MAOS and Medicinal Chemistry: Some Important Examples from the Last Years

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
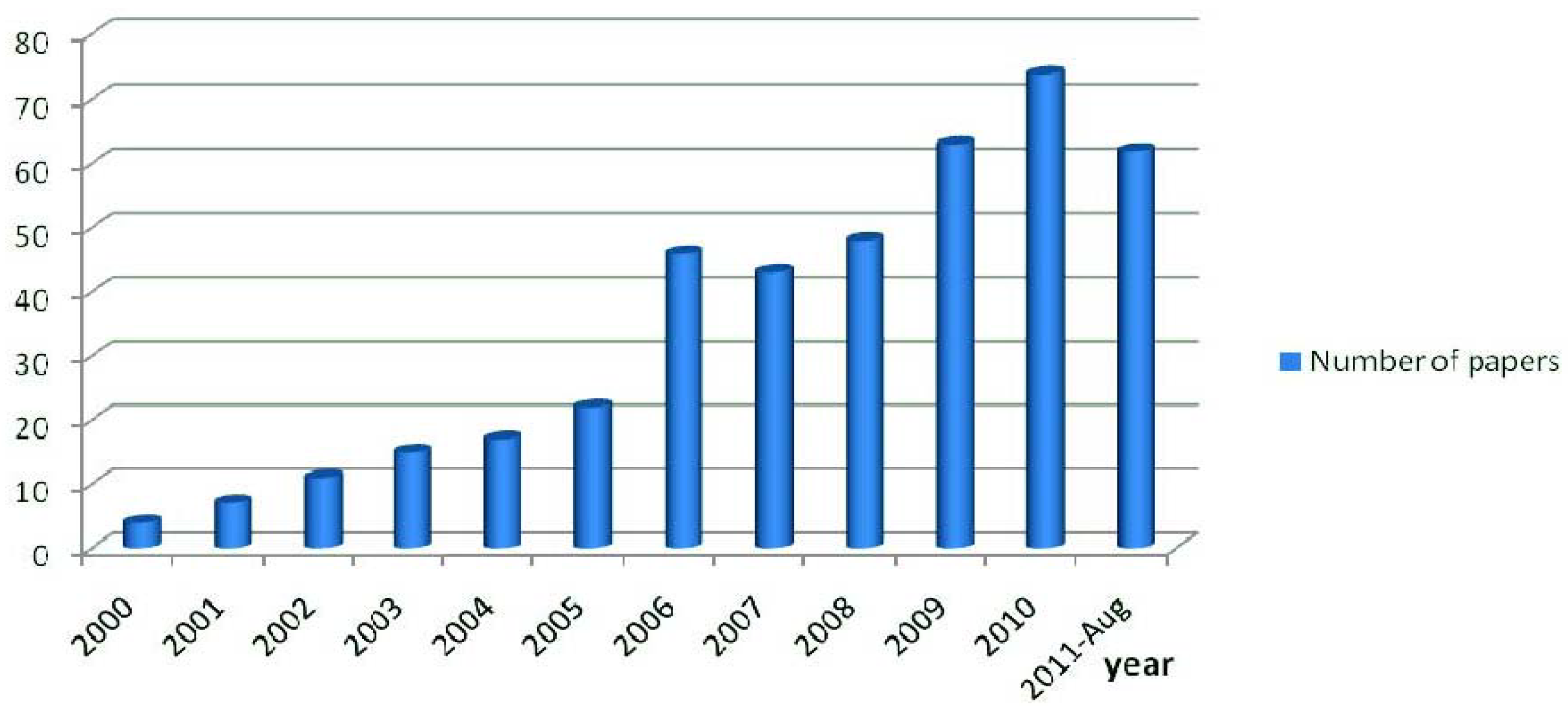
:1. Introduction
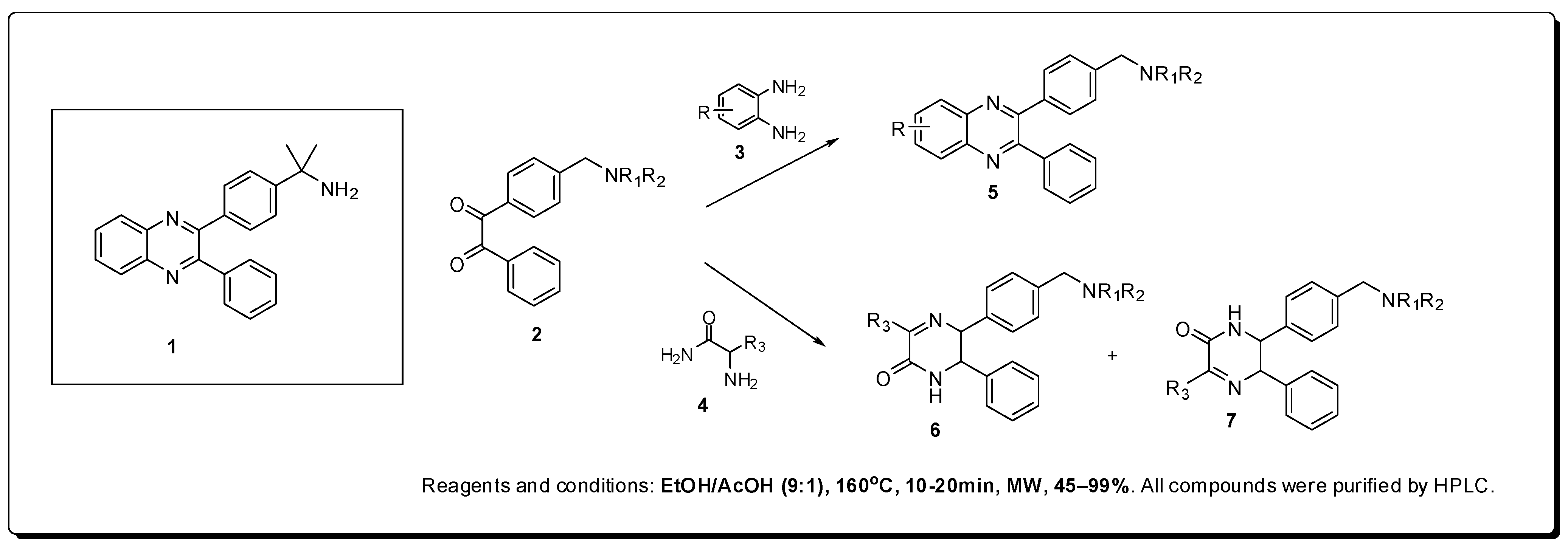
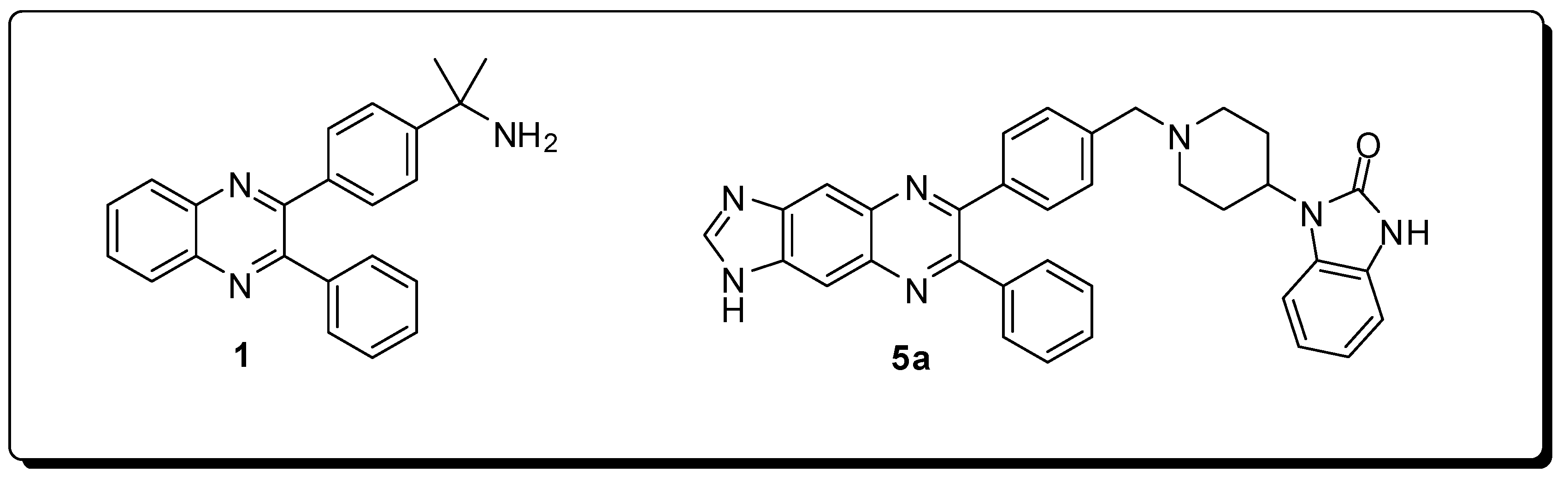
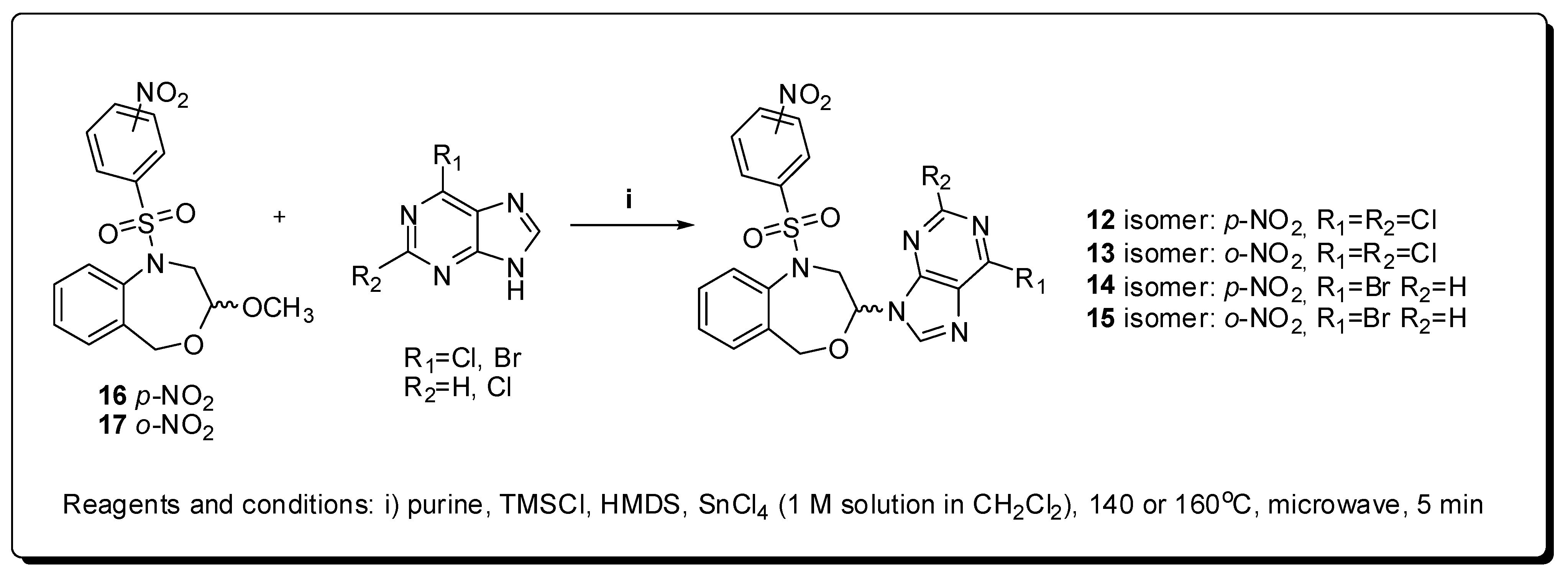
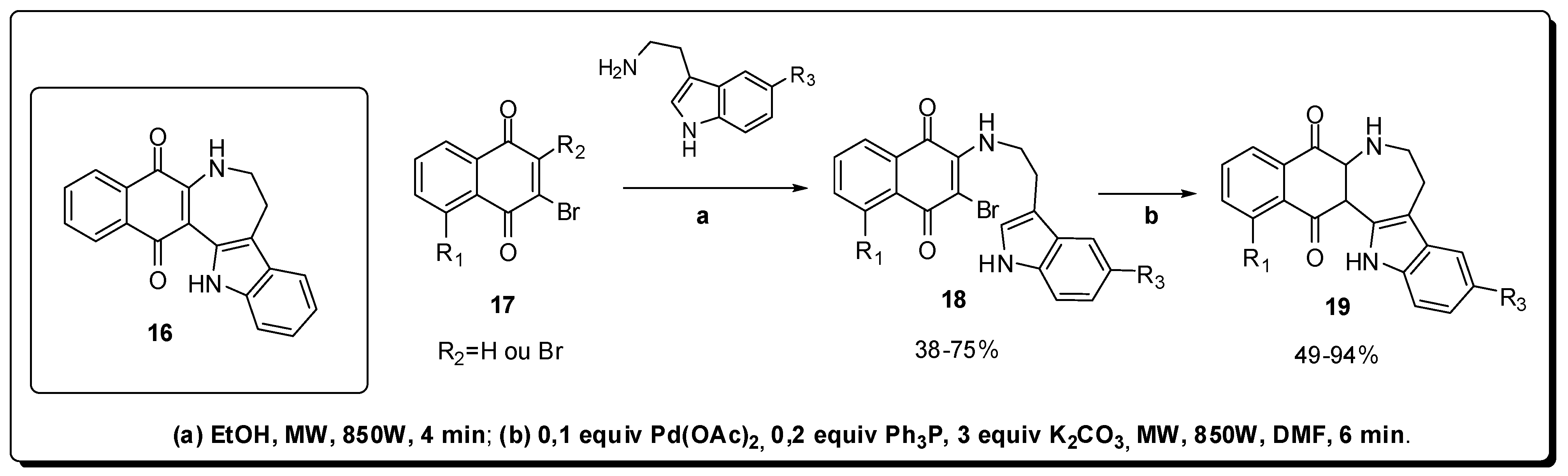
2. Antineoplastics
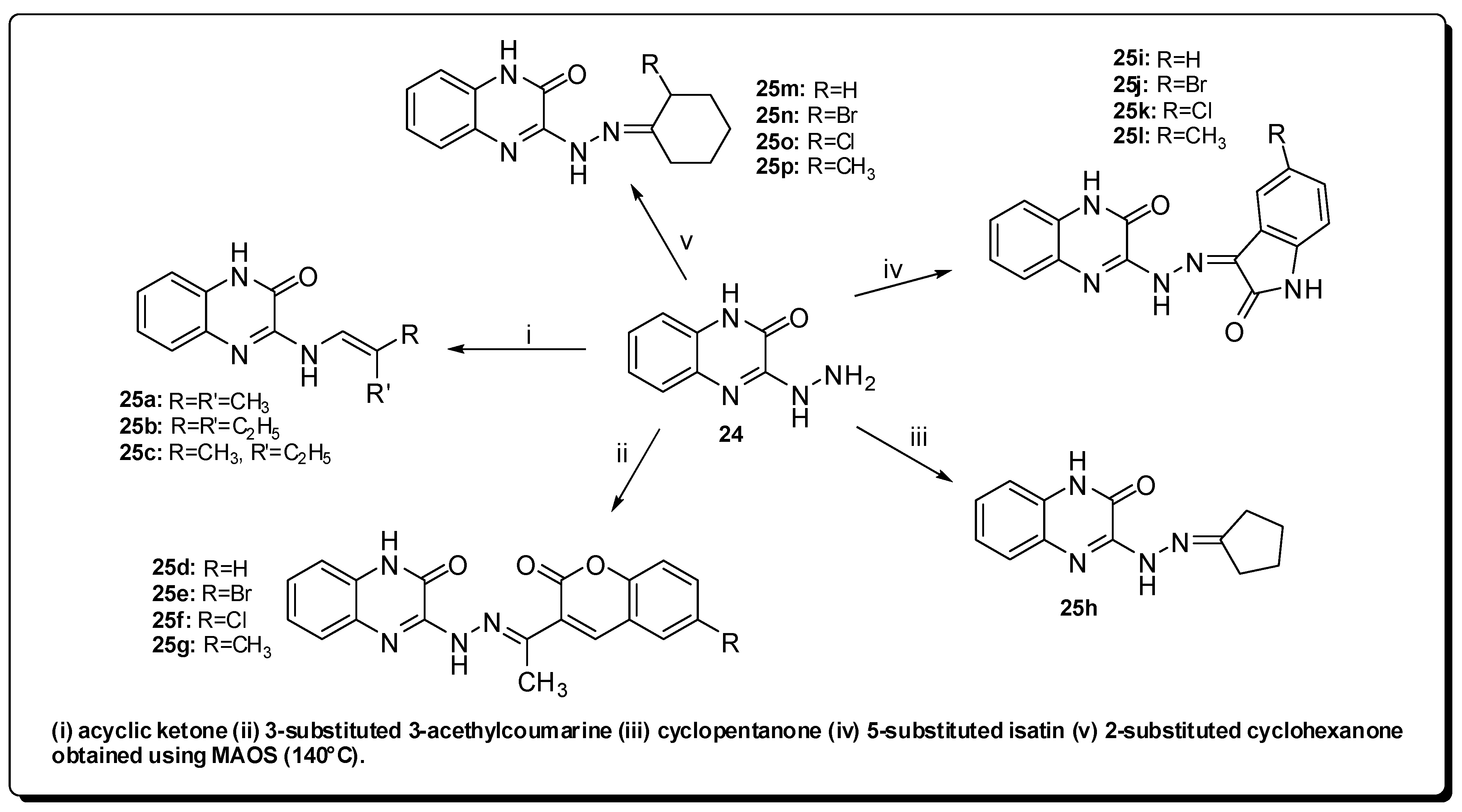
3. Anti-Inflammatory
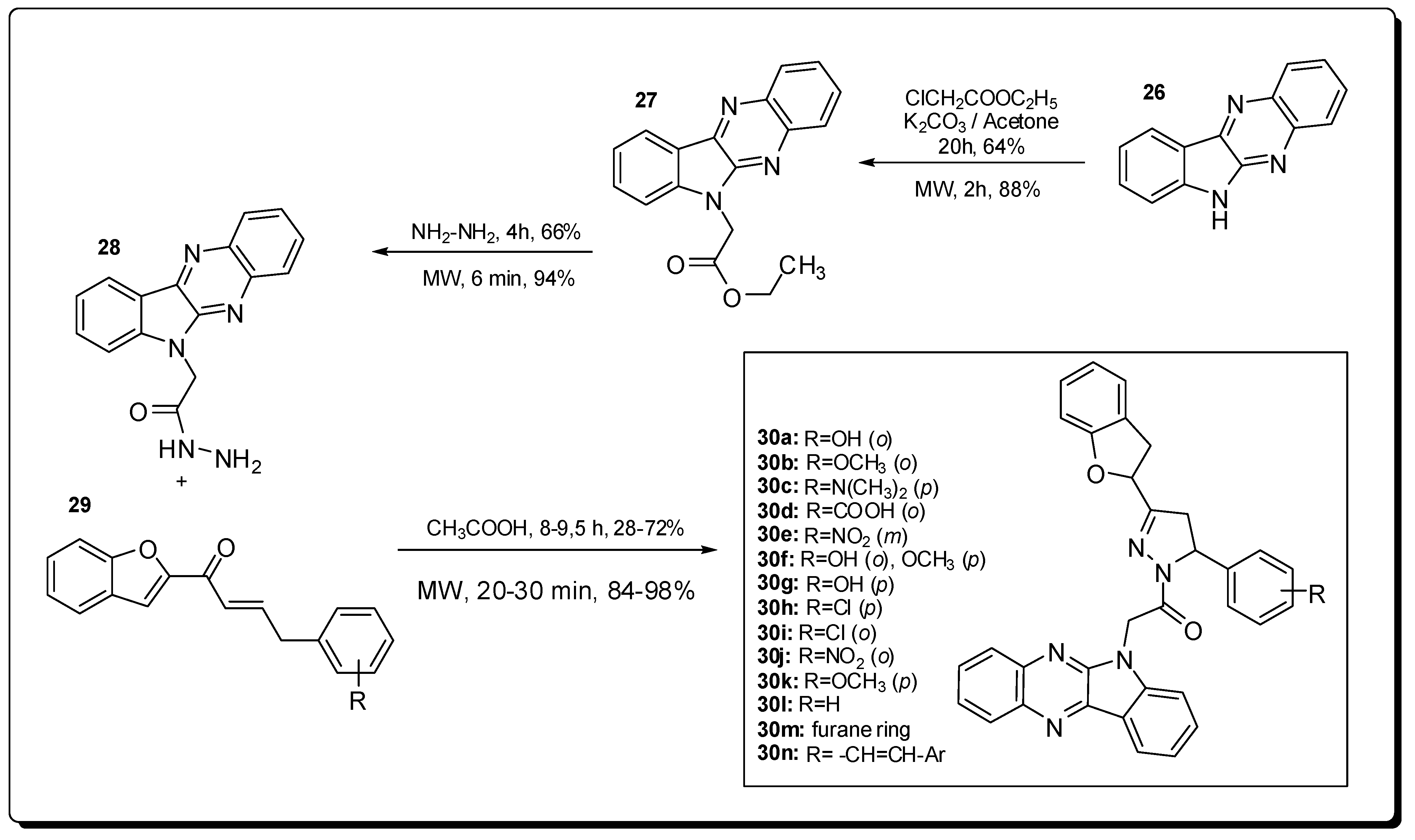
4. Antimicrobial Agents
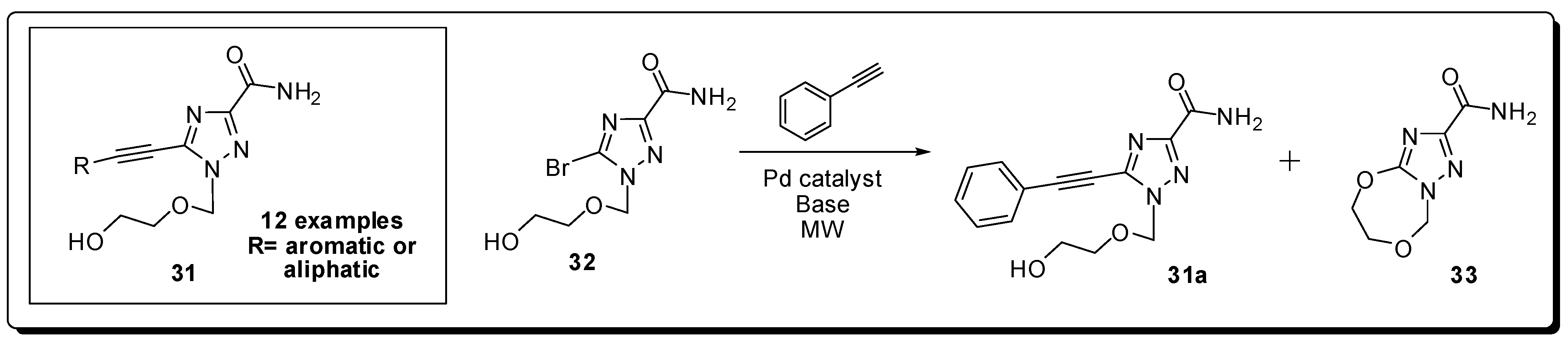
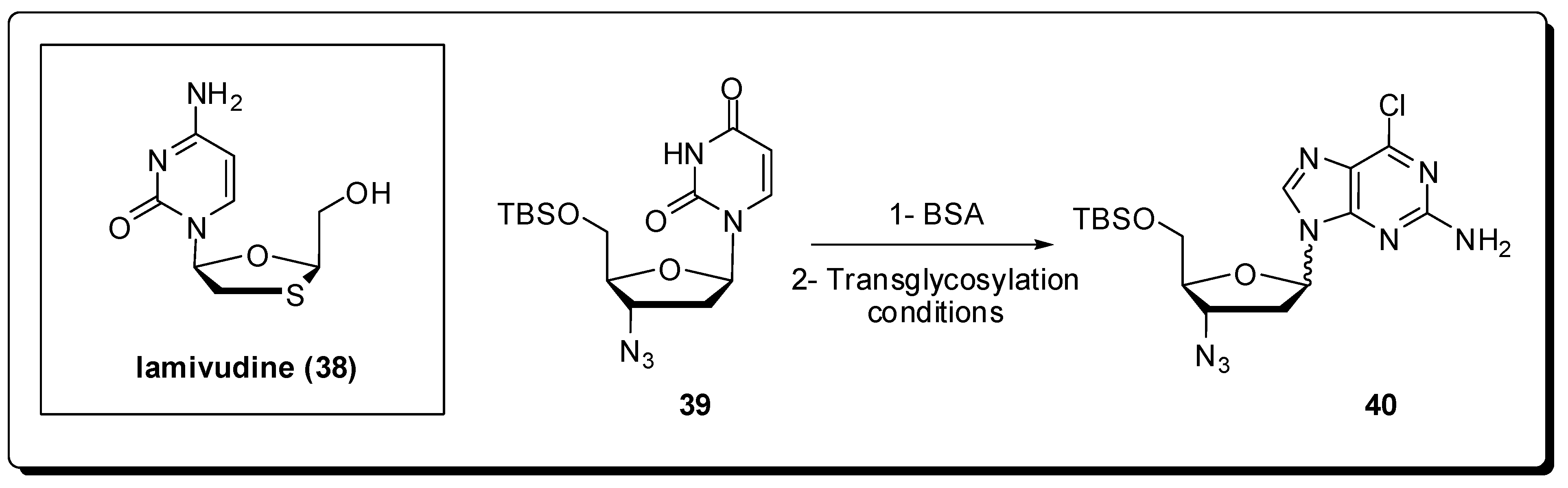
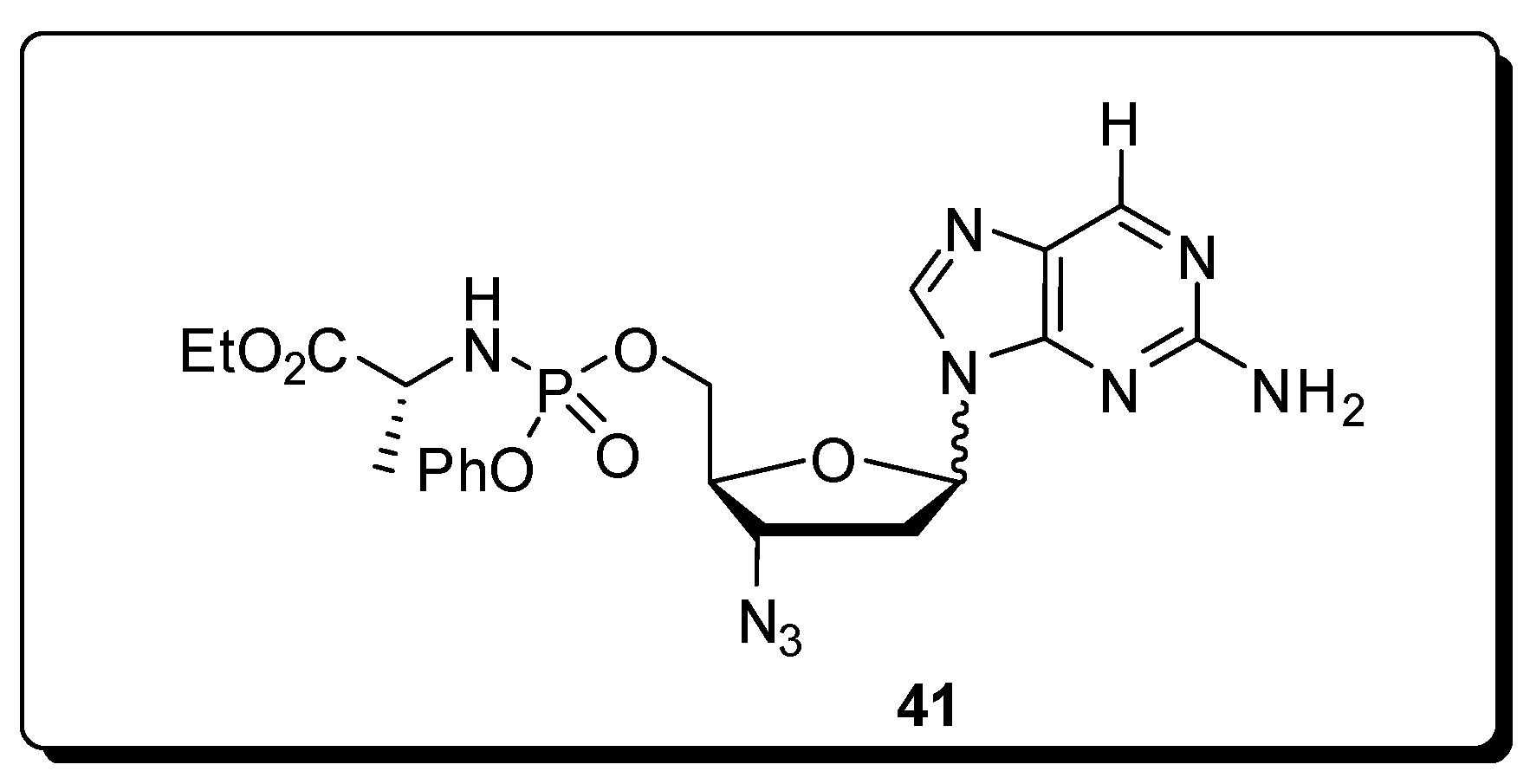
5. Antivirals
6. Neglected Diseases
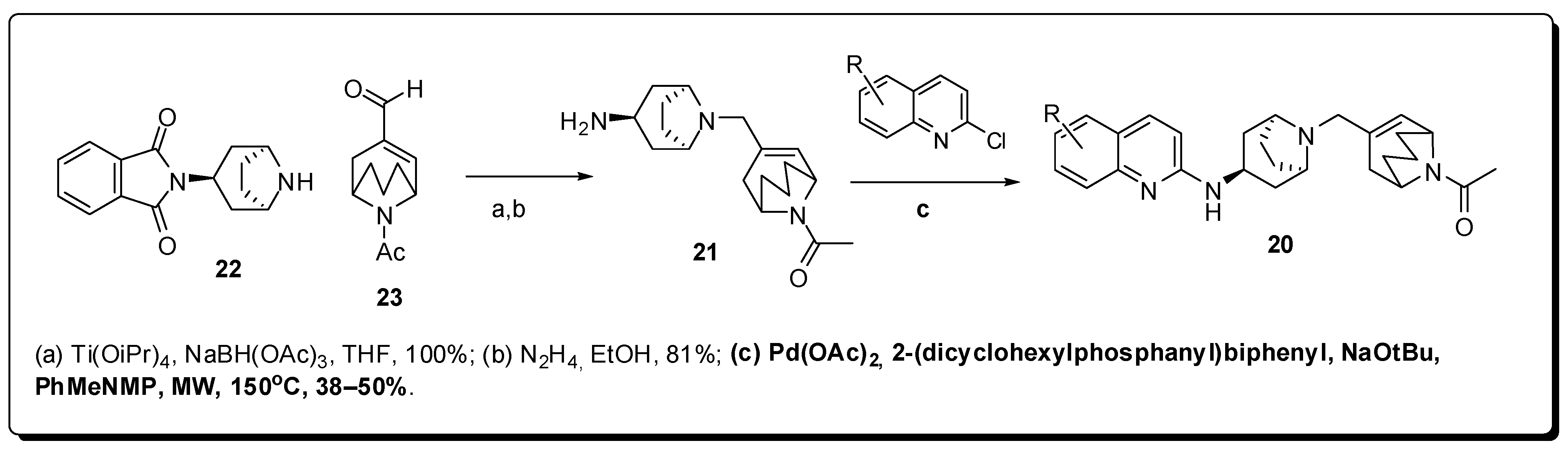
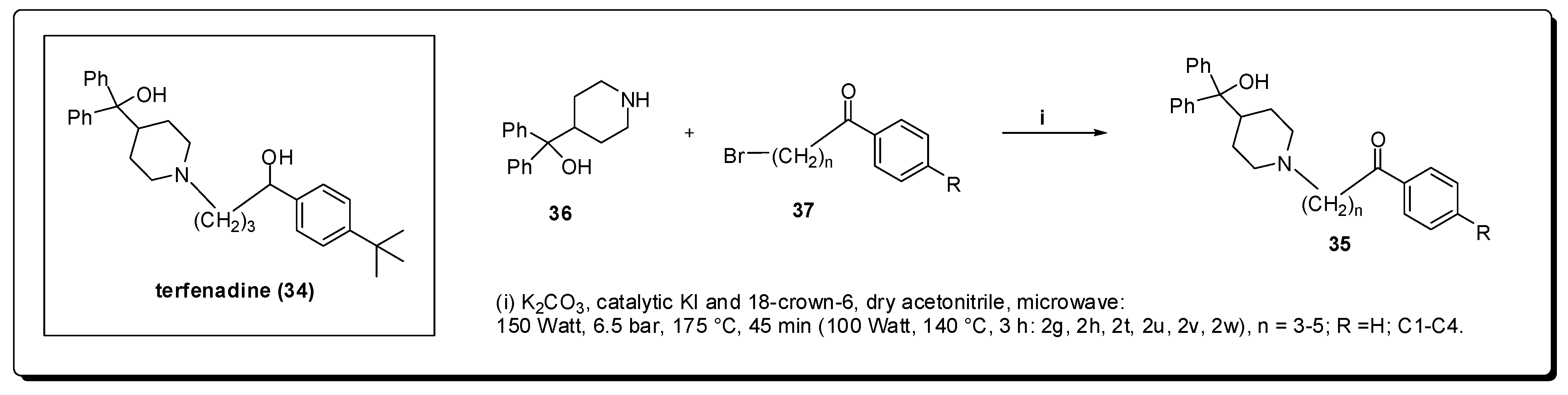
7. Central Nervous System
8. Conclusions
Acknowledgments
References
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis – a review. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Bougrin, K.; Loupy, A.; Soufiaoui, M. Microwave-assisted solvent-free heterocyclic synthesis. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 139–167. [Google Scholar] [CrossRef]
