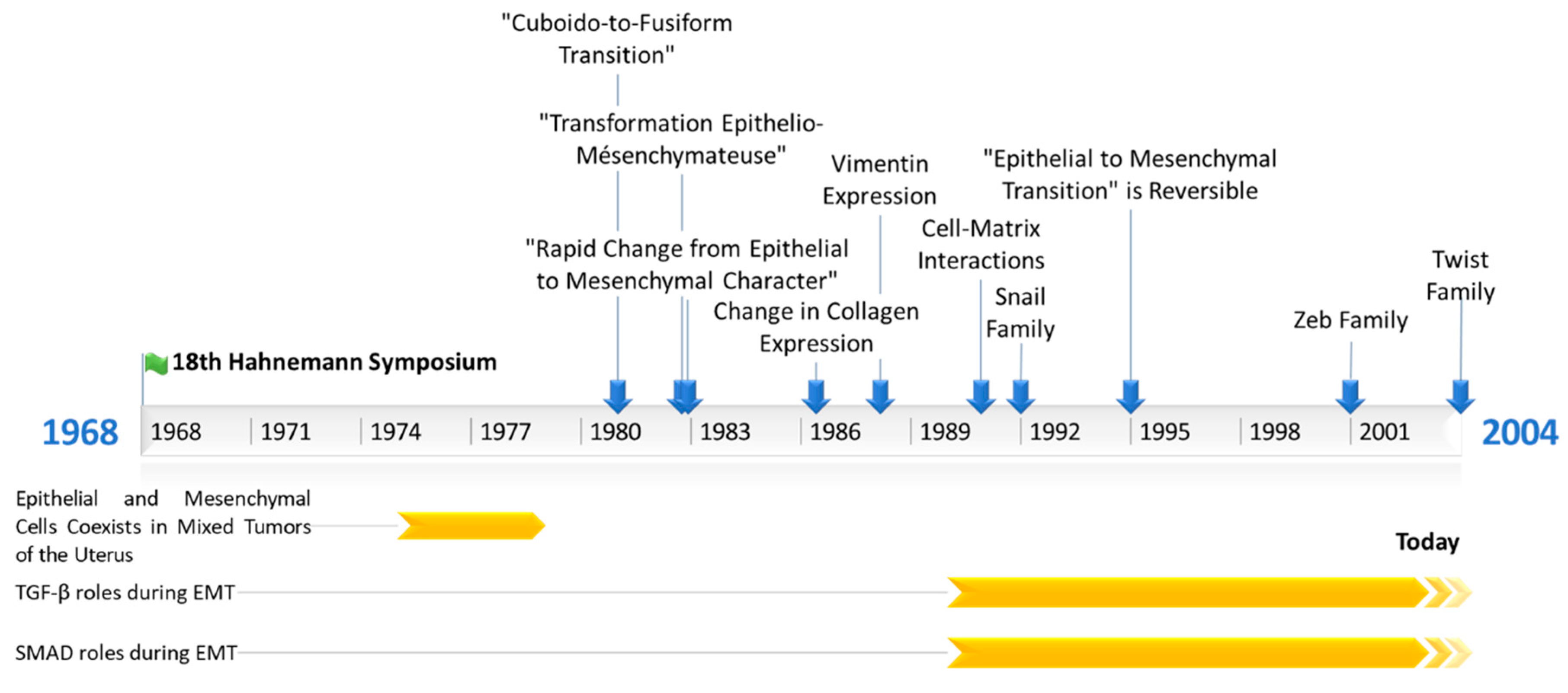
2. The First Steps of Research on EMT
2.1. The First Phenotypical Observations of an EMT
2.2. The First Mechanistical and Molecular Descriptions of EMT
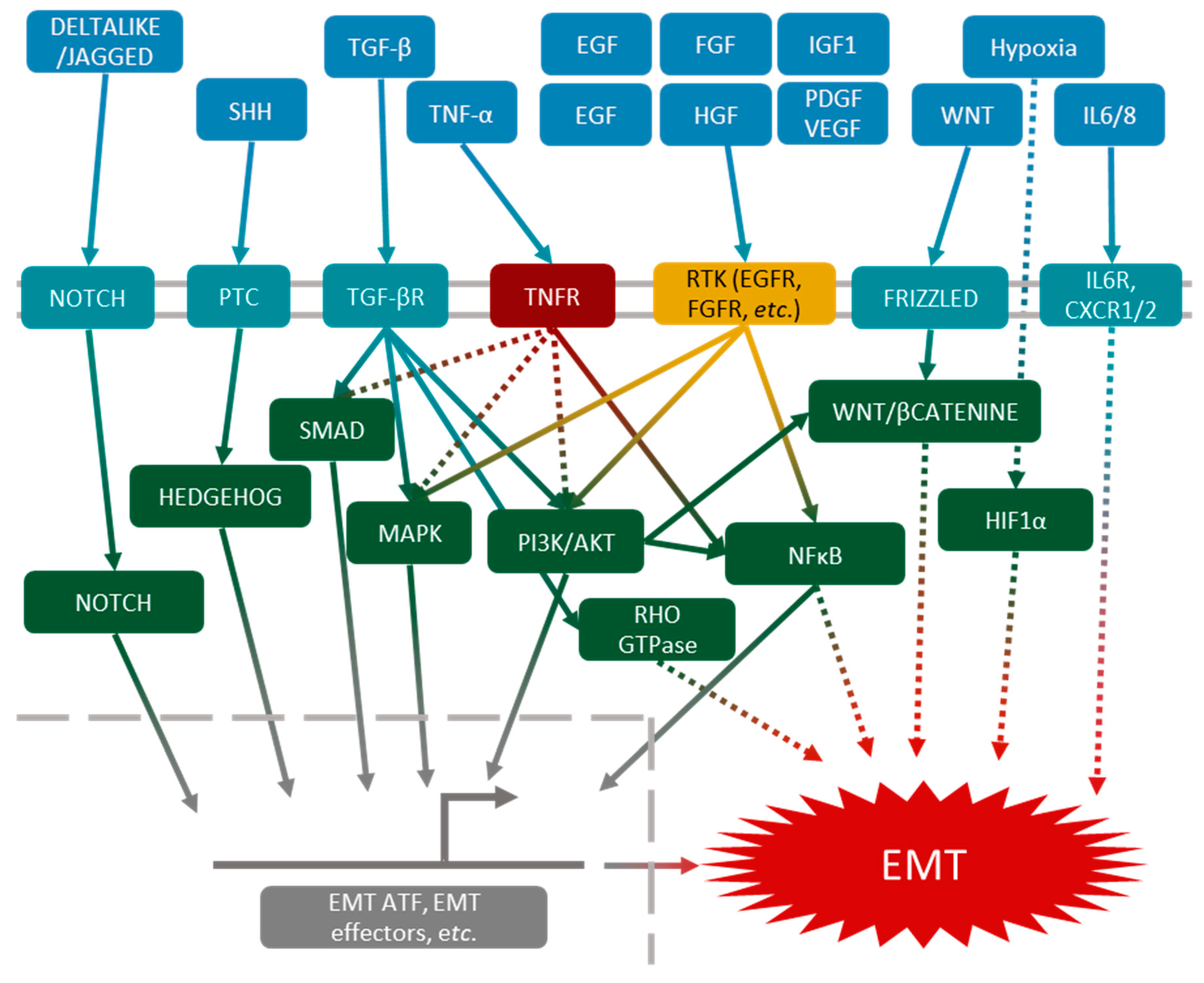
3. Main Molecular Aspects of EMT
3.1. Molecular Pathways Leading to EMT
3.2. Transcriptional Regulation of EMT
3.3. Non-Coding RNA Regulation of EMT
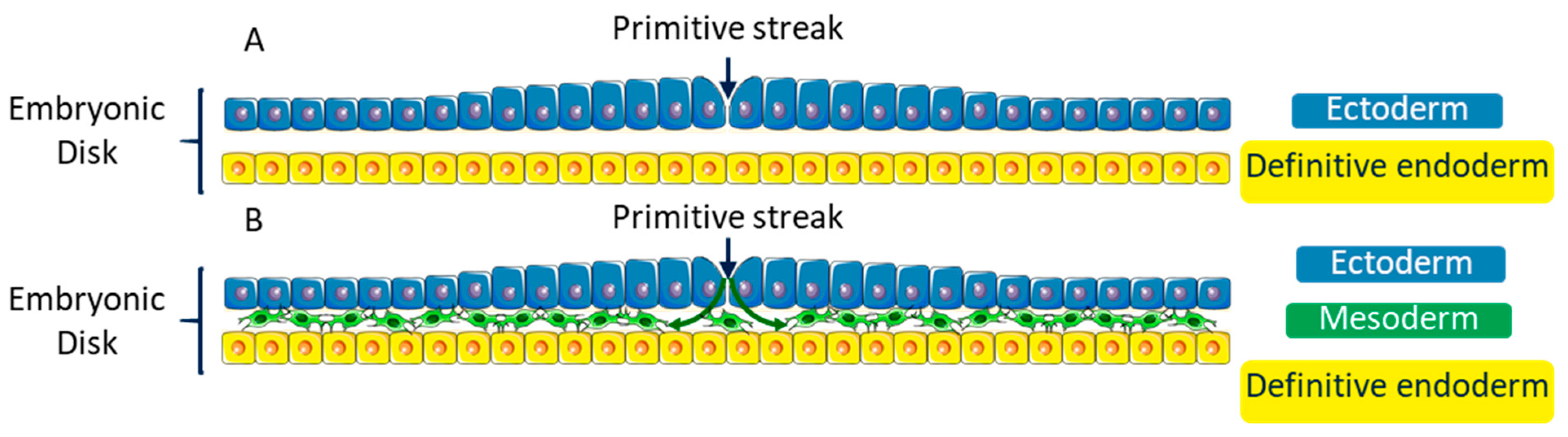
4. EMT during Embryonic Development
4.1. Gastrulation
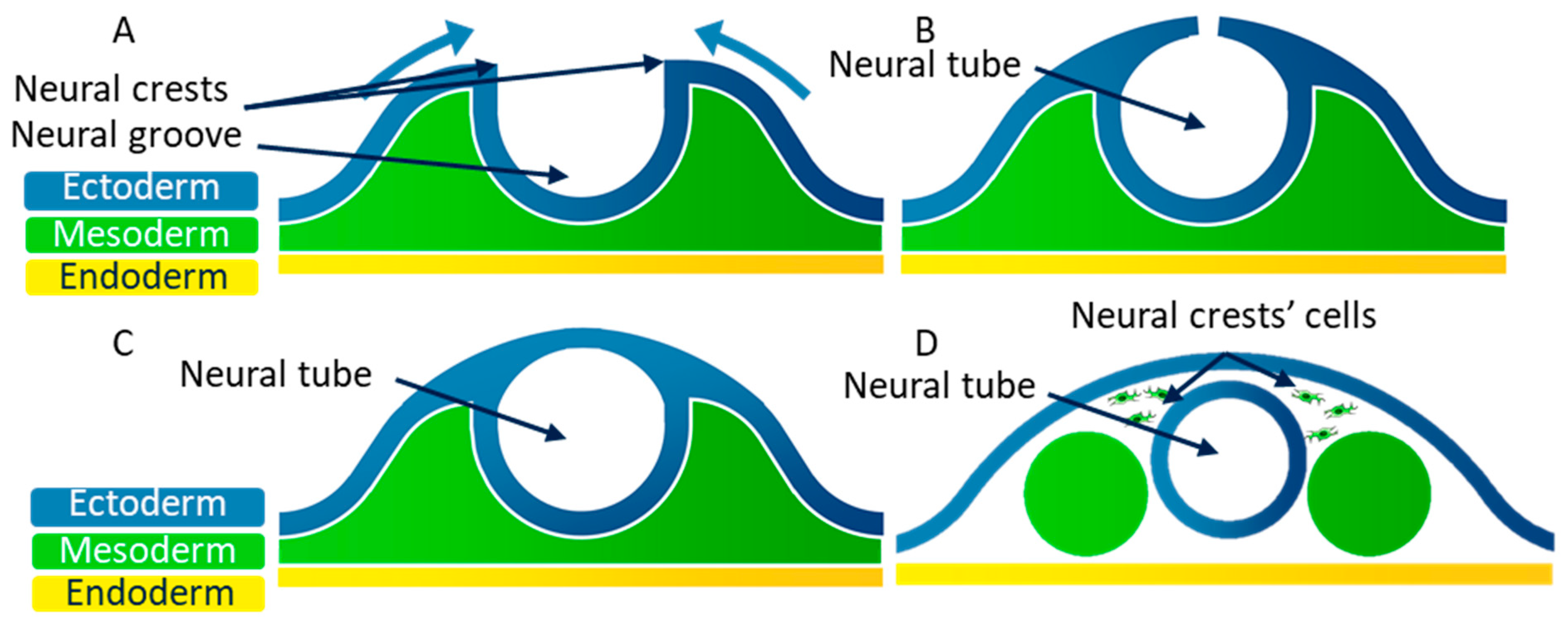
4.2. Neural Tube Formation
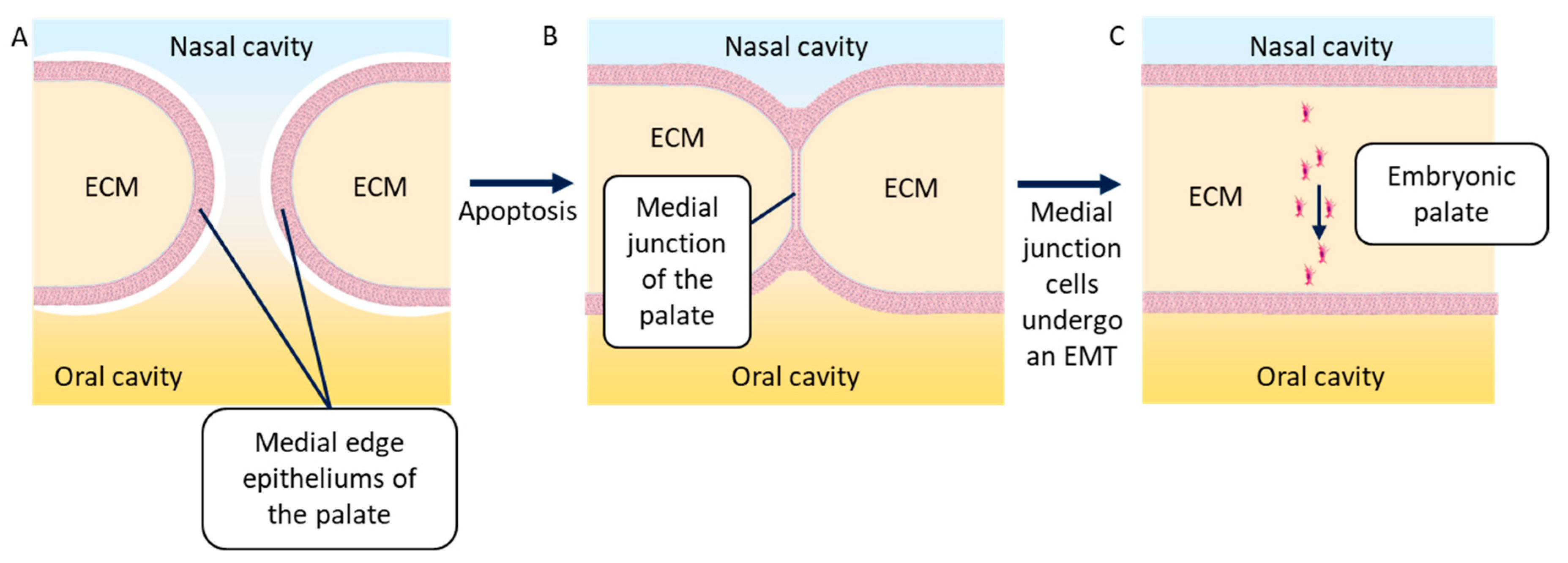
4.3. Embryonic Palatal Fusion
5. EMT in Fibrosis
6. EMT in Cancers
6.1. EMT Induction Factors of Cancer Cells
6.2. EMT and Anticancer Therapy Resistance
6.3. Immune Escape
6.4. Metastasis Formation
6.5. Cancer Stem Cells
6.6. Anticancer Treatments Targeting EMT
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | Adenosine triphosphate-binding cassette |
| ADAM19 | Adam metallopeptidase 19 |
| ALDH1 | Aldehyde dehydrogenase 1 |
| CTC | Circulating tumor cells |
| DDR | DNA damage response |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EMT | Epithelial–mesenchymal transition |
| EMT-ATF | Epithelial to mesenchymal transition-activated transcription factor |
| ESA | Epidermal surface antigen |
| FGF | Fibroblast growth factor |
| GAG | Glycosaminoglycans |
| HER2 | Human EGF receptor 2 |
| HGF | Hepatocyte growth factor |
| HIF1-α | Hypoxia inducible factor 1-α |
| IGF1 | Insluin-like growth factor 1 |
| IL | Interleukin |
| MDSC | Myeloid-derived suppressor cells |
| MET | Mesenchymal to epithelial transition |
| MHC | Major histocompatibility complex |
| MMP9 | Matrix metalloproteinase 9 |
| NK | Natural killer |
| PD1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| PDGF | Platelet-derived growth factor |
| SMAD | Mothers against decapentaplegic homolog |
| TGF | Tranforming growth gactor |
| TGF-βR | TGF-β receptor |
| TNF | Tumor necrosis factor |
| Treg | Regulatory T cell |
| VEGF | Vascular endothelial growth factor |
| WB | Western blotting |
| ZA | Zonula adherens |
References
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Hay, E.D. The Fine Structure of Blastema Cells and Differentiating Cartilage Cells in Regenerating Limbs of Amblystoma Larvae. J. Cell Biol. 1958, 4, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Meier, S.; Hay, E.D. Control of corneal differentiation by extracellular materials. Collagen as a promoter and stabilizer of epithelial stroma production. Dev. Biol. 1974, 38, 249–270. [Google Scholar] [CrossRef]
- Hay, E.D. Organization and Fine Structure of Epithelium and Mesenchyme in the Developing Chick Embryo. In Epithelial-Mesenchymal Intercations. 18th Hahnemann Symposium; Fleischmajer, R., Billingham, R., Eds.; The Williams and Wilkins Company: Baltimore, MD, USA, 1968; pp. 31–55. [Google Scholar]
