Consecutive Three-Component Synthesis of 3-(Hetero)Aryl-1H-pyrazoles with Propynal Diethylacetal as a Three-Carbon Building Block


Abstract
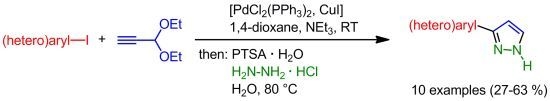
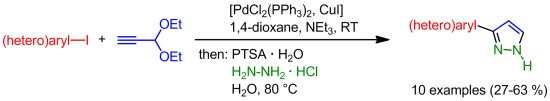
:
1. Introduction
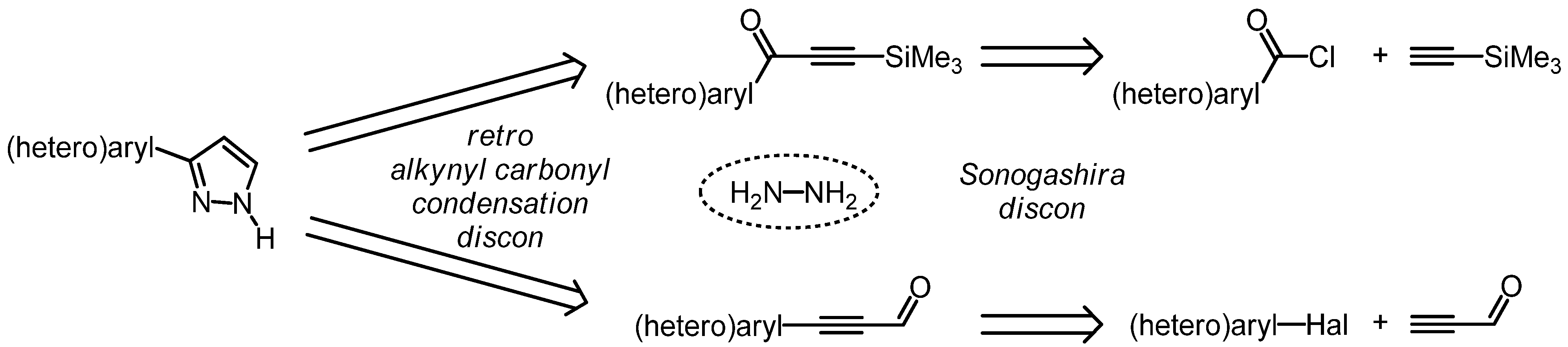
2. Results and Discussion