- Pérez-Picaso, L.; Escalante, J.; Olivo, H.F.; Rios, M.Y. Efficient microwave assisted syntheses of 2,5-Diketopiperazines in aqueous media. Molecules 2009, 14, 2836–2849. [Google Scholar] [CrossRef] [PubMed]
- Hügel, H.M. Microwave multicomponent synthesis. Molecules 2009, 14, 4936–4972. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O. High-speed combinatorial synthesis utilizing microwave irradiation. Curr. Opin. Chem. Biol. 2002, 6, 314–320. [Google Scholar] [CrossRef]
- Kong, L.; Lv, X.; Lin, K.; Liu, X.; Zhou, Y.; Jia, Y. Efficient synthesis of imidazoles from aldehydes ans 1,2-Diketones under superheating continuous by using a continuous flow microreactor system under pressure. Org. Proc. Res. Dev. 2010, 14, 902–904. [Google Scholar] [CrossRef]
- Schmink, J.R.; Kormos, C.M.; Devine, W.G.; Leadbeater, N.E. Exploring the scope for scale up of organic chemistry using a large batch microwave reactor. Org. Proc. Res. Dev. 2010, 14, 205–214. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Miller, D.D. Microwave-assisted synthesis of medicinally relevant indoles. Curr. Med. Chem. 2011, 18, 615–637. