Microwave Assisted Reactions of Some Azaheterocylic Compounds


Abstract
:Introduction
Results and Discussion



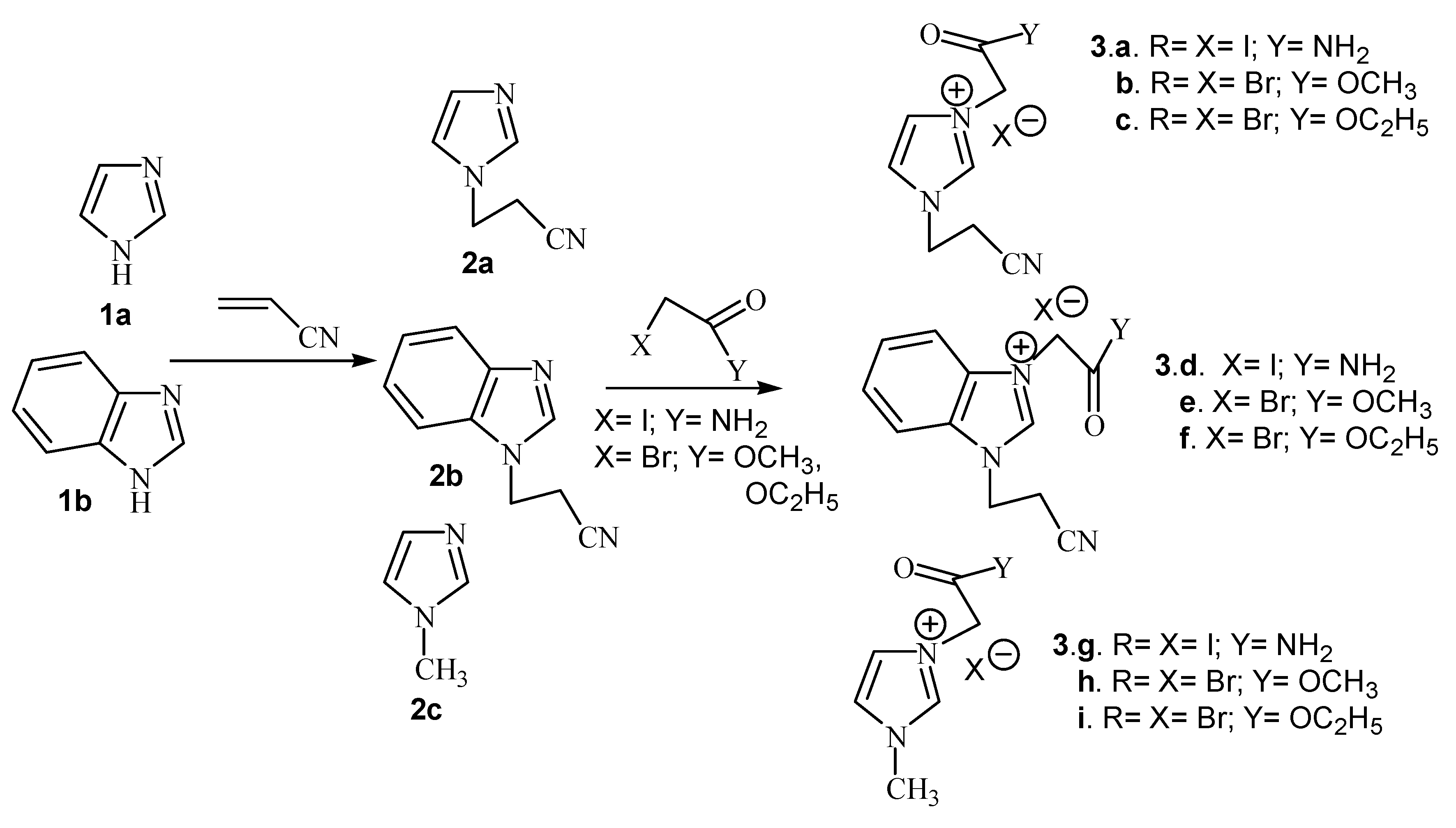{kind=link}
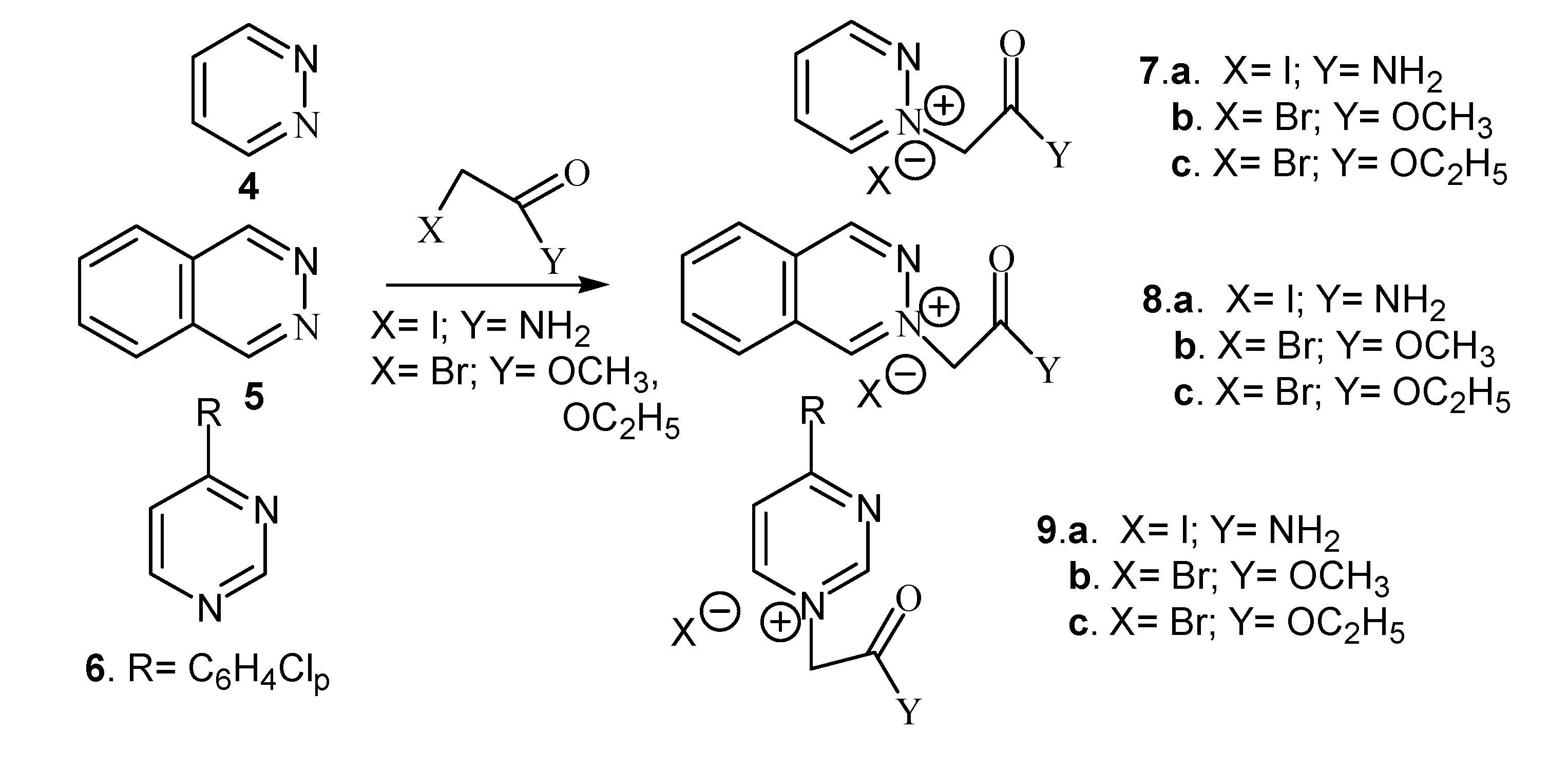{kind=link}
| Compound | Classical | Microwaves | ||||
|---|---|---|---|---|---|---|
| Reaction time | Reaction temp., oC | Yields, % | Reaction time | Reaction temp., oC | Yields, % | |
| 3a. X=I;Y= NH2 | 20 | 56 | 92 | 10 min. | 56 | 92 |
| 3b. X=Br;Y= OCH3 | 20 | 56 | 84 | 10 min. | 56 | 87 |
| 3c. X=Br;Y= OC2H5 | 20 | 56 | 92 | 10 min. | 56 | 94 |
| 3d. X=I;Y= NH2 | 25 | 56 | 60 | 15 min. | 56 | 82 |
| 3e. X=Br;Y= OCH3 | 25 | 56 | 59 | 15 min. | 56 | 86 |
| 3f. X=Br;Y= OC2H5 | 25 | 56 | 87 | 15 min. | 56 | 88 |
| 3g. X=I;Y= NH2 | 20 | 56 | 88 | 10 min. | 56 | 88 |
| 3h. X=Br;Y= OCH3 | 20 | 56 | 70 | 10 min. | 56 | 87 |
| 3i. X=Br;Y= OC2H5 | 20 | 56 | 90 | 10 min. | 56 | 91 |
| 7a. X=I;Y= NH2 | 3 h | 80 | 86 | 5 min. | 70 | 89 |
| 7b. X=Br;Y= OCH3 | 4 h | 80 | 91 | 5 min. | 70 | 92 |
| 7c. X=Br;Y= OC2H5 | 3 h | 80 | 94 | 5 min. | 70 | 94 |
| 8a. X=I;Y= NH2 | 24 h | r.t. | 74 | 5 min. | 65 | 78 |
| 8b. X=Br;Y= OCH3 | 32 h | r.t. | 89 | 5 min. | 65 | 89 |
| 8c.X=Br;Y= OC2H5 | 24 h | r.t | 83 | 5 min. | 65 | 86 |
| 9a. X=I;Y= NH2 | 32 h | 112 | 70 | 10 min. | 80 | 74 |
| 9b. X=Br;Y= OCH3 | 41 h | 112 | 68 | 10 min. | 80 | 81 |
| 9c. X=Br;Y= OC2H5 | 32 h | 112 | 66 | 10 min. | 80 | 80 |
Conclusions
Experimental
General
General procedure for synthesis of imidazolium salts 3 under classical heating