- Böcker, W.; Stegner, H.-E. A light and electron microscopic study of endometrial sarcomas of the uterus. Virchows Arch. A Path. Anat. Histol. 1975, 368, 141–156. [Google Scholar] [CrossRef]
- Ishikawa, S.; Kaneko, H.; Sumida, T.; Sekiya, M. Ultrastructure of Mesodermal Mixed Tumor of the Uterus. Pathol. Int. 1979, 29, 801–809. [Google Scholar] [CrossRef]
- Dulbecco, R.; Henahan, M.; Bowman, M.; Okada, S.; Battifora, H.; Unger, M. Generation of fibroblast-like cells from cloned epithelial mammary cells in vitro: A possible new cell type. Proc. Natl. Acad. Sci. USA 1981, 78, 2345–2349. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.C.; Peachey, L.A.; Durbin, H.; Rudland, P.S. A possible mammary stem cell line. Cell 1978, 15, 283–298. [Google Scholar] [CrossRef]
- Franke, W.W.; Grund, C.; Kuhn, C.; Jackson, B.W.; Illmensee, K. Formation of cytoskeletal elements during mouse embryogenesis. III. Primary mesenchymal cells and the first appearance of vimentin filaments. Differentiation 1982, 23, 43–59. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E. Cytodifferentiation and tissue phenotype change during transformation of embryonic lens epithelium to mesenchyme-like cells in vitro. Dev. Biol. 1986, 115, 363–379. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E. Cytoskeleton and thyroglobulin expression change during transformation of thyroid epithelium to mesenchyme-like cells. Development 1988, 102, 605–622. [Google Scholar] [CrossRef]
- Bilozur, M.E.; Hay, E.D. Cell migration into neural tube lumen provides evidence for the “fixed cortex” theory of cell motility. Cell Motil. 1989, 14, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Bilozur, M.E.; Hay, E.D. Neural crest migration in 3D extracellular matrix utilizes laminin, fibronectin, or collagen. Dev. Biol. 1988, 125, 19–33. [Google Scholar] [CrossRef]
- Hay, E.D. Role of cell-matrix contacts in cell migration and epithelial-mesenchymal transformation. Cell Differ. Dev. 1990, 32, 367–375. [Google Scholar] [CrossRef]
- Fitchett, J.E.; Hay, E.D. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 1989, 131, 455–474. [Google Scholar] [CrossRef]
- Gavrilovic, J.; Moens, G.; Thiery, J.P.; Jouanneau, J. Expression of transfected transforming growth factor alpha induces a motile fibroblast-like phenotype with extracellular matrix-degrading potential in a rat bladder carcinoma cell line. Cell Regul. 1990, 1, 1003–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potts, J.D.; Dagle, J.; Walder, J.A.; Weeks, D.L.; Runyan, R.B. Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta 3. Proc. Natl. Acad. Sci. USA 1991, 88, 1516–1520. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Vanderburg, C.; Odierna, G.; Hay, E. TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development 1998, 125, 95–105. [Google Scholar] [CrossRef]