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wisnoski, D.D.; Wolkenberg, S.E.; Leister, W.H.; Wang, Y.; Lindsley, C.W. General microwave-assisted protocols for the expedient synthesis of quinoxalines and heterocyclic pyrazines. Tetrahedron Lett. 2004, 45, 4873–4876. [Google Scholar] [CrossRef]
- Chilin, A.; Marzaro, G.; Zanatta, S.; Guiotto, A. A microwave improvement in the synthesis of the quinazoline scaffold. Tetrahedron Lett. 2007, 48, 3229–3231. [Google Scholar] [CrossRef]
- Reichelt, A.; Filsey, J.R.; Rzasa, R.M.; Thiel, O.R.; Achmatowicz, M.M.; Larsen, R.D.; Zhang, D. Palladium-catalyzed chemoselective monoarylation of hydrazides for the synthesis of [1,2,4]triazolo[4,3-a]pyridines. Org. Lett. 2009, 12, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Vergnaud, J.; Teste, K.; Zerrouk, R.; Sol, V.; Krausz, P. A facile and rapid iodine-catalyzed meso-tetraphenylporphyrin synthesis using microwave activation. Tetrahedron Lett. 2008, 49, 5537–5539. [Google Scholar] [CrossRef]
- Yeung, K.-S.; Farkas, M.E.; Kadow, J.F.; Meanwell, N.A. A base-catalyzed, direct synthesis of 3,5-disubstituted 1,2,4-triazoles from nitriles and hidrazides. Tetrahedron Lett. 2005, 46, 3429–3432. [Google Scholar] [CrossRef]