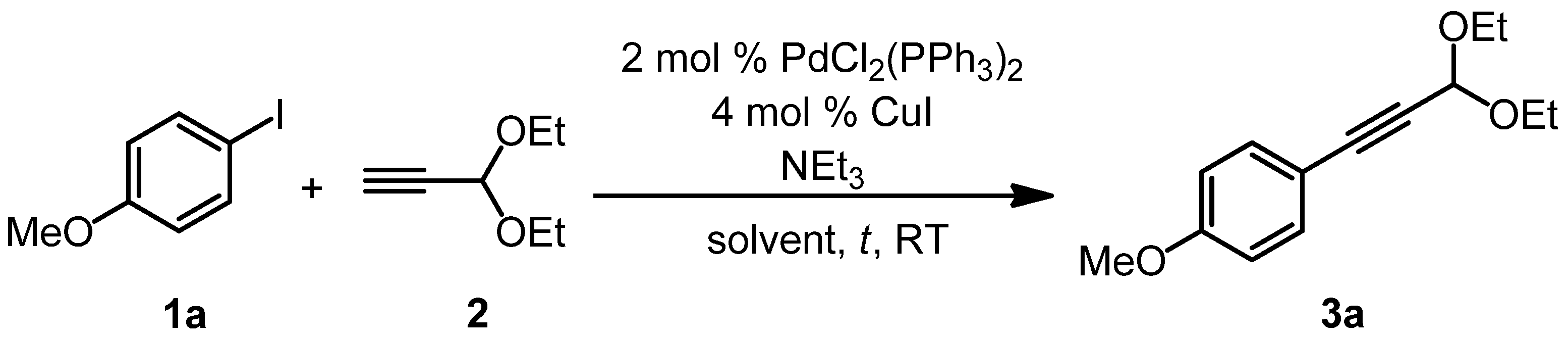

{kind=link}
{kind=link}
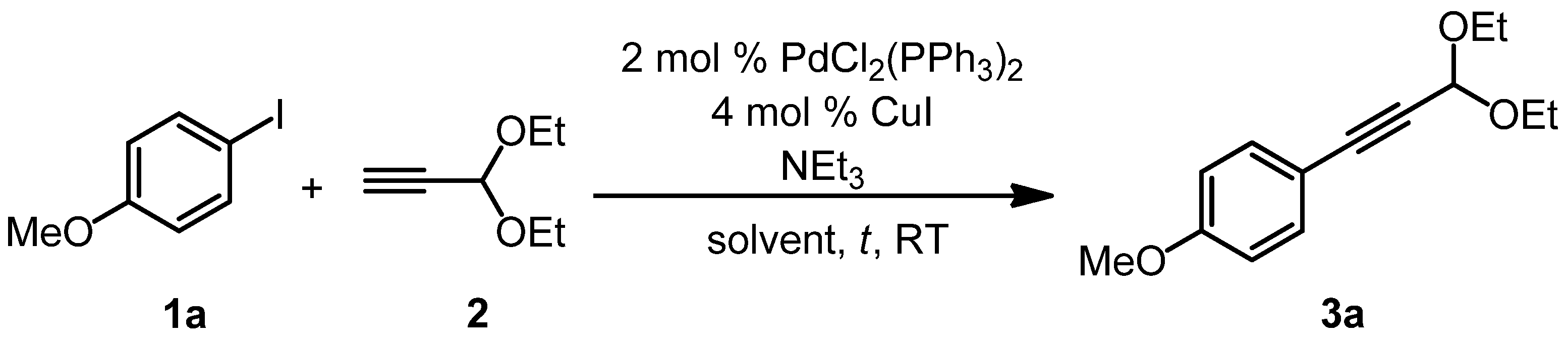{kind=link}
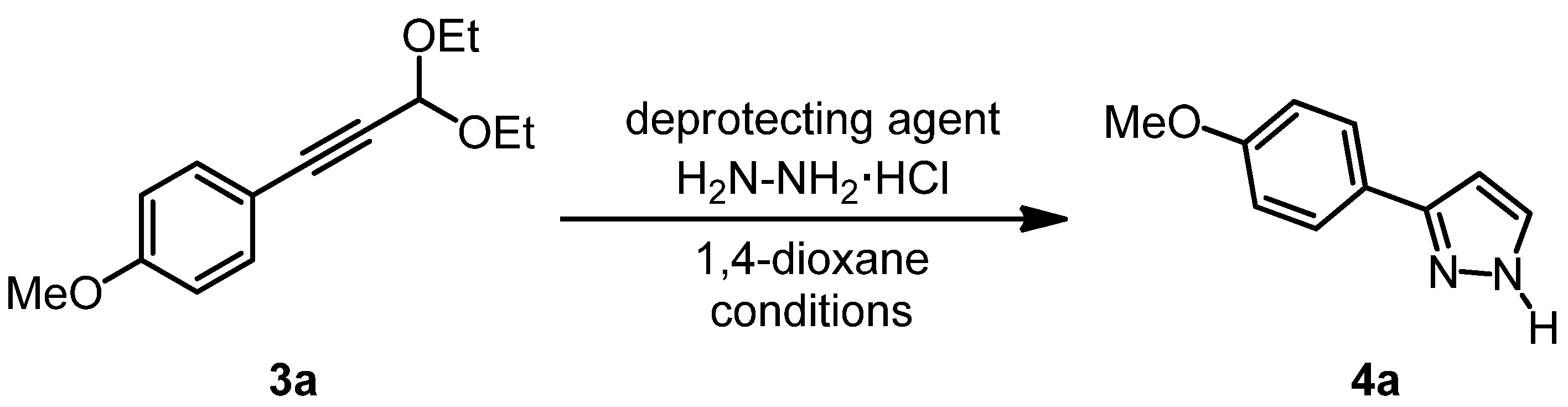{kind=link}
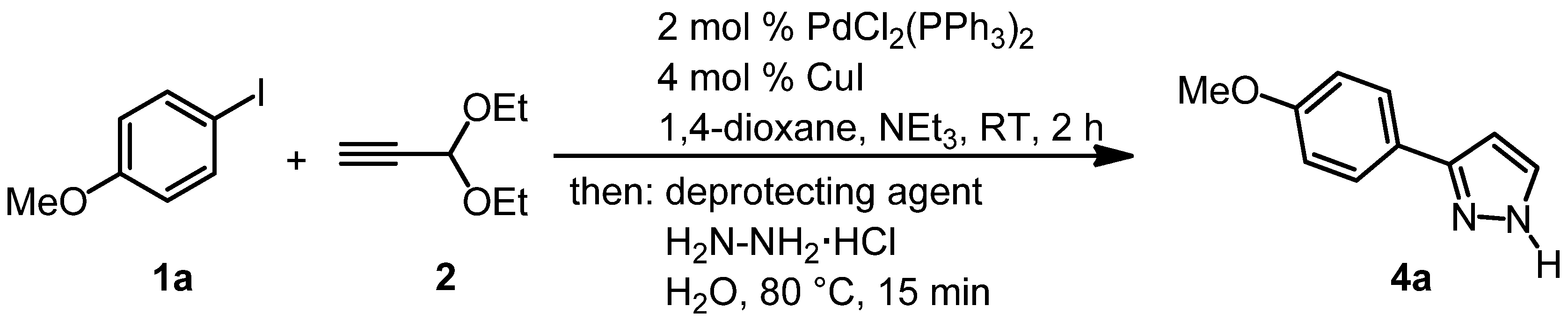{kind=link}
{kind=link}
| Entry 1 | Alkyne 2 | NEt3 | Solvent | Reaction time | Yield of 3a 2 |
|---|---|---|---|---|---|
| 1 | 1.0 equiv. | 2.0 equivs. | THF | 2 h | 64% |
| 2 | 1.0 equiv. | 2.0 equivs. | 1,4-dioxane | 2 h | 80% |
| 3 | 1.1 equivs. | 1.1 equivs. | 1,4-dioxane | 2 h | 64% |
| 4 3 | 1.1 equivs. | 2.0 equivs. | 1,4-dioxane | 2 h | 89% |