- Nawshad, A.; LaGamba, D.; Hay, E. Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch. Oral Biol. 2004, 49, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Nawshad, A.; Medici, D.; Liu, C.-C.; Hay, E.D. TGFβ3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J. Cell Sci. 2007, 120, 1646–1653. [Google Scholar] [CrossRef] [Green Version]
- Piek, E.; Moustakas, A.; Kurisaki, A.; Heldin, C.; Dijke, P.T. TGF-(beta) type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 1999, 112, 4557–4568. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug Promote Epithelial-Mesenchymal Transition through β-Catenin–T-Cell Factor-4-dependent Expression of Transforming Growth Factor-β3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Dijke, P.T. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Tavares, A.L.P.; Mercado-Pimentel, M.E.; Runyan, R.B.; Kitten, G.T. TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev. Dyn. 2006, 235, 1589–1598. [Google Scholar] [CrossRef]
- Cho, H.J.; Yoo, J. Rho activation is required for transforming growth factor-beta-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol. Int. 2007, 31, 1225–1230. [Google Scholar] [CrossRef]
- Kattla, J.J.; Carew, R.M.; Heljić, M.; Godson, C.; Brazil, D.P. Protein kinase B/Akt activity is involved in renal TGF-β1-driven epithelial-mesenchymal transition in vitro and in vivo. Am. J. Physiol. Ren. Physiol. 2008, 295, F215–F225. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-β1-Induced EMT In Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002, 115, 3193–3206. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-J.; Luo, J.; Li, D.; Zhou, Y.-H.; Yan, B.; Wei, J.-J.; Tu, J.-C.; Li, Y.-R.; Zhang, G.-M.; Feng, Z.-H. TGF-β1 and TNF-α synergistically induce epithelial to mesenchymal transition of breast cancer cells by enhancing TAK1 activation. J. Cell Commun. Signal. 2019, 13, 369–380. [Google Scholar] [CrossRef]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar]
- Sun, X.; Meyers, E.N.; Lewandoski, M.; Martin, G.R. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev. 1999, 13, 1834–1846. [Google Scholar] [CrossRef] [Green Version]
- Grotegut, S.; Von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like Growth Factor-I–Dependent Up-regulation of ZEB1 Drives Epithelial-to-Mesenchymal Transition in Human Prostate Cancer Cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Liu, Z.-R. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.J.; et al. Vascular Endothelial Growth Factor Receptor-1 Activation Mediates Epithelial to Mesenchymal Transition in Human Pancreatic Carcinoma Cells. Cancer Res. 2006, 66, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.; del Amo, F.F.; Gridley, T. Isolation of Sna, a mouse gene homologous to the Drosophila genes snail and escargot: Its expression pattern suggests multiple roles during postimplantation development. Development 1992, 116, 1033–1039. [Google Scholar] [CrossRef]