- Adib, M.; Jahromi, A.H.; Tavoosi, N.; Mahdavi, H.; Bijanzadeh, H.R. Microwave-assisted one efficient, one pot, three-multicomponent synthesis of 3,5-disubstituted 1,2,4-oxadiazoles under free-solvent conditions. Tetrahedron Lett. 2006, 47, 2965–2967. [Google Scholar] [CrossRef]
- Andrade, M.M.; Barros, M.T. Fast synthesis of N-Acylhydrazones employing a microwave assisted neat protocol. J. Comb. Chem. 2010, 12, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Baxendale, I.R.; Ley, S.V. Polymer-supported reagents for multi-step organic synthesis: Application to the synthesis of sidenafil. Bioorg. Med. Chem. Lett. 2000, 10, 1983–1986. [Google Scholar] [CrossRef]
- SCOPUS. Available online: http://www.scopus.com (accessed on 13 September 2011).
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Larhed, M.; Hallberg, A. Microwave-assisted high-speed chemistry: A new technique in drug discovery. Drug Discov. Today 2001, 6, 406–416. [Google Scholar] [CrossRef]
- Shipe, W.D.; Wolkenberg, S.E.; Lindsley, C.W. Accelerating lead development by microwave-enhanced medicinal chemistry. Drug Discov. Today Technol. 2005, 2, 155–161. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://www.who.int/cancer/en/ (accessed on 1 February 2011).
