
mRNA-Based Anti-TCR CDR3 Tumour Vaccine for T-Cell Lymphoma

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Production of Ivt mRNA
2.2. Cells and FACS
2.3. Patient and T Cell Receptor (TCR) Clonality Assessment by Flow Cytometry
2.4. Animals and In Vivo Experiments
3. Results
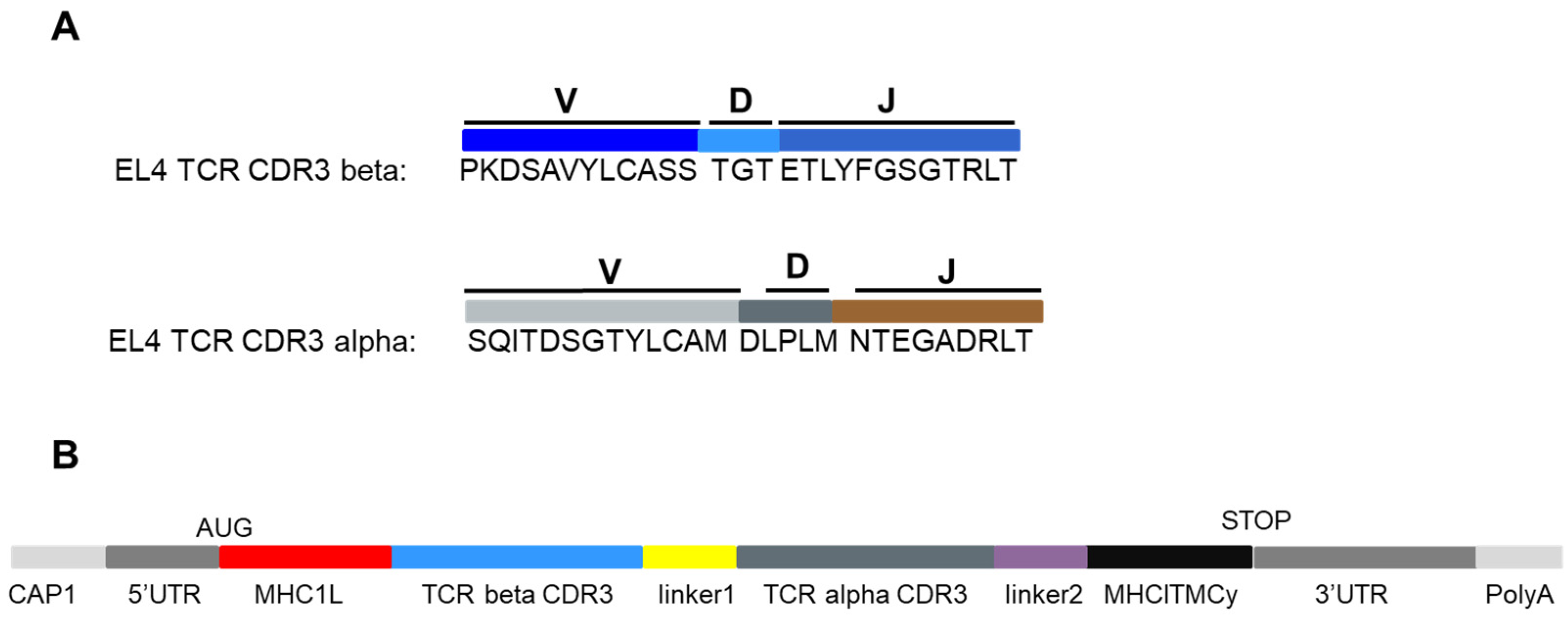
3.1. Design of an Ivt mRNA Coding for the TCR CDR3 Regions
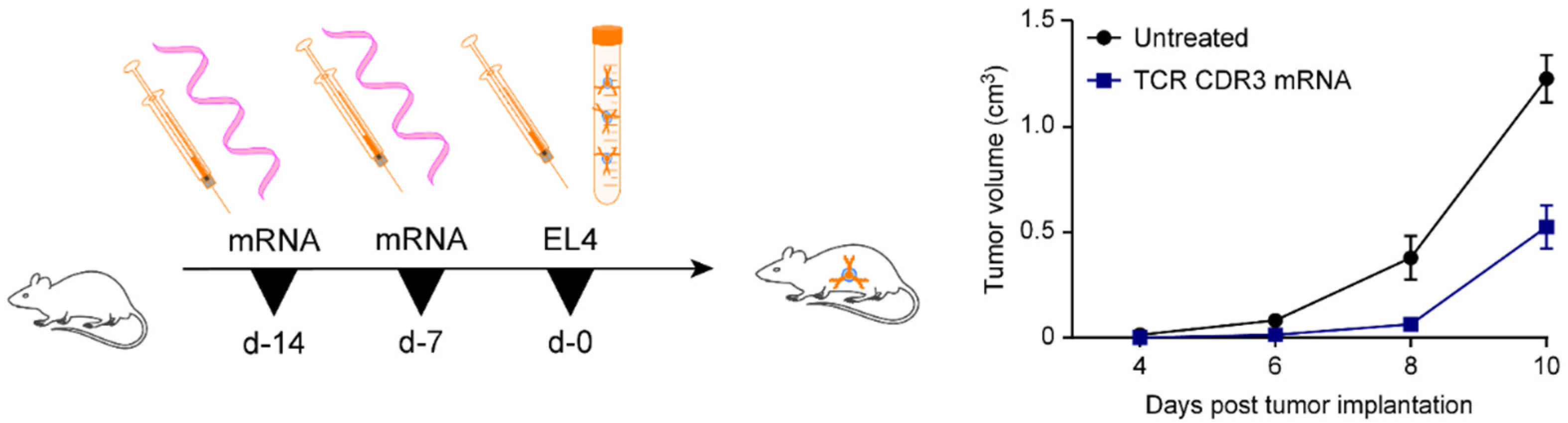
3.2. Administration of CDR3-Encoding mRNAs Induces Resistance towards EL4
3.3. Retention of the TCR by CTCL
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A



References
- Pascolo, S. Messenger RNA-based vaccines. Expert Opin. Biol. Ther. 2004, 4, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S. Vaccination with messenger RNA. In DNA Vaccines; Methods in Molecular Medicine; Humana Press: Totowa, NJ, USA, 2006; Volume 127, pp. 23–40. [Google Scholar] [CrossRef]
- Pascolo, S. Vaccination with messenger RNA (mRNA). In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 221–235. [Google Scholar] [CrossRef]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Carralot, J.P.; Probst, J.; Hoerr, I.; Scheel, B.; Teufel, R.; Jung, G.; Rammensee, H.G.; Pascolo, S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 2004, 61, 2418–2424. [Google Scholar] [CrossRef]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In Vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Rittig, S.M.; Haentschel, M.; Weimer, K.J.; Heine, A.; Muller, M.R.; Brugger, W.; Horger, M.S.; Maksimovic, O.; Stenzl, A.; Hoerr, I.; et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol. Ther. 2011, 19, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Rittig, S.M.; Haentschel, M.; Weimer, K.J.; Heine, A.; Müller, M.R.; Brugger, W.; Horger, M.S.; Maksimovic, O.; Stenzl, A.; Hoerr, I.; et al. Long-term survival correlates with immunological responses in renal cell carcinoma patients treated with mRNA-based immunotherapy. Oncoimmunology 2016, 5, e1108511. [Google Scholar] [CrossRef] [Green Version]
- Weide, B.; Carralot, J.P.; Reese, A.; Scheel, B.; Eigentler, T.K.; Hoerr, I.; Rammensee, H.G.; Garbe, C.; Pascolo, S. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J. Immunother. 2008, 31, 180–188. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. (Hagerstown, Md.: 1997). 2009, 32, 498–507. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; Van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Probst, J.; Weide, B.; Scheel, B.; Pichler, B.J.; Hoerr, I.; Rammensee, H.G.; Pascolo, S. Spontaneous cellular uptake of exogenous messenger RNA In Vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther. 2007, 14, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Pascolo, S. Messenger RNA: The Inexpensive Biopharmaceutical. J. Multidiscip. Eng. Sci. Technol. 2017, 4, 6937–6941. [Google Scholar]