| 5 | 1.1 equivs. | 2.0 equivs. | 1,4-dioxane | 1 h | 87% |
| 6 | 1.1 equivs. | 2.0 equivs. | 1,4-dioxane | 3 h | 91% |
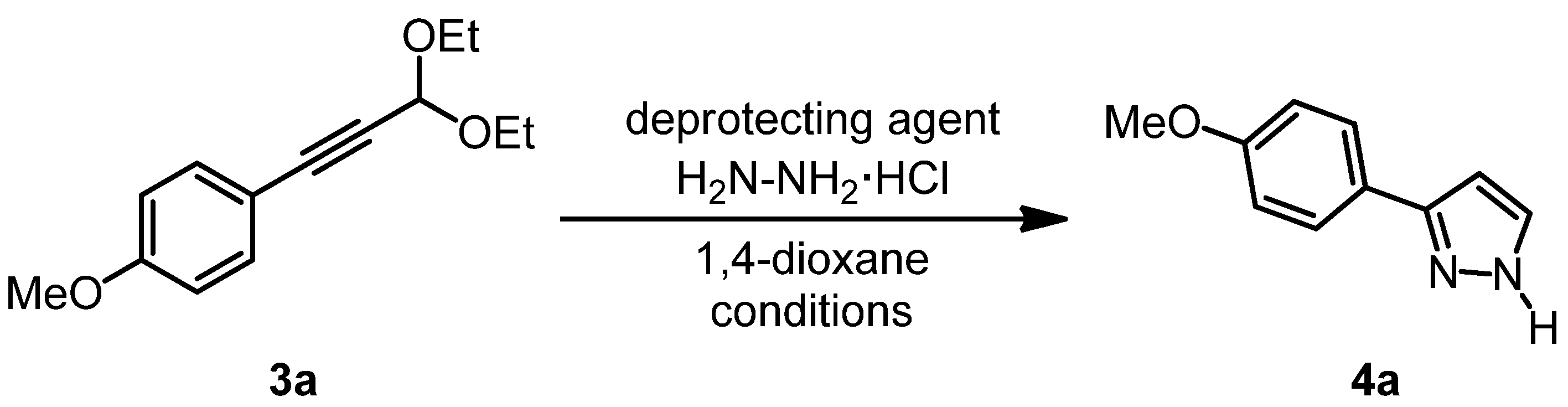
| Entry | Deprotecting agent 1 | H2N-NH2·HCl | Reaction temperature | Reaction time | Yield of 4a 2 |
|---|---|---|---|---|---|
| 1 3 | H2O | 2.0 equivs. | 80 °C (MW) | 15 min | 53% |
| 2 4 | H2O | 2.0 equivs. | 80 °C 5 | 15 min | 65% |
| 3 3 | H2O | 2.0 equivs. | 80 °C 5 | 1 h | 62% |
| 4 4 | H2O | 1.0 equiv. | 80 °C 5 | 15 min | 22% |
| 5 3 | HClaq (1 N) | 2.0 equivs. | 130 °C 5 | 10 min | 55% |