- Nieto, M.; Bennett, M.; Sargent, M.; Wilkinson, D. Cloning and developmental expression of Sna, a murine homologue of the Drosophila snail gene. Development 1992, 116, 227–237. [Google Scholar] [CrossRef]
- Nieto, M.Á.; Sargent, M.G.; Wilkinson, D.; Cooke, J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 1994, 264, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The Mouse Snail Gene Encodes a Key Regulator of the Epithelial-Mesenchymal Transition. Mol. Cell. Biol. 2001, 21, 8184–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 Expression Is Necessary for Metastatic Competence in Breast Cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The Two-Handed E Box Binding Zinc Finger Protein SIP1 Downregulates E-Cadherin and Induces Invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Girelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; de Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Birchmeier, C.; Brand-Saheri, B.; Birchmeier, W.; Brand-Saberi, B. Epithelial-Mesenchymal Transitions in Cancer Progression. Cells Tissues Organs 1996, 156, 217–226. [Google Scholar] [CrossRef]
- Perl, A.T.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nat. Cell Biol. 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Xu, L.-Y.; Li, E.-M. A Family of Pleiotropically Acting MicroRNAs in Cancer Progression, miR-200: Potential Cancer Therapeutic Targets. Curr. Pharm. Des. 2014, 20, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, P.P.L.; Williams, E.A.; Chan, W.Y. Gastrulation in the mouse embryo: Ultrastructural and molecular aspects of germ layer morphogenesis. Microsc. Res. Tech. 1993, 26, 301–328. [Google Scholar] [CrossRef]
- Nakaya, Y.; Sheng, G. Epithelial to mesenchymal transition during gastrulation: An embryological view. Dev. Growth Differ. 2008, 50, 755–766. [Google Scholar] [CrossRef]
- Tam, P.; Beddington, R. The formation of mesodermal tissues in the mouse embryo during gastrulation and early organogenesis. Development 1987, 99, 109–126. [Google Scholar] [CrossRef]
- Rogers, C.D.; Saxena, A.; Bronner, M.E. Sip1 mediates an E-cadherin-to-N-cadherin switch during cranial neural crest EMT. J. Cell Biol. 2013, 203, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Bronner, M.E. Formation and migration of neural crest cells in the vertebrate embryo. Histochem. Cell Biol. 2012, 138, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Gibbins, J.R.; Manthey, A.; Tazawa, Y.M.; Scott, B.; Bloch-Zupan, A.; Hunter, N. Midline fusion in the formation of the secondary palate anticipated by upregulation of keratin K5/6 and localized expression of vimentin mRNA in medial edge epithelium. Int. J. Dev. Biol. 1999, 43, 237–244. [Google Scholar] [PubMed]