General procedure for syntheses of diazine salts 7-9 under classical heating
General procedure for syntheses of imidazolium (3) and diazine salts (7-9) under MW irradiation
Acknowledgements
References
- Loupy, A. Microwaves in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Kappe, O.C.; Stadler, A. Microwaves in Organic and Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Tierney, J.T.; Lindström, P. Microwave-assisted Organic Synthesis; Blackwell: Oxford, UK, 2005. [Google Scholar]
- Kappe, O.C. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6248. [Google Scholar] [CrossRef]
- Bogdal, D. Fast solvent-free alkylation of amides and lactams under microwave irradiation. Molecules 1999, 4, 333–337. [Google Scholar] [CrossRef]
- Bogdal, D.; Pielichowski, J.; Jaskott, K. Remarkable Fast N-Alkylation of Azaheterocycles Under Microwave Irradiation in Dry Media. Heterocycles 1997, 45, 715–722. [Google Scholar] [CrossRef]
- Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J.L.; Petit, A. Microwave activation in phase transfer catalysis. Tetrahedron 1999, 55, 10851–10870. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis-A review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Zbancioc, G.N.; Mangalagiu, I.I. Microwave-Assisted Synthesis of Highly Fluorescent Pyrrolopyridazine Derivatives. Synlett. 2006, 5, 804–806. [Google Scholar] [CrossRef]