| Entry | Deprotecting agent | Yield of 4a 1 |
|---|---|---|
| 1 | not isolated | |
| 2 | 1.0 equiv. of HClaq (1 N) | 50% |
| 3 | 1.0 equiv. of CH3COOH | 25% |
| 4 | 1.0 equiv. of PTSA·H2O | 56% |
| 5 | 2.0 equivs. of PTSA·H2O | 55% |
3. Experimental
3.1. General
3.2. Experimental Procedure for the Synthesis of 1-(3,3-Diethoxyprop-1-yn-1-yl)-4-methoxybenzene (3a)

3.3. Typical Experimental Procedure (Synthesis of Pyrazole 4b)
| Entry | (Hetero)Aryl iodide 1 | Reaction time t | Pyrazole 4 | Chromatographic |
|---|---|---|---|---|
| (2.00 mmol) | 2nd step | (isolated yield) | purification | |
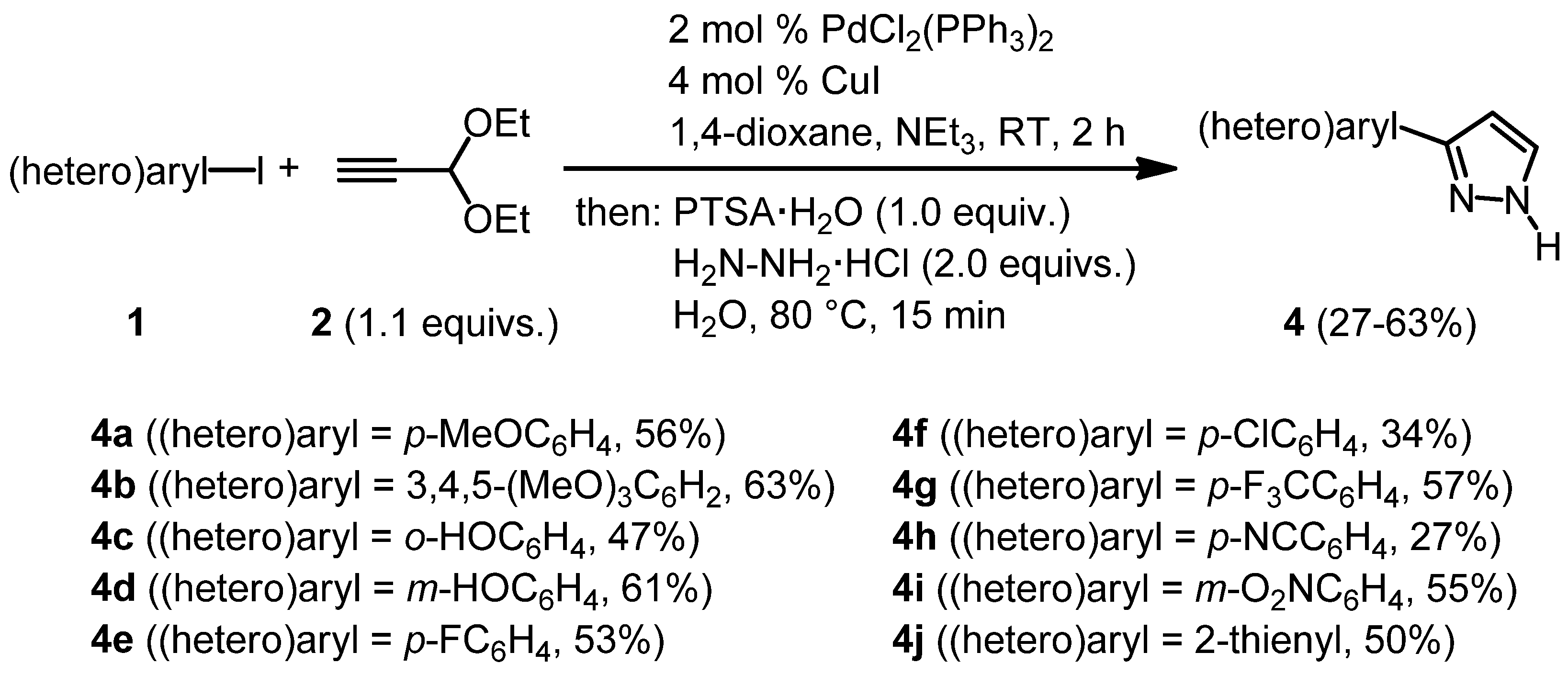
| 1 | 1a, 1-Iodo-4-methoxybenzene | 15 min | 195 mg | CH2Cl2/MeOH/NH3 |
| Merck | (1.12 mmol, 56%) | |||
| 478 mg | 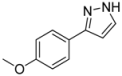 | |||
| 4a | 100:2:1 | |||
| 2 | 1b, 1-Iodo-3,4,5-trimethoxybenzene | 15 min | 295 mg | CH2Cl2/MeOH/NH3 |
| Alfa Aesar | (1.26 mmol, 63%) | |||
| 600 mg | 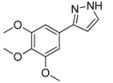 | |||
| 4b | 100:2:1 | |||
| 3 | 1c, 1-Iodo-2-hydroxybenzene | 15 min | 150 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (0.94 mmol, 47%) | |||
| 449 mg | 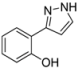 | |||
| 4c | 100:1:1 | |||
| 4 | 1d, 1-Iodo-3-hydroxybenzene | 15 min | 196 mg | CH2Cl2/MeOH/NH3 |
| Alfa Aesar | (1.23 mmol, 61%) | |||
| 449 mg | 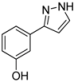 | |||
| 4d | 100:1:1 → 100:3:1 → 100:7:1 | |||