- Cuervo, R.; Covarrubias, L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 2004, 131, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, C.; Nakamura, N.; Okamoto, Y.; Osawa, M.; Shiota, K. Cytochemical identification of programmed cell death in the fusing fetal mouse palate by specific labelling of DNA fragmentation. Anat. Embryol. 1994, 190, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.M.; Benson, M.D. Medial epithelial seam cell migration during palatal fusion. J. Cell. Physiol. 2020, 235, 1417–1424. [Google Scholar] [CrossRef]
- Duband, J.L.; Dufour, S.; Hatta, K.; Takeichi, M.; Edelman, G.M.; Thiery, J.P. Adhesion molecules during somitogenesis in the avian embryo. J. Cell Biol. 1987, 104, 1361–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, Y.; Kuroda, S.; Katagiri, Y.T.; Kaibuchi, K.; Takahashi, Y. Mesenchymal-Epithelial Transition during Somitic Segmentation Is Regulated by Differential Roles of Cdc42 and Rac1. Dev. Cell 2004, 7, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Sato, Y.; Suetsugu, R.; Nakaya, Y. Mesenchymal-to-Epithelial Transition during Somitic Segmentation: A Novel Approach to Studying the Roles of Rho Family GTPases in Morphogenesis. Cells Tissues Organs 2005, 179, 36–42. [Google Scholar] [CrossRef]
- Person, A.D.; Klewer, S.E.; Runyan, R.B. Cell Biology of Cardiac Cushion Development. Int. Rev. Cytol. 2005, 243, 287–335. [Google Scholar]
- Runyan, R.B.; Heimark, R.L.; Camenisch, T.D.; Klewer, S.E. Epithelial-Mesenchymal Transformation in the Embryonic Heart. In Rise and Fall of Epithelial Phenotype: Concepts of Epithelial-Mesenchymal Transition; Savagner, P., Ed.; Springer: Boston, MA, USA, 2005; pp. 40–55. [Google Scholar]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts Derive from Hepatocytes in Liver Fibrosis via Epithelial to Mesenchymal Transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; de Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M. Ángela Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Borges, F.T.; Melo, S.A.; Özdemir, B.C.; Kato, N.; Revuelta, I.; Miller, C.A.; Ii, V.H.G.; LeBleu, V.S.; Kalluri, R. TGF-β1–Containing Exosomes from Injured Epithelial Cells Activate Fibroblasts to Initiate Tissue Regenerative Responses and Fibrosis. J. Am. Soc. Nephrol. 2012, 24, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Strippoli, R.; Moreno-Vicente, R.; Battistelli, C.; Cicchini, C.; Noce, V.; Amicone, L.; Marchetti, A.; del Pozo, M.A.; Tripodi, M. Molecular Mechanisms Underlying Peritoneal EMT and Fibrosis. Stem Cells Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Khoo, B.L.; Lee, S.C.; Kumar, P.; Tan, T.Z.; Warkiani, M.E.; Ow, S.G.; Nandi, S.; Lim, C.T.; Thiery, J.P. Short-term expansion of breast circulating cancer cells predicts response to anti-cancer therapy. Oncotarget 2015, 6, 15578–15593. [Google Scholar] [CrossRef] [Green Version]