- Dima, St.; Zbancioc, G.N.; Mangalagiu, I.I. The 1,3-dipolar cycloaddition of 1-methyl-phtalazinium ylides to non-symmetrically activated alkenes on a solid support under microwave irradiation. J. Serb. Chim. Soc. 2006, 71, 103–110. [Google Scholar] [CrossRef]
- Nogrady, T. Medicinal Chemistry; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Dy, G.K.; Adjei, A.A. Systemic cancer therapy: Evolution over the last 60 years. Cancer 2008, 113, 1857–1887. [Google Scholar] [CrossRef]
- Keating, M.J.; Yasothan, U.; Bach, C.; Kirkpatric, P. Bendamustine. Nat. Rev. Drug Discov. 2008, 7, 473–474. [Google Scholar] [CrossRef]
- Andreani, A.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Giorgi, G.; Salvini, L.; Garaliene, V. Synthesis and antitumor activity of substituted 3-(5-imidazo[2,1-b]thiazolylmethylene)-2-indolinones. Anticancer Drug Des. 2001, 16, 167–174. [Google Scholar]
- Dubey, S.; Satyanarayana, Y.D.; Lavania, H. Development of integrase inhibitors for treatment of AIDS: An overview. Eur. J. Med. Chem. 2007, 42, 1159–1168. [Google Scholar] [CrossRef]
- De Clercq, E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [Google Scholar] [CrossRef]
- Ungureanu, M.; Moldoveanu, C.; Poeata, A.; Drochioiu, G.; Petrovanu, M.; Mangalagiu, I.I. New pyrimidine compounds with in vitro antimicrobial or antifungal activity. Ann. Pharm. Fr. 2006, 64, 287–288. [Google Scholar] [CrossRef]
- Caprosu, M.; Butnariu, R.; Mangalagiu, I.I. Synthesis and antimicrobial activity of some new pyridazine derivatives. Heterocycles 2005, 65, 1871–1879. [Google Scholar] [CrossRef]