| 5 | 1e, 1-Iodo-4-fluorobenzene | 15 min | 170 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (1.05 mmol, 53%) | |||
| 0.32 mL | 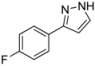 | |||
| 4e | 100:1:1 → 100:2:1 | |||
| 6 | 1f, 1-Iodo-4-chlorobenzene | 30 min | 121 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (0.68 mmol, 34%) | |||
| 482 mg | 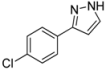 | |||
| 4f | 100:2:1 | |||
| 7 | 1g, 1-Iodo-4-trifluoromethylbenzene | 15 min | 244 mg | CH2Cl2/MeOH/NH3 |
| Alfa Aesar | (1.15 mmol, 57%) | |||
| 0.30 mL | 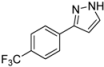 | |||
| 4g | 100:1:1 → 100:2:1 | |||
| 8 | 1h, 1-Iodo-4-cyanobenzene | 15 min | 90 mg | CH2Cl2/NH3 100:1 → CH2Cl2/MeOH/NH3 |
| ABCR | (0.53 mmol, 27%) | |||
| 467 mg | 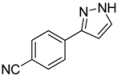 | |||
| 4h | 100:1:1 | |||
| 9 | 1i, 1-Iodo-3-nitrobenzene | 15 min | 209 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (1.10 mmol, 55%) | |||
| 508 mg | 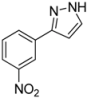 | |||
| 4i | 100:2:1 | |||
| 10 | 1j, 2-Iodothiophene | 1 h | 151 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (1.00 mmol, 50%) | |||
| 429 mg | 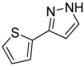 | |||
| 4j | 100:1:1 |
3.4. Spectroscopic Data of 1H-Pyrazoles 4
3.4.1. 3-(4-Methoxyphenyl)-1H-pyrazole (4a)

3.4.2. 3-(3,4,5-Trimethoxyphenyl)-1H-pyrazole (4b)

3.4.3. 2-(1H-Pyrazol-3-yl)phenol (4c)