- Eskiizmir, G.; Özgür, E. Epithelial-Mesenchymal Transition in Tumor Microenvironment Induced by Hypoxia. Cancer Metastasis 2018. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Mittal, V. Tumor Microenvironment Regulates Epithelial—Mesenchymal Transitions in Mestastasis. Expert. Rev. Anticancer Ther. 2012, 12, 857–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.-C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.-P.; Abastado, J.-P. Mesenchymal Transition and Dissemination of Cancer Cells Is Driven by Myeloid-Derived Suppressor Cells Infiltrating the Primary Tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ieda, T.; Tazawa, H.; Okabayashi, H.; Yano, S.; Shigeyasu, K.; Kuroda, S.; Ohara, T.; Noma, K.; Kishimoto, H.; Nishizaki, M.; et al. Visualization of epithelial-mesenchymal transition in an inflammatory microenvironment–colorectal cancer network. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Lyons, J.G.; Patel, V.; Roue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail Up-regulates Proinflammatory Mediators and Inhibits Differentiation in Oral Keratinocytes. Cancer Res. 2008, 68, 4525–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Wang, J.; Li, J.; Post, M. Mouse Snail Is a Target Gene for HIF. Mol. Cancer Res. 2011, 9, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.; Choi, H.; Rayes, T.E.; Ryu, S.; Troeger, J.; et al. EMT is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. EMT Program is Dispensable for Metastasis but Induces Chemoresistance in Pancreatic Cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.S.-S.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Chang, S.-Y.; Tsai, T.-L.; Chang, C.-C.; Tzeng, C.-H.; Wu, K.-J.; Kao, J.-Y.; et al. Regulation of Excision Repair Cross-Complementation Group 1 by Snail Contributes to Cisplatin Resistance in Head and Neck Cancer. Clin. Cancer Res. 2010, 16, 4561–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; A Stephens, M.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [Green Version]
- De Mattos-Arruda, L.; Bottai, G.; Nuciforo, P.G.; di Tommaso, L.; Giovannetti, E.; Peg, V.; Losurdo, A.; Pérez-Garcia, J.; Masci, G.; Corsi, F.; et al. MicroRNA-21 links epithelial-to-mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2-positive breast cancer patients. Oncotarget 2015, 6, 37269–37280. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; de Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell–Mediated Lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. OncoImmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Eaton, E.N.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nat. Cell Biol. 2015, 525, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celià-Terrassa, T.; Meca-Cortés, Ó.; Mateo, F.; de Paz, A.M.