- Noble, M.E.M.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, G.D. Targeting serine/threonine protein kinase B/Akt and cell-cycle checkpoint kinases for treating cancer. Curr. Top. Med. Chem. 2002, 2, 939–971. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W.; Zhao, Z.; Leister, W.H.; Robinson, R.G.; Barnett, S.F.; Defeo-Jones, D.; Jones, R.E.; Hartman, G.D.; Huff, J.R.; Huber, H.E.; et al. Allosteric Akt (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Lindsley, C.W.; Cheng, G.Z.; Yang, H.; Nicosia, S.V. The Akt/PKB pathway: Molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.F.; Bilodeau, M.T.; Lindsley, C.W. The Akt/PKB family of protein kinases: A review of small molecule inhibitors and progress towards target validation. Curr. Top. Med. Chem. 2005, 5, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.J.; Davies, T.G.; Donald, A.; McHardy, T.; Rowlands, M.G.; Aherne, G.W.; Hunter, L.K.; Taylor, K.; Ruddle, R.; Raynaud, F.I.; et al. Identification of 4-(4-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as selective inhibitors of protein kinase B through fragment elaboration. J. Med. Chem. 2008, 51, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.J.; Cheung, K.-M.; Collins, I. Synthesis of 4-(cyclic dialkylamino)-7-azaindoles by microwave heating of 4-halo-7-azaindoles and cyclic secondary amines. Tetrahedron Lett. 2007, 48, 1527–1529. [Google Scholar] [CrossRef]
- Hajdúch, M.; Havlíèek, L.; Veselỳ, J.; Novotnỳ, R.; Mihàl, V.; Strnad, M. Synthetic cyclin-dependent kinase inhibitors: New generation of potent anti-cancer drugs. Adv. Exp. Med. Biol. 1999, 457, 341–353. [Google Scholar] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- López-Cara, L.C.; Conejo-García, A.; Marchal, J.A.; MacChione, G.; Cruz-López, O.; Boulaiz, H.; García, M.A.; Rodríguez-Serrano, F.; Ramírez, A.; Cativiela, C.; et al. New (RS)-benzoxazepin-purines with antitumour activity: The chiral switch from (RS)-2,6-dichloro-9-[1-(p-nitrobenzenesulfonyl)-1,2,3,5-tetrahydro-4,1-benzoxazepin-3-yl]-9H-purine. Eur. J. Med. Chem. 2011, 46, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gavilán, M.; Rodríguez-Serrano, F.; Gómez-Vidal, J.A.; Marchal, J.A.; Aránega, A.; Gallo, M.A.; Espinosa, A.; Campos, J.M. Synthesis of tetrahydrobenzoxazepine acetals with electron-withdrawing groups on the nitrogen atom. Novel scaffolds endowed with anticancer activity against breast cancer cells. Tetrahedron 2004, 60, 11547–11557. [Google Scholar] [CrossRef]
- Rang, H.P.; Dale, M.M.; Ritter, J.M.; Flower, R.J. (Trad. Do Nascimento, A.P.). Farmacologia, 6ª ed; Elsevier: Rio de Janeiro, Brazil, 2007. [Google Scholar]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503. [Google Scholar] [CrossRef] [PubMed]
- Tillie-Leblond, I.; Pugin, J.; Marquette, C.-H.; Lamblin, C.; Saulnier, F.; Brichet, A.; Wallaert, B.; Tonnel, A.-B.; Gosset, P. Balance between proinflammatory cytokinesand their inhibitors in bronchial lavage from patients with status asthmaticus. Am. J. Respir. Crit. Care Med. 1999, 159, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Checker, R.; Sharma, D.; Sandur, S.K.; Khanam, S.; Poduval, T.B. Anti-inflammatory effects of plungabin are mediated by inhibition of NF-kappaB activation in lymphocytes. Int. Immunopharmacol. 2009, 9, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.D.; Nunes, O.D.R.H.; Fontenele, J.B.; Pessoa, O.D.L.; Lemos, T.L.G.; Viana, G.S.B. Analgesic and anti-inflammatory activities of a fraction rich in oncocalyxone A isolated from Auxemma oncocalyx. Phytomedicine 2004, 11, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Phutdhawong, W.S.; Ruensamran, W.; Phutdhawong, W.; Taechowisan, T. Synthesis of 1,6,7,8-tetrahydro-naphtho[2,3-d]-azepino[4,5b]indole-9,14-diones and their inhibitory effects on pro-inflammatory cytokines. Bioorg. Med. Chem. Lett. 2009, 19, 5753. [Google Scholar] [CrossRef] [PubMed]
- Phutdhawong, W.; Buddhasukh, D. Facile microwave-assisted synthesis of 9,10-Dihydro-9,10-ethanoanthracene-11-carboxylic acid methyl ester. Molecules 2005, 10, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Mattioli, I.; Gong, J.H.; Loetscher, P. Structure-function relationship between the human chemokine receptor CXCR3 and its ligands. J. Biol. Chem. 2003, 278, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Rottman, J.B.; Myers, P.; Kassam, N.; Weinblatt, M.; Loetscher, M.; Koch, A.E.; Moser, B.; Mackay, C.R. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Invest. 1998, 101, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Romagnani, P. CXCR3-bindind chemokines: Novel multifunctional therapeutic targets. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Salomon, I.; Netzer, N.; Wildbaum, G.; Schif-Zuck, S.; Maor, G.; Karin, N. Targeting the function of IFN-γ-Inducible protein 10 suppresses ongoing adjuvant arthritis. J. Immunol. 2002, 169, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Hancock, W.W.; Lu, B.; Gao, W.; Csizmadia, V.; Faia, K.; King, J.A.; Smiley, S.T.; Ling, M.; Gerard, N.P.; Gerard, C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 2000, 192, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.E.; Barnes, P.J. Chemokine receptors as therapeutic targets in chronic obstructive pulmonary disease. Trends Pharmacol. Sci. 2006, 27, 546–553. [Google Scholar] [CrossRef] [PubMed]
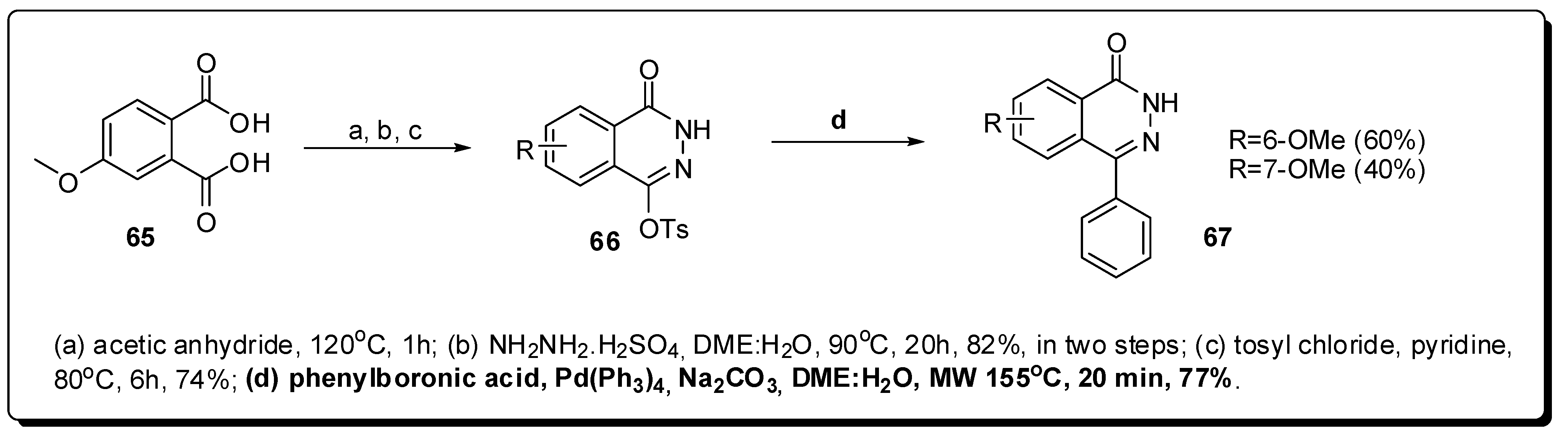
- Knight, R.L.; Allen, D.R.; Birch, H.L.; Chapman, G.A.; Galvin, F.C.; Jopling, L.A.; Lock, C.J.; Meissner, J.W.G.; Owen, D.A.; Raphy, G.; et al. Development of CXCR3 antagonists. Part 4: Discovery of 2-amino-(4-tropinyl)quinolines. Bioorg. Med. Chem. Lett. 2008, 18, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Maes, B.U.W.; Loones, K.T.J.; Lemière, G.L.F.; Dommisse, R.A. Rapid palladium-catalyzed aminations of aryl chlorides with aliphatic amines under temperature-controlled microwave heating. Synlett 2003, 12, 1822–1825. [Google Scholar] [CrossRef]
- Hardman, J.; Limbird, L.E. Goodman & Gilman’s the Pharmacological Basis of Therapeutcs, 10th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Schmid, M.B. Novel approaches to the discovery of antimicrobial agents. Curr. Opin. Chem. Biol. 1998, 2, 529–534. [Google Scholar] [CrossRef]
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.A.M.; Barreiro, E.J. Medicinal chemistry of N-Acylhydrazones: New lead-compounds of analgesic, antiinflammatory and antithrombotic drugs. Curr. Med. Chem. 2006, 13, 167–198. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Obafemi, C.A.; Nwinyi, O.C.; Akinpelu, D.A. Microwave assisted synthesis and antimicrobial activity of 2-quinoxalinone-3-hydrazone derivatives. Bioorg. Med. Chem. 2010, 18, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Kumar, H.; Khan, S.A. Synthesis and pharmacological evaluation of pyrazoline derivatives as new anti-inflammatory and analgesic agents. Bioorg. Med. Chem. Lett. 2008, 18, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarthi, K.J.E.; Richard, J.G.; Peter, T.; Janette, H.; Terrence, H.A. 5-Acyl-3-substituted benzofuran-2(3H)-ones as potential antiinflammatory agents. J. Med. Chem. 1987, 30, 1663–1668. [Google Scholar] [CrossRef]