3.4.4. 3-(1H-Pyrazol-3-yl)phenol (4d)

3.4.5. 3-(4-Fluorophenyl)-1H-pyrazole (4e)

3.4.6. 3-(4-Chlorophenyl)-1H-pyrazole (4f)

3.4.7. 3-[4-(Trifluoromethyl)phenyl]-1H-pyrazole (4g)

3.4.8. 4-(1H-Pyrazol-3-yl)benzonitrile (4h)

3.4.9. 3-(3-Nitrophenyl)-1H-pyrazole (4i)

3.4.10. 3-(Thiophen-2-yl)-1H-pyrazole (4j)

4. Conclusions
Acknowledgments
References
- Kost, A.N.; Grandberg, I.I. Progress in pyrazole chemistry. Adv. Heterocycl. Chem. 1966, 6, 347–429. [Google Scholar] [CrossRef]
- Elguero, J. Pyrazoles and their benzo derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon: Oxford, UK, 1984; Volume 5, pp. 167–303. [Google Scholar]
- Elguero, J. Pyrazoles. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Elsevier: Oxford, UK, 1996; Volume 3, pp. 1–75. [Google Scholar]
- Wustrow, D.J.; Capiris, T.; Rubin, R.; Knobelsdorf, J.A.; Akunne, H.; Davis, M.D.; MacKenzie, R.; Pugsley, T.A.; Zoski, K.T.; Heffner, T.G.; et al. Pyrazolo[1,5-a]pyrimidine CRF-1 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2067–2070. [Google Scholar]
- Menozzi, G.; Mosti, L.; Fossa, P.; Mattioli, F.; Ghia, M. ω-Dialkylaminoalkyl ethers of phenyl-(5-substituted 1-phenyl-1H-pyrazol-4-yl)methanols with analgesic and anti-inflammatory activity. J. Heterocycl. Chem. 1997, 34, 963–968. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Manna, F.; Chimenti, P.; Secci, D.; Befani, O.; Turini, P.; Ortuso, F.; Alcaro, S. Monoamine oxidase isoform-dependent tautomeric influence in the recognition of 3,5-diaryl pyrazole inhibitors. J. Med. Chem. 2007, 50, 425–428. [Google Scholar] [CrossRef]
- Walworth, B.L.; Klingsberg, E. Neue herbicide Zusammensetzungen. DE 2260485 19730628, 1973. [Google Scholar]
- Trofimenko, S. Coordination chemistry of pyrazole-derived ligands. Chem. Rev. 1972, 72, 497–509. [Google Scholar] [CrossRef]
- Mukherjee, R. Coordination chemistry with pyrazole-based chelating ligands: Molecular structural aspects. Coord. Chem. Rev. 2000, 203, 151–218. [Google Scholar]
- Trofimenko, S. Scorpionates: Genesis, milestones, prognosis. Polyhedron 2004, 23, 197–203. [Google Scholar] [CrossRef]
- Ward, M.D.; McCleverty, J.A.; Jeffery, J.C. Coordination and supramolecular chemistry of multinucleating ligands containing two or more pyrazolyl-pyridine ‘arms’. Coord. Chem. Rev. 2001, 222, 251–272. [Google Scholar]
- Harb, A.-F.A.; Abbas, H.H.; Mostafa, F.H. Pyrazoles as building blocks in heterocyclic synthesis: Synthesis of some new substituted 1-triazinylpyrazolo[3,4-d]pyrimidine and 1-triazinylpyrazolo[3,4-b]pyridine derivatives. Chem. Pap. 2005, 59, 187–195. [Google Scholar]
- Harb, A.-F.A.; Abbas, H.H.; Mostafa, F.H. Pyrazoles as building blocks in heterocyclic synthesis: Synthesis of pyrazolo[3,4-d]pyrimidine, pyrazolo[3,4-e][1,4]diazepine, pyrazolo[3,4-d][1,2,3]-triazine and pyrrolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives. J. Iranian Chem. Soc. 2005, 2, 115–123. [Google Scholar] [CrossRef]
- Sachse, A.; Penkova, L.; Noël, G.; Dechert, S.; Varzatskii, O.A.; Fritsky, I.O.; Meyer, F. Efficient syntheses of some versatile 3,5-bifunctional pyrazole building blocks. Synthesis 2008, 800–806. [Google Scholar]
- Maeda, H.; Ito, Y.; Kusunose, Y.; Nakanishi, T. Dipyrrolylpyrazoles: Anion receptors in protonated form and efficient building blocks for organized structures. Chem. Commun. 2007, 1136–1138. [Google Scholar]