- Heesen, L.; Frenzel, K.; Bolte, S.; Bukur, V.; Diken, M.; Derhovanessian, E.; Kreiter, S.; Kuhn, A.; Kühlcke, K.; Löwer, M.; et al. Mutanome engineered RNA immuno-therapy (MERIT) for patients with triple negative breast cancer (TNBC). Ann. Oncol. 2018, 29, viii86. [Google Scholar] [CrossRef]
- Vormehr, M.; Schrörs, B.; Boegel, S.; Löwer, M.; Türeci, Ö.; Sahin, U. Mutanome Engineered RNA Immunotherapy: Towards Patient-Centered Tumor Vaccination. J. Immunol. Res. 2015, 595363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vormehr, M.; Tureci, O.; Sahin, U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu. Rev. Med. 2019, 70, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, C.; Ilias, W.; Tahar, O.; Güler, Y.; Miguet, L.; Mayeur-Rousse, C.; Mauvieux, L.; Fornecker, L.M.; Toussaint, E.; Herbrecht, R.; et al. BCR-associated factors driving chronic lymphocytic leukemia cells proliferation ex vivo. Sci. Rep. 2019, 9, 701. [Google Scholar] [CrossRef]
- Lopez-Requena, A.; Burrone, O.R.; Cesco-Gaspere, M. Idiotypes as immunogens: Facing the challenge of inducing strong therapeutic immune responses against the variable region of immunoglobulins. Front. Oncol. 2012, 2, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tusup, M.; French, L.E.; Guenova, E.; Kundig, T.M.; Pascolo, S. Optimizing the functionality of In Vitro—Transcribed mRNA. Biomed. J. Sci. Tech. Res. 2018, 1, 6. [Google Scholar]
- Scheel, B.; Braedel, S.; Probst, J.; Carralot, J.P.; Wagner, H.; Schild, H.; Jung, G.; Rammensee, H.G.; Pascolo, S. Immunostimulating capacities of stabilized RNA molecules. Eur. J. Immunol. 2004, 34, 537–547. [Google Scholar] [CrossRef]
- Scheel, B.; Teufel, R.; Probst, J.; Carralot, J.P.; Geginat, J.; Radsak, M.; Jarrossay, D.; Wagner, H.; Jung, G.; Rammensee, H.G.; et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Tusup, M.; Kundig, T.; Pascolo, S. An eIF4G-recruiting aptamer increases the functionality of In Vitro transcribed mRNA. EPH Int. J. Med. Health Sci. 2018, 4. [Google Scholar]
- von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3′ UTRs identified by cellular library screening. Mol. Ther. 2018, 27, 824–836. [Google Scholar] [CrossRef] [Green Version]
- van Hall, T.; van Bergen, J.; van Veelen, P.A.; Kraakman, M.; Heukamp, L.C.; Koning, F.; Melief, C.J.; Ossendorp, F.; Offringa, R. Identification of a novel tumor-specific CTL epitope presented by RMA, EL-4, and MBL-2 lymphomas reveals their common origin. J. Immunol. 2000, 165, 869–877. [Google Scholar] [CrossRef]