- Akbas, E.; Berber, I. Antibacterial and antifungal activities of new pyrazolo[3,4-d]pyridazin derivatives. Eur. J. Med. Chem. 2005, 40, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.F.; Shalaby, M.A.; El-Demellawy, M.A. Polysubstituted pyrazoles, part 5. Synthesis of new 1-(4-chlorophenyl)-4-hydroxy-1H-pyrazole-3-carboxylic acid hydrazide analogs and some derived ring systems. A novel class of potential antitumor and anti-HVC agents. Eur. J. Med. Chem. 2003, 38, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Manna, K.; Agrawal, Y.K. Microwave assisted synthesis of new indophenazine 1,3,5-trisubstruted pyrazoline derivatives of benzofuran and their antimicrobial activity. Bioorg. Med. Chem. Lett. 2009, 19, 2688–2692. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U. Hepatitis C. Nature 2005, 436, 929–978. [Google Scholar] [CrossRef]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Wang, M.; Xia, Y.; Qu, F.; Neyts, J.; Peng, L. Arylethynyltriazole acyclonucleosides inhibit hepatitis C virus replication. Bioorg. Med. Chem. Lett. 2008, 18, 3321–3327. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Qu, F.; Quéléver, G.; Peng, L. Direct synthesis of 5-aryltriazole acyclonucleosides via Suzuki coupling in aqueous solution. Tetrahedron Lett. 2007, 48, 2389–2393. [Google Scholar] [CrossRef]
- van Compernolle, S.E.; Wiznycia, A.V.; Rush, J.R.; Dhanasekaran, M.; Baures, P.W.; Todd, S.C. Small molecule inhibition of hepatitis C virus E2 binding to CD81. Virology 2003, 314, 371–380. [Google Scholar] [CrossRef]
- Holzer, M.; Ziegler, S.; Albrecht, B.; Kronenberger, B.; Kaul, A.; Bartenschlager, R.; Kattner, L.; Klein, C.D.; Hartmann, R.W. Identification of terfenadine as an inhibitor of human CD81-receptor HCV-E2 interaction: Synthesis and structure optimization. Molecules 2008, 13, 1081–1110. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.; Kronenberger, B.; Albrecht, B.A.-M.; Kaul, A.; Gamer, A.-L.; Klein, C.D.; Hartmann, R.W. Development and evaluation of a FACS-based medium-throughput assay for HCV entry inhibitors. J. Biomol. Screen. 2009, 14, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Clyde, R. Piperidinderivate. D.E. Patent 25.06.770, 1975. [Google Scholar]
- de Clercq, E. Recent highlights in the development of new antiviral drugs. Curr. Opin. Microb. 2005, 8, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D. Antiviral drug resistance. Antivir. Res. 2006, 71, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Smejkal, J.; Sorm, F. Nucleic acid components and their analogues. 53. Preparation of 1-(2-deoxy-β-l-ribofuranosyl)thymine (l-thymidine). Collect. Czech. Chem. Commun. 1964, 29, 2809–2813. [Google Scholar] [CrossRef]
- Maury, G. The enantioselectivity of enzymes involved in current antiviral therapy using nucleoside analogues: A new strategy? Antivir. Chem. Chemother. 2000, 11, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Focher, F.; Spadari, S.; Maga, G. Antivirals at the mirror: The lack of stereospecificity of some viral and human enzymes offers novel opportunities in antiviral drug development. Curr. Drug Targets Infect. Disord. 2003, 3, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Beach, J.W.; Jeong, L.S.; Alves, A.J.; Pohl, D.; Kim, H.O.; Chang, C.-N.; Doong, S.-L.; Schinazi, R.F.; Cheng, Y.-C.; Chu, C.K. Synthesis of enantiomerically pure (2’R,5’S)-(-)-1-[2-(hydroxymethyl)oxathiolan-5-yl]cytosine as a potent antiviral agent against hepatitis B virus (HBV) and human immunodeficiency virus (HIV). J. Org. Chem. 1992, 57, 2217–2219. [Google Scholar] [CrossRef]
- Cameron, J.M.; Collins, P.; Daniel, M.; Storer, R.; Wilcox, P. Lamivudine. Drugs Future 1993, 18, 319–323. [Google Scholar] [CrossRef]
- Schinazi, R.F.; Lloyd, R.M., Jr.; Nguyen, M.-H.; Cannon, D.L.; McMillan, A.; Ilksoy, N.; Chu, C.K.; Liotta, D.C.; Bazmi, H.Z.; Mellors, J.W. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob. Agents Chemother. 1993, 37, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-W.; Detorio, M.; Herman, B.D.; Solomon, S.; Bassit, L.; Nettles, J.H.; Obikhod, A.; Tao, S.-J.; Mellors, J.W.; Sluis-Cremer, N.; et al. Synthesis, antiviral activity, cytotoxicity and cellular pharmacology of l-3’-azido-2’,3’-dideoxypurine nucleosides. Eur. J. Med. Chem. 2011, 46, 3832–3844. [Google Scholar] [CrossRef] [PubMed]
- Bookser, B.C.; Raffaele, N.B. High-throughput five minute microwave accelerated glycosylation approach to the synthesis of nucleoside libraries. J. Org. Chem. 2007, 72, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Pepin, J.; Meda, H.A. The epidemiology and control of human African trypanosomiasis. Adv. Parasitol. 2001, 49, 71–132. [Google Scholar] [PubMed]
- Barrett, M.P.; Burchmore, R.J.S.; Stich, A.; Lazzari, J.O.; Frasch, A.C.; Cazzulo, J.J.; Krishna, S. The trypanosomiases. Lancet 2003, 362, 1469–1480. [Google Scholar] [CrossRef]
- Rodgers, J.J. Human African trypanosomiasis, chemotherapy and CNS disease. J. Neuroimmunol. 2009, 211, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E. The continuing problem of human African trypanosomiasis (sleeping sickness). Ann. Neurol. 2008, 64, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Denise, H.; Barrett, M.P. Uptake and mode of action of drugs used against sleeping sickness. Biochem. Pharmacol. 2001, 61, 1–5. [Google Scholar] [CrossRef]
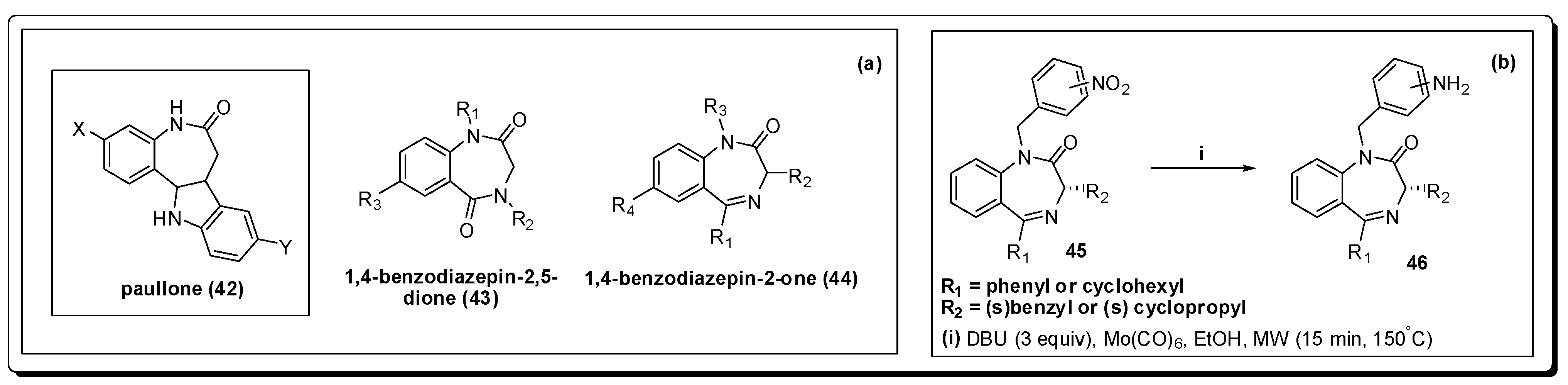
- Knockaert, M.; Wieking, K.; Schmitt, S.; Leost, M.; Grant, K.M.; Mottram, J.C.; Kunick, C.; Meijer, L. Intracellular targets of paullones: Identification following affinity purification on immobilized inhibitor. J. Biol. Chem. 2002, 277, 25493–25501. [Google Scholar] [CrossRef] [PubMed]
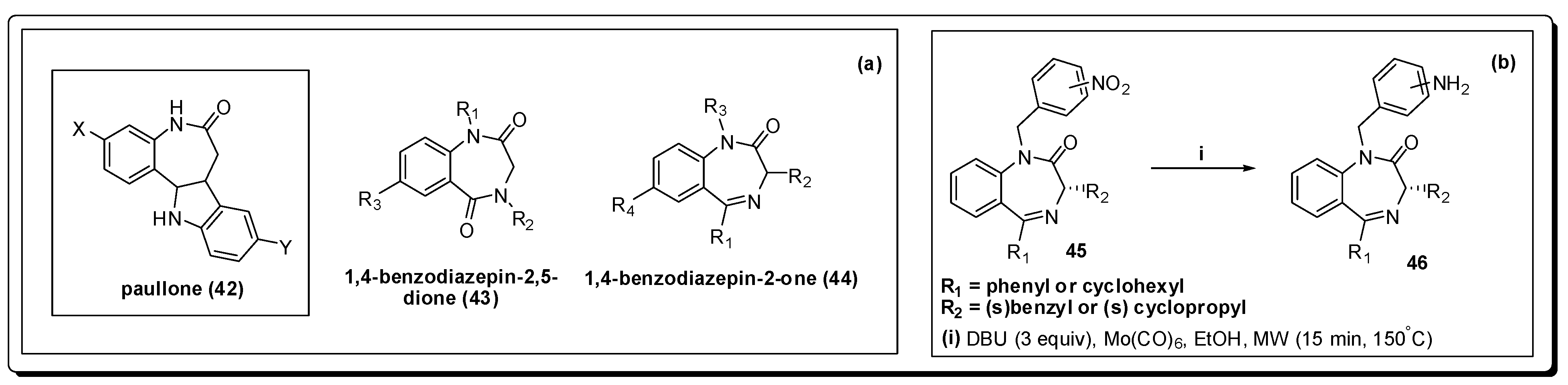
- Clark, R.L.; Carter, K.C.; Mullen, A.B.; Coxon, G.D.; Owusu-Dapaah, G.; McFarlane, E.; Duong Thi, M.D.; Mackay, S.P. Identification of the benzodiazepines as a new class of antileishmanial agent. Bioorg. Med. Chem. Lett. 2007, 17, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Rathnam, R.P.; Harvey, A.L.; Clements, C.J.; Clark, R.L.; Barrett, M.P.; Wong, P.E.; Male, L.; Coles, S.J.; MacKay, S.P. Synthesis and biological evaluation of 1,4-benzodiazepin-2-ones with antitrypanosomal activity. Bioorg. Med. Chem. 2011, 19, 1802–1815. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.; Anjum, N.; Patel, H.; Rathnam, R.P.; Verma, J. Molybdenum hexacarbonyl and DBU reduction of nitro compounds under microwave irradiation. Synlett 2007, 13, 2557–2558. [Google Scholar] [CrossRef]
- Spencer, J.; Rathnam, R.P.; Patel, H.; Anjum, N. Microwave mediated reduction of heterocycle and fluorine containing nitroaromatics with Mo(CO)6 and DBU. Tetrahedron 2008, 64, 10195–10200. [Google Scholar] [CrossRef]
- World Malaria Report 2008; World Health Organization: Geneva, Switzerland, 2008.