- Gemming, S.; Schreiber, M.; Thiel, W.; Heine, T.; Seifert, G.; Avelino de Abreu, H.; Anderson Duarte, H. Tunable discotic building blocks for liquid crystalline displays. J. Luminescence 2004, 108, 143–147. [Google Scholar] [CrossRef]
- Dolars, A.; Schellhammer, C.-W.; Schroeder, J. Heterocycles as structural units in new optical brighteners. Angew. Chem. Int. Ed. Engl. 1975, 14, 665–679. [Google Scholar] [CrossRef]
- Catalan, J.; Fabero, F.; Claramunt, R.M.; Santa Maria, M.D.; Foces-Foces, M.C.; Hernandez Cano, F.; Martinez-Ripoll, M.; Elguero, J.; Sastre, R. New ultraviolet stabilizers: 3- and 5-(2'-Hydroxyphenyl)pyrazoles. J. Am. Chem. Soc. 1992, 114, 5039–5048. [Google Scholar]
- Karatsu, T.; Shiochi, N.; Aono, T.; Miyagawa, N.; Kitamura, A. Photoinduced electron transfer reactions of 3H-pyrazole derivatives. Formation of solvent adduct by specific sensitizer. Bull. Chem. Soc. Jpn. 2003, 76, 1227–1231. [Google Scholar] [CrossRef]
- Yen, Y.-P.; Huang, T.-M.; Tseng, Y.-P.; Lin, H.-Y.; Lai, C.-C. Photoinduced electron transfer reactions of 3,3-dialkylated 4,5-diphenyl-3H-pyrazoles: A new route to the formation of the solvent adducts. J. Chin. Chem. Soc. 2004, 51, 393–398. [Google Scholar]
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Touré, B.B.; Hall, D.G. Natural product synthesis using multicomponent reaction strategies. Chem. Rev. 2009, 109, 4439–4486. [Google Scholar] [CrossRef]
- Sunderhaus, J.D.; Martin, S.F. Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chem. Eur. J. 2009, 15, 1300–1308. [Google Scholar] [CrossRef]
- Isambert, N.; Lavilla, R. Heterocycles as key substrates in multicomponent reactions: The fast lane towards molecular complexity. Chem. Eur. J. 2008, 14, 8444–8454. [Google Scholar] [CrossRef]
- D’Souza, D.M.; Müller, T.J.J. Multi-component syntheses of heterocycles by transition-metal catalysis. Chem. Soc. Rev. 2007, 36, 1095–1108. [Google Scholar]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar]
- Orru, R.V.A.; de Greef, M. Recent advances in solution-phase multicomponent methodology for the synthesis of heterocyclic compounds. Synthesis 2003, 1471–1499. [Google Scholar]
- Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Maximizing synthetic efficiency: Multi-component transformations lead the way. Chem. Eur. J. 2000, 6, 3321–3329. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. Engl. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Ugi, I.; Dömling, A.; Werner, B. Since 1995 the new chemistry of multicomponent reactions and their libraries, including their heterocyclic chemistry. J. Heterocycl. Chem. 2000, 37, 647–658. [Google Scholar] [CrossRef]
- Weber, L.; Illgen, K.; Almstetter, M. Discovery of new multi-component reactions with combinatorial methods. Synlett 1999, 366–374. [Google Scholar]
- Armstrong, R.W.; Combs, A.P.; Tempest, P.A.; Brown, S.D.; Keating, T.A. Multiple-component condensation strategies for combinatorial library synthesis. Acc. Chem. Res. 1996, 29, 123–131. [Google Scholar] [CrossRef]
- Ugi, I.; Dömling, A.; Hörl, W. Multicomponent reactions in organic chemistry. Endeavour 1994, 18, 115–122. [Google Scholar] [CrossRef]
- Posner, G.H. Multicomponent one-pot annulations forming 3 to 6 bonds. Chem. Rev. 1986, 86, 831–844. [Google Scholar]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar]