- Reddy Chichili, V.P.; Kumar, V.; Sivaraman, J. Linkers in the structural biology of protein-protein interactions. Protein Sci. 2013, 22, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug. Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Türeci, Ö.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lu, Y.; Polk, A.; Chowdhury, P.; Zamalloa, C.M.; Fujiwara, H.; Suemori, K.; Beyersdorf, N.; Hristov, A.C.; Lim, M.S.; et al. T-cell receptor signaling activates an ITK/NF-kappaB/GATA-3 axis in T-cell lymphomas facilitating resistance to chemotherapy. Clin. Cancer Res. 2017, 23, 2506–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenova, E.; Ignatova, D.; Chang, Y.T.; Contassot, E.; Mehra, T.; Saulite, I.; Navarini, A.A.; Mitev, V.; Dummer, R.; Kazakov, D.V.; et al. Expression of CD164 on malignant T cells in Sezary syndrome. Acta Derm. Venereol. 2016, 96, 464–467. [Google Scholar] [CrossRef]
- De Masson, A.; O’Malley, J.T.; Elco, C.P.; Garcia, S.S.; Divito, S.J.; Lowry, E.L.; Tawa, M.; Fisher, D.C.; Devlin, P.M.; Teague, J.E.; et al. High-throughput sequencing of the T cell receptor beta gene identifies aggressive early-stage mycosis fungoides. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Moczko, A.; Dimitriou, F.; Kresbach, H.; Amarov, B.; Hoetzenecker, W.; Pascolo, S.; Anzengruber, F.; Koch, T.; Duda, A.; Guenova, E. Sensitivity and specificity of T-cell receptor PCR BIOMED-2 clonality analysis for the diagnosis of cutaneous T-cell lymphoma. Eur. J. Dermatol. 2020, 30, 12–15. [Google Scholar] [CrossRef]
- Gonthier, M.; Llobera, R.; Arnaud, J.; Rubin, B. Self-reactive T cell receptor-reactive CD8+ T cells inhibit T cell lymphoma growth In Vivo. J. Immunol. 2004, 173, 7062–7069. [Google Scholar] [CrossRef] [Green Version]
- Khodadoust, M.S.; Rook, A.H.; Porcu, P.; Foss, F.; Moskowitz, A.J.; Shustov, A.; Shanbhag, S.; Sokol, L.; Fling, S.P.; Ramchurren, N.; et al. Pembrolizumab in relapsed and refractory mycosis fungoides and Sezary syndrome: A multicenter mhase II study. J. Clin. Oncol. 2020, 38, 20–28. [Google Scholar] [CrossRef]
- Anzengruber, F.; Ignatova, D.; Schlaepfer, T.; Chang, Y.T.; French, L.E.; Pascolo, S.; Contassot, E.; Bobrowicz, M.; Hoetzenecker, W.; Guenova, E. Divergent LAG-3 versus BTLA, TIGIT, and FCRL3 expression in Sezary syndrome. Leuk. Lymphoma 2019, 60, 1899–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrowicz, M.; Zagozdzon, R.; Domagala, J.; Vasconcelos-Berg, R.; Guenova, E.; Winiarska, M. Monoclonal antibodies in dermatooncology—State of the art and future perspectives. Cancers 2019, 11, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saulite, I.; Ignatova, D.; Chang, Y.T.; Fassnacht, C.; Dimitriou, F.; Varypataki, E.; Anzengruber, F.; Nägeli, M.; Cozzio, A.; Dummer, R.; et al. Blockade of programmed cell death protein 1 (PD-1) in Sézary syndrome reduces Th2 phenotype of non-tumoral T lymphocytes but may enhance tumor proliferation. OncoImmunology 2020, 9, 1738797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerschner, M.; Pekar-Lukacs, A.; Messerli-Odermatt, O.; Dommann-Scherrer, C.; Rütti, M.; Müller, A.M.; Nair, G.; Kamarachev, J.; Kerl, K.; Beer, M.; et al. Interferon alfa-2a maintenance after salvage autologous stem cell transplantation in atypical mycosis fungoides with central nervous system involvement. Br. J. Dermatol. 