- White, N.J.J. Antimalarial drug resistance. Clin. Invest. 2004, 113, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
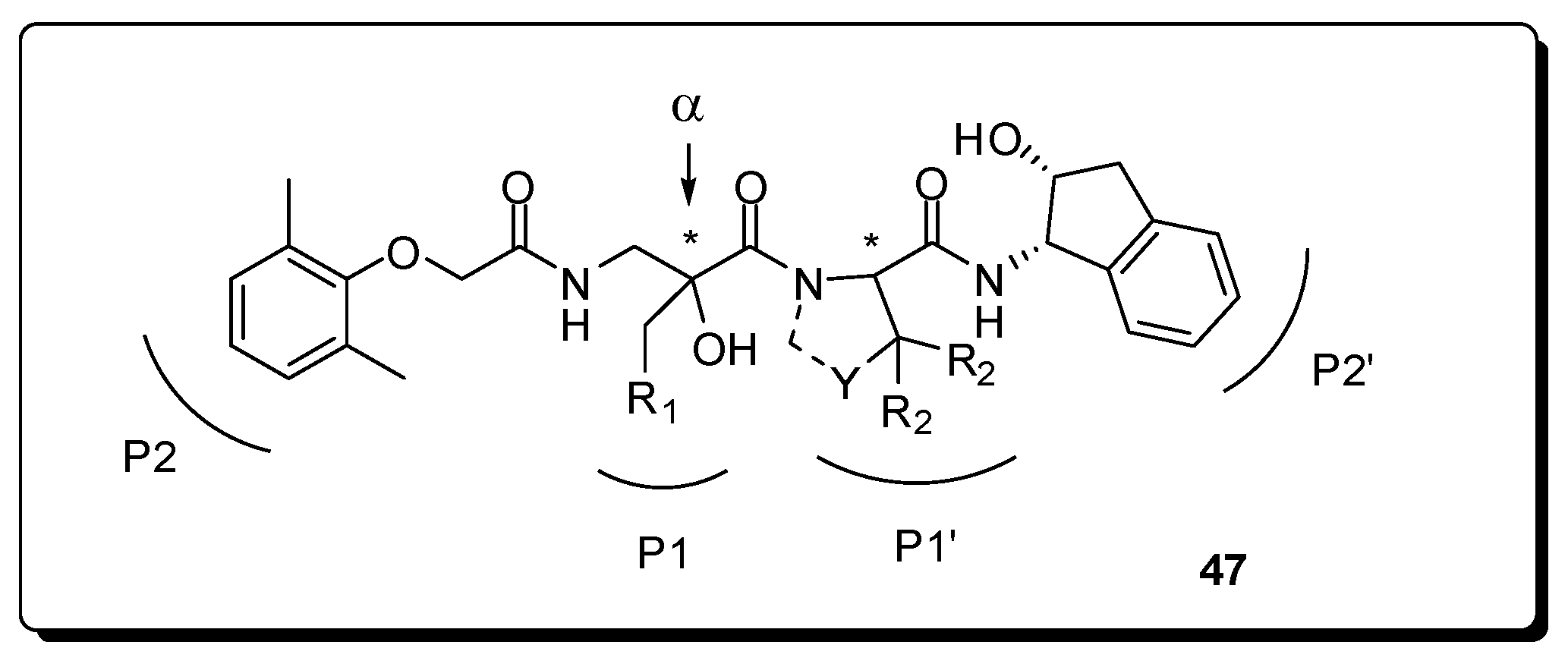
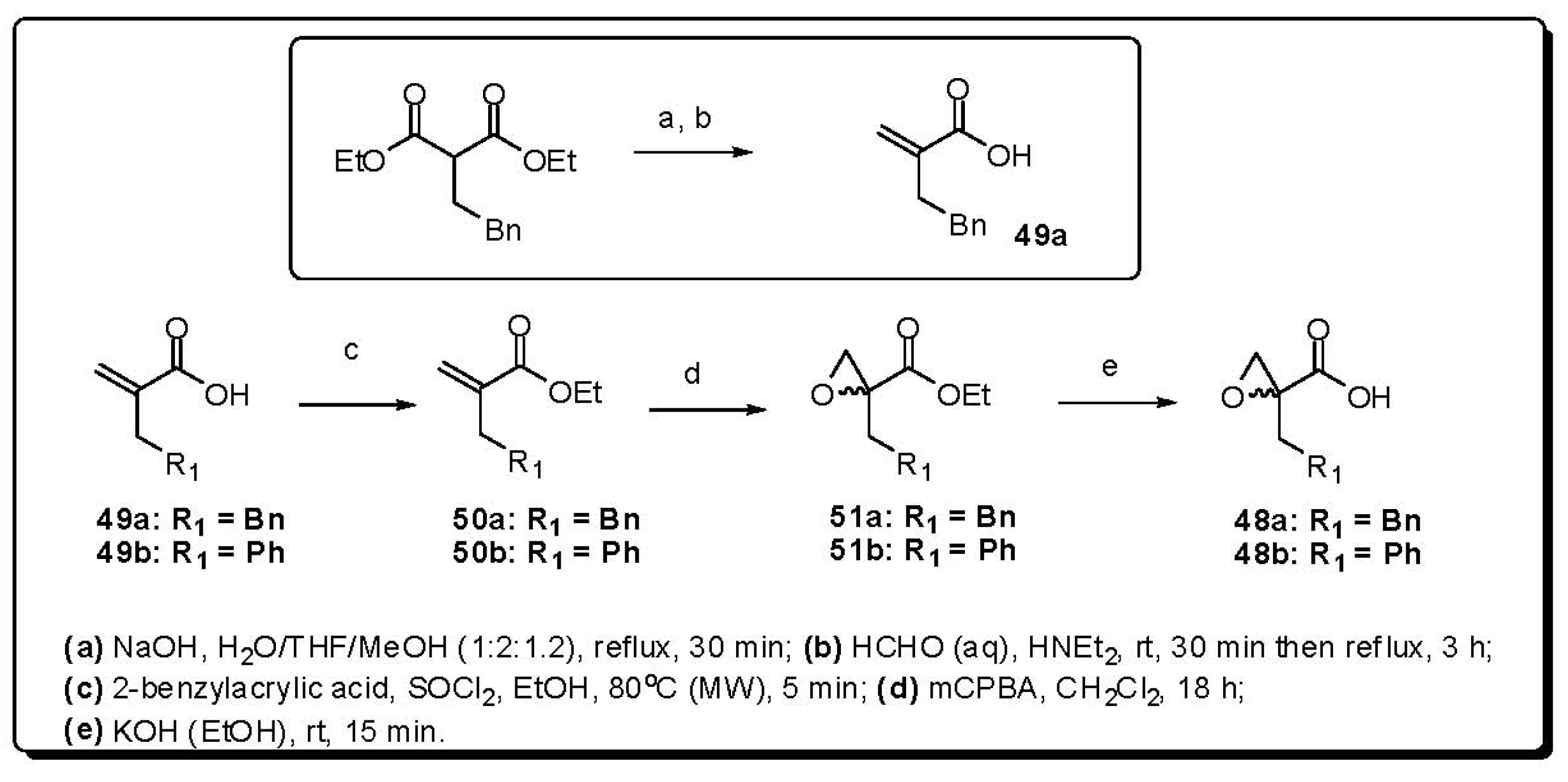
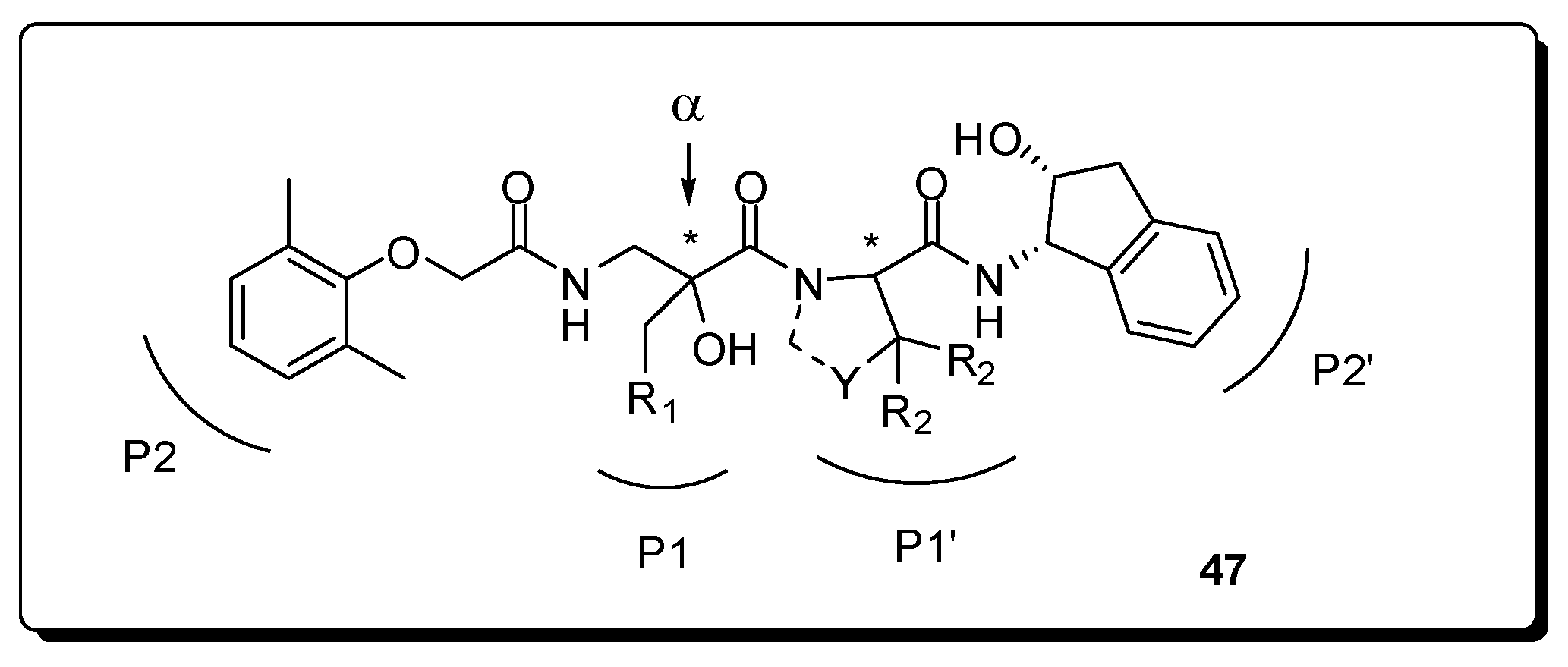
- Orrling, K.M.; Marzahn, M.R.; Gutiérrez-de-Terán, H.; Åqvist, J.; Dunn, B.M.; Larhed, M. α-Substituted norstatines as the transition-state mimic in inhibitors of multiple digestive vacuole malaria aspartic proteases. Bioorg. Med. Chem. 2009, 17, 5933–5949. [Google Scholar] [CrossRef] [PubMed]
- Ekegren, J.K.; Gising, J.; Wallberg, H.; Larhed, M.; Samuelsson, B.; Hallberg, A. Variations of the P2 group in HIV-1 protease inhibitors containing a tertiary alcohol in the transition-state mimicking scaffold. Org. Biomol. Chem. 2006, 4, 3040–3043. [Google Scholar] [CrossRef] [PubMed]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007, 5, S7–S16. [Google Scholar] [CrossRef]
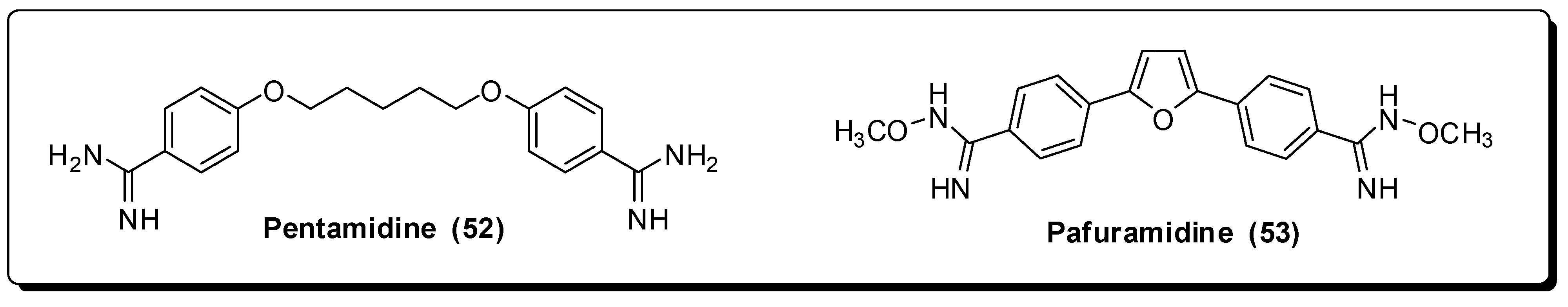
- Sands, M.; Kron, M.A.; Brown, R.B. Pentamidine: A review. Rev. Infect. Dis. 1985, 7, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; de Koning, H.P. Pentamidine uptake and resistance in pathogenic protozoa: Past, present and future. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef]
- Berman, J.D. Human leishmaniasis: Clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin. Infect. Dis. 1997, 24, 684–703. [Google Scholar] [CrossRef] [PubMed]
- Castro, M. Treatment and prophylaxis of pneumocystis carinii pneumonia. Semin. Respir. Infect. 1998, 13, 296–303. [Google Scholar] [PubMed]
- Burri, C. Chemotherapy against human African trypanosomiasis: Is there a road to success? Parasitology 2010, 137, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.F.; Schubach, A.O.; Martins, M.M.; Passos, S.L.; Oliveira, R.V.; Marzochi, M.C.; Andrade, C.A. Systematic review of the adverse effects of cutaneous leishmaniasis treatment in the new world. Acta Trop. 2011, 118, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Fairlamb, A.H. Chemotherapy of human African trypanosomiasis: Current and future prospects. Trends Parasitol. 2003, 19, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Zolek, T.; Maciejewska, D. Theoretical models of pentamidine analogs activity based on their DNA minor groove complexes. Eur. J. Med. Chem. 2010, 45, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Moreno, T.; Pous, J.; Subirana, J.A.; Campos, J.L. Coiled-coil conformation of a pentamidine-DNA complex. Acta Crystallorgr. D Biol. Crystallogr. 2010, 66, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Thuita, J.K.; Karanja, S.M.; Wenzler, T.; Mdachi, R.E.; Ngotho, J.M.; Kagira, J.M.; Tidwell, R.; Brun, R. Efficacy of the diamidine DB75 and its prodrug DB289, against murine models of human African trypanosomiasis. Acta Trop. 2008, 108, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Paloque, L.; Bouhlel, A.; Curti, C.; Dumtre, A.; Verhaeghe, P.; Azas, N.; Vanelle, P. Synthesis and evaluation of monoamidoxime derivatives: Toward new antileishmanial compounds. Eur. J. Med. Chem. 2011, 46, 2984–2991. [Google Scholar] [CrossRef] [PubMed]
- Knol – A unit of knowledge. Available online: http://knol.google.com/k/krishan-maggon/global-brain-drugs-market-review-2008/3fy5eowy8suq3/1# (accessed on 15 July 2011).
- Korzh, M. The efficacy and safety of zolpidem over 12 months in patient with low back pain and chronic insomnia. Sleep Med. 2007, 8, 578. [Google Scholar] [CrossRef]
- Yogeeswari, P.; Menon, N.; Semwal, A.; Arjun, M.; Sriram, D. Discovery of mole cules for the treatment of neuropathic pain: Synthesis, antiallodynic and antihyperalgesic activities of 5-(4-nitrophenyl0furoic-2-acid hydrazones. Eur. J. Med. Chem. 2011, 46, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