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef] [Green Version]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudernaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nat. Cell Biol. 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nat. Cell Biol. 2017, 547, E7–E8. [Google Scholar] [CrossRef]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat. Commun. 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, H.; Huang, Y.; Xiang, L.; Chen, Z.; Fang, Y.; Lin, Y.; Cui, Z.; Yu, S.; Li, X.; Yang, D. Circulating Tumor Cell Phenotype Indicates Poor Survival and Recurrence After Surgery for Hepatocellular Carcinoma. Dig. Dis. Sci. 2018, 63, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Polioudaki, H.; Agelaki, S.; Chiotaki, R.; Politaki, E.; Mavroudis, D.; Matikas, A.; Georgoulias, V.; Theodoropoulos, P.A. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer 2015, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to Mesenchymal Transition Is Mechanistically Linked with Stem Cell Signatures in Prostate Cancer Cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef]
- Wellner, U.F.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Hwang, W.-L.; Jiang, J.-K.; Yang, S.-H.; Huang, T.-S.; Lan, H.-Y.; Teng, H.-W.; Yang, C.-Y.; Tsai, Y.-P.; Lin, C.-H.; Yang, M.-H. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef]
- Biddle, A.; Liang, X.; Gammon, L.; Fazil, B.; Harper, L.J.; Emich, H.; Costea, D.E.; MacKenzie, I.C. Cancer Stem Cells in Squamous Cell Carcinoma Switch between Two Distinct Phenotypes That Are Preferentially Migratory or Proliferative. Cancer Res. 2011, 71, 5317–5326. [Google Scholar] [CrossRef] [Green Version]
- Malek, R.; Wang, H.; Taparra, K.; Tran, P.T. Therapeutic Targeting of Epithelial Plasticity Programs: Focus on the Epithelial-Mesenchymal Transition. Cells Tissues Organs 2017, 203, 114–127. [Google Scholar] [CrossRef]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFβ receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Giannelli, G.; Santoro, A.; Kelley, R.K.; Gane, E.; Paradis, V.; Cleverly, A.; Smith, C.; Estrem, S.T.; Man, M.; Wang, S.; et al. Biomarkers and overall survival in patients with advanced hepatocellular carcinoma treated with TGF-βRI inhibitor galunisertib. PLoS ONE 2020, 15, e0222259. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncology 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formenti, S.C.; Hawtin, R.E.; Dixit, N.; Evensen, E.; Lee, P.; Goldberg, J.D.; Li, X.; Vanpouille-Box, C.; Schaue, D.; McBride, W.H.; et al. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J. Immunother. Cancer 2019, 7, 177. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Szczésna, A.; Juhász, E.; Esteban, E.; Molinier, O.; Brugger, W.; Melezínek, I.; et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: A multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010, 11, 521–529. [Google Scholar] [CrossRef]