- Tietze, L.F.; Beifuss, U. Sequential transformations in organic chemistry: A synthetic strategy with a future. Angew. Chem. Int. Ed. Engl. 1993, 32, 131–163. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino-reactions: The tandem-Knoevenagel-hetero-Diels-Alder reaction and its application in natural product synthesis. J. Heterocycl. Chem. 1990, 27, 47–69. [Google Scholar] [CrossRef]
- Coquerel, Y.; Boddaert, T.; Presset, M.; Mailhol, D.; Rodriguez, J. Multiple bond-forming transformations: The key concept toward eco-compatible synthetic organic chemistry. In Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 187–202. [Google Scholar]
- Vries, J.G. The Heck reaction in the production of fine chemicals. Can. J. Chem. 2001, 79, 1086–1092. [Google Scholar] [CrossRef]
- Pommer, H. The Wittig reaction in industrial practice. Angew. Chem. Int. Ed. Engl. 1977, 16, 423–429. [Google Scholar] [CrossRef]
- Müller, T.J.J. Palladium-copper catalyzed alkyne activation as an entry to multicomponent syntheses of heterocycles. Top. Heterocycl. Chem. 2010, 25, 25–94. [Google Scholar] [CrossRef]
- Willy, B.; Müller, T.J.J. Multi-component heterocycle syntheses via catalytic generation of alkynones. Curr. Org. Chem. 2009, 13, 1777–1790. [Google Scholar] [CrossRef]
- Willy, B.; Müller, T.J.J. Consecutive multi-component syntheses of heterocycles via palladium-copper catalyzed generation of alkynones. ARKIVOC 2008, 195–208. [Google Scholar]
- Müller, T.J.J. Multi-component syntheses of heterocycles by virtue of palladium catalyzed generation of alkynones and chalcones. Targets Heterocycl. Syst. 2006, 10, 54–65. [Google Scholar]
- Willy, B.; Müller, T.J.J. Regioselective three-component synthesis of highly fluorescent 1,3,5-trisubstituted pyrazoles. Eur. J. Org. Chem. 2008, 4157–4168. [Google Scholar]
- Willy, B.; Müller, T.J.J. Rapid one-pot, four-step synthesis of highly fluorescent 1,3,4,5-tetrasubstituted pyrazoles. Org. Lett. 2011, 13, 2082–2085. [Google Scholar]
- Bagley, M.C.; Lubinu, M.C.; Mason, C. Regioselective microwave-assisted synthesis of substituted pyrazoles from ethynyl ketones. Synlett 2007, 704–708. [Google Scholar]
- Evans, G.B.; Furneaux, R.H.; Gainsford, G.J.; Hanson, J.C.; Kicska, G.A.; Sauve, A.A.; Schramm, V.L.; Tyler, P.C. 8-Aza-immucillins as transition-state analogue inhibitors of purine nucleoside phosphorylase and nucleoside hydrolases. J. Med. Chem. 2003, 46, 155–160. [Google Scholar]
- Zhou, J.; Yang, M.; Akdag, A.; Wang, H.; Schneller, S.W. Carbocyclic 4′-epi-formycin. Tetrahedron 2008, 64, 433–438. [Google Scholar]
- Zhou, J.; Yang, M.; Schneller, S.W. A model study to carbocyclic formycin A and B analogues. Tetrahedron Lett. 2004, 45, 8233–8234. [Google Scholar]
- Buchanan, J.G.; Edgar, A.R.; Hutchison, R.J.; Stobie, A.; Wightman, R.H. C-nucleoside studies. Part 10. A new synthesis of 3-(2,3,5-tri-O-benzyl-β-D-ribofuranosyl)pyrazole and its conversion into 4-nitro-3(5)-β-D-ribofuranosylpyrazole. J. Chem. Soc. Perkin Trans. 1 1980, 2567–2571. [Google Scholar]
- Buchanan, J.G.; Dunn, A.D.; Edgar, A.R.; Hutchison, R.J.; Power, M.J.; Williams, G.C. C-nucleoside studies. Part 6. Synthesis of 3-[2,3,5-tri-O-benzyl-β-(and α)-D-ribofuranosyl]prop-2-yn-1-ol and related compounds; a new synthesis of 3(5)-(2,3,5-tri-O-benzyl-β-D-ribofuranosyl)