2019, 181, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilson, D.; Whittaker, S.J.; Child, F.J.; Scarisbrick, J.J.; Illidge, T.M.; Parry, E.J.; Mohd Mustapa, M.F.; Exton, L.S.; Kanfer, E.; Rezvani, K.; et al. British Association of Dermatologists and UK Cutaneous Lymphoma Group guidelines for the management of primary cutaneous lymphomas 2018. Br. J. Dermatol. 2019, 180, 496–526. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.L.; Longley, J.; Hanlon, D.; Girardi, M.; Edelson, R. The clonotypic T cell receptor is a source of tumor-associated antigens in cutaneous T cell lymphoma. Ann. N. Y. Acad. Sci. 2001, 941, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Müller-Berghaus, J.; Nguyen, X.D.; Klüter, H.; Schönhaber, H.; Song, M.; Schwinn, N.; Schadendorf, D.; Goerdt, S.; Eichmüller, S.; et al. Identification of HLA class I dependent immunogenic peptides from clonotypic TCRbeta expressed in cutaneous T-cell lymphoma. Int. J. Cancer 2006, 119, 2476–2480. [Google Scholar] [CrossRef] [PubMed]
- Winter, D.; Fiebiger, E.; Meraner, P.; Auer, H.; Brna, C.; Strohal, R.; Trautinger, F.; Knobler, R.; Fischer, G.F.; Stingl, G.; et al. Definition of TCR epitopes for CTL-mediated attack of cutaneous T cell lymphoma. J. Immunol. 2003, 171, 2714–2724. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Recher, M.; Junt, T.; Navarini, A.A.; Harris, N.L.; Freigang, S.; Odermatt, B.; Conrad, C.; Ittner, L.M.; Bauer, S.; et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat. Med. 2005, 11, 138–145. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tusup, M.; Läuchli, S.; Jarzebska, N.T.; French, L.E.; Chang, Y.-T.; Vonow-Eisenring, M.; Su, A.; Kündig, T.M.; Guenova, E.; Pascolo, S. mRNA-Based Anti-TCR CDR3 Tumour Vaccine for T-Cell Lymphoma. Pharmaceutics 2021, 13, 1040. https://doi.org/10.3390/pharmaceutics13071040
Tusup M, Läuchli S, Jarzebska NT, French LE, Chang Y-T, Vonow-Eisenring M, Su A, Kündig TM, Guenova E, Pascolo S. mRNA-Based Anti-TCR CDR3 Tumour Vaccine for T-Cell Lymphoma. Pharmaceutics. 2021; 13(7):1040. https://doi.org/10.3390/pharmaceutics13071040
Chicago/Turabian StyleTusup, Marina, Severin Läuchli, Natalia Teresa Jarzebska, Lars E. French, Yun-Tsan Chang, Maya Vonow-Eisenring, Andreas Su, Thomas M. Kündig, Emmanuella Guenova, and Steve Pascolo. 2021. "mRNA-Based Anti-TCR CDR3 Tumour Vaccine for T-Cell Lymphoma" Pharmaceutics 13, no. 7: 1040. https://doi.org/10.3390/pharmaceutics13071040
APA StyleTusup, M., Läuchli, S., Jarzebska, N. T., French, L. E., Chang, Y.-T., Vonow-Eisenring, M., Su, A., Kündig, T. M., Guenova, E., & Pascolo, S. (2021). mRNA-Based Anti-TCR CDR3 Tumour Vaccine for T-Cell Lymphoma. Pharmaceutics, 13(7), 1040. https://doi.org/10.3390/pharmaceutics13071040