- Kummerle, A.E.; Vieira, M.M.; Schmitt, M.; Miranda, A.L.P.; Fraga, C.A.M.; Bourguignon, J.-J.; Barreiro, E.J. Design, synthesis and analgesic properties of novel conformationally-restricted N-acylhydrazones. Eur. J. Med. Chem. 2009, 19, 4963–4966. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, R.; Silva, G.M.S.; Zapata-Sudo, G.; Raimundo, J.M.; Sudo, R.T.; Barreiro, E.J.; Fraga, C.A.M. Design, synthesis and pharmacological evaluation of new neuroactive pyrazolo[3,4-b]pyrrolo[3,4-d]pyridine derivatives with in vivo hypnotic and analgesic profile. Bioorg. Med. Chem. 2006, 14, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Mendes, T.C.F.; Raimundo, J.M.; Nascimento-Junior, N.M.; Fraga, C.A.M.; Barreiro, E.J.; Sudo, R.T.; Zapata-Sudo, G. Sedation and antinociception induced by a new pyrazolo[3,4-b]pyrrolo[3,4-d]pyridine derivative (LASSBio-873) is modulated by activation of muscarinic receptors. Pharmacol. Biochem. Behav. 2009, 94, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, R.; Isobe, N.; Kawasaki, H. Mechanism of prolongation of pentobarbital-induced sleeping time by empenthrin in mice. Toxicology 1996, 108, 185–190. [Google Scholar] [CrossRef]
- Mora, S.; Diaz-Veliz, G.; Lungentrass, H.; García-Gonzáles, M.; Coto-Morales, T.; Poletti, C.; de Lima, T.C.; Herrera-Ruiz, M.; Tortoriello, J. Central nervous system activity of the hydroalcocholic extract of Casimiroa edulis in rat and mice. J. Ethnopharmacol. 2005, 97, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Dundee, J.W.; Halliday, N.J.; Harper, K.W.; Brogden, R.N. Midazolan a review of its pharmacological properties and therapeutic use. Drugs 1984, 28, 519–543. [Google Scholar] [CrossRef] [PubMed]
- Kuraishi, Y.; Harada, Y.; Aratani, S.; Satoh, M.; Takagi, H. Separate involvement of the spinal noradrenergic and serotoninergic system in morphine analgesia: The differences in mechanical and thermal algesic tests. Brain Res. 1983, 273, 245–252. [Google Scholar] [CrossRef]
- Nascimento-Júnior, N.M.; Mendes, T.C.F.; Leal, D.M.; Corrêa, C.M.N.; Sudo, R.T.; Zapata-Sudo, G.; Bareiro, E.J.; Fraga, C.A.M. Microwave assisted-synthesis and structure-activity relatioships of neuroactive pyrazolo[3,4-b]pyrrolo[3,4-d]pyridine derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Ortiz, A.; Carrilo, J.R.; Gómez-Escalonilla, M.J.; de La Hoz, A.; Moreno, A.; Prieto, P. First diels-alder reaction of pyrazolil imides under microwave irradiation. Synlett 1998, 10, 1069–1070. [Google Scholar] [CrossRef]
- Díaz-Ortiz, A.; de La Hoz, A.; Langa, F. Microwave irradiation in free-solvent conditions: An eco-friendly methodology to prepare indazoles, pyrazolopyridines and bipyrazoles by cycloaddition reactions. Green Chem. 2000, 2, 165–172. [Google Scholar] [CrossRef]
- Díaz-Ortiz, A.; Carrilo, J.R.; Cossio, F.P.; Gómez-Escalonila, M.J.; de La Hoz, A.; Moreno, A.; Prieto, P. Synthesis of pyrazolo[3,4-b]pyridines by cycloaddition reactions under microwave irradiation. Tetrahedron 2000, 56, 1569–1577. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |



















© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nascimento-Júnior, N.M.; Kümmerle, A.E.; Barreiro, E.J.; Fraga, C.A.M. MAOS and Medicinal Chemistry: Some Important Examples from the Last Years. Molecules 2011, 16, 9274-9297. https://doi.org/10.3390/molecules16119274
Nascimento-Júnior NM, Kümmerle AE, Barreiro EJ, Fraga CAM. MAOS and Medicinal Chemistry: Some Important Examples from the Last Years. Molecules. 2011; 16(11):9274-9297. https://doi.org/10.3390/molecules16119274
Chicago/Turabian StyleNascimento-Júnior, Nailton M., Arthur E. Kümmerle, Eliezer J. Barreiro, and Carlos A. M. Fraga. 2011. "MAOS and Medicinal Chemistry: Some Important Examples from the Last Years" Molecules 16, no. 11: 9274-9297. https://doi.org/10.3390/molecules16119274
APA StyleNascimento-Júnior, N. M., Kümmerle, A. E., Barreiro, E. J., & Fraga, C. A. M. (2011). MAOS and Medicinal Chemistry: Some Important Examples from the Last Years. Molecules, 16(11), 9274-9297. https://doi.org/10.3390/molecules16119274