- Goto, Y.; Zeng, L.; Yeom, C.J.; Zhu, Y.; Morinibu, A.; Shinomiya, K.; Kobayashi, M.; Hirota, K.; Itasaka, S.; Yoshimura, M.; et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1α. Nat. Commun. 2015, 6, 6153. [Google Scholar] [CrossRef] [Green Version]
- Arima, Y.; Hayashi, H.; Sasaki, M.; Hosonaga, M.; Goto, T.M.; Chiyoda, T.; Kuninaka, S.; Shibata, T.; Ohata, H.; Nakagama, H.; et al. Induction of ZEB Proteins by Inactivation of RB Protein Is Key Determinant of Mesenchymal Phenotype of Breast Cancer. J. Biol. Chem. 2012, 287, 7896–7906. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of STAT3 in cancer. Sci. Rep. 2016, 5, 17663. [Google Scholar] [CrossRef] [Green Version]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W.; et al. Phase I study of OPB -51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef]
- Ardiani, A.; Gameiro, S.R.; Palena, C.; Hamilton, D.H.; Kwilas, A.; King, T.H.; Schlom, J.; Hodge, J.W. Vaccine-mediated immunotherapy directed against a transcription factor driving the metastatic process. Cancer Res. 2014, 74, 1945–1957. [Google Scholar] [CrossRef] [Green Version]
- Heery, C.R.; Singh, B.H.; Rauckhorst, M.; Marté, J.L.; Donahue, R.N.; Grenga, I.; Rodell, T.C.; Dahut, W.L.; Arlen, P.M.; Madan, R.A.; et al. Phase I Trial of a Yeast-Based Therapeutic Cancer Vaccine (GI-6301) Targeting the Transcription Factor Brachyury. Cancer Immunol. Res. 2015, 3, 1248–1256. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Ezzeldin, H.H.; Diasio, R.B. Histone deacetylase inhibitors: Current status and overview of recent clinical trials. Drugs 2009, 69, 1911–1934. [Google Scholar] [CrossRef]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes That Promote | ||
|---|---|---|
| EMT | MET | |
| Cell surface programs | α5β1 integrin | L-CAM; E-cadherin; α6 integrin; Syndecan 1; Laminin/nidogen |
| Oncogenes | v-src; c-fos; v-ras; v-mos | c-met |
| Growth factors | TGF-β1-3; MIF; TGF-α; aFGF | HGF/SF |
| Other genes | wnt-1; wnt-4; pax 2; E1a | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachat, C.; Peixoto, P.; Hervouet, E. Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules 2021, 11, 782. https://doi.org/10.3390/biom11060782
Lachat C, Peixoto P, Hervouet E. Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules. 2021; 11(6):782. https://doi.org/10.3390/biom11060782
Chicago/Turabian StyleLachat, Camille, Paul Peixoto, and Eric Hervouet. 2021. "Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers" Biomolecules 11, no. 6: 782. https://doi.org/10.3390/biom11060782
APA StyleLachat, C., Peixoto, P., & Hervouet, E. (2021). Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules, 11(6), 782. https://doi.org/10.3390/biom11060782