- Demirayak, S.; Karaburun, A.C.; Beis, R. Some pyrrole substituted aryl pyridazinone and phthalazinone derivatives and their antihypertensive activities. Eur. J. Med. Chem. 2004, 39, 1089–1095. [Google Scholar] [CrossRef]
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Macaev, F.; Munteanu, V.; Stingaci, E.; Barba, A.; Pogrebnoi, S. New room temperature liquids: synthesis and characterization. Chem. J. Moldova 2007, 2, 119–122. [Google Scholar]
- Sample Availability: Samples of all prepared compounds are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zbancioc, G.; Bejan, V.; Risca, M.; Moldoveanu, C.; Mangalagiu, I.I. Microwave Assisted Reactions of Some Azaheterocylic Compounds. Molecules 2009, 14, 403-411. https://doi.org/10.3390/molecules14010403
Zbancioc G, Bejan V, Risca M, Moldoveanu C, Mangalagiu II. Microwave Assisted Reactions of Some Azaheterocylic Compounds. Molecules. 2009; 14(1):403-411. https://doi.org/10.3390/molecules14010403
Chicago/Turabian StyleZbancioc, Gheorghita, Vasilichia Bejan, Marian Risca, Costel Moldoveanu, and Ionel I. Mangalagiu. 2009. "Microwave Assisted Reactions of Some Azaheterocylic Compounds" Molecules 14, no. 1: 403-411. https://doi.org/10.3390/molecules14010403
APA StyleZbancioc, G., Bejan, V., Risca, M., Moldoveanu, C., & Mangalagiu, I. I. (2009). Microwave Assisted Reactions of Some Azaheterocylic Compounds. Molecules, 14(1), 403-411. https://doi.org/10.3390/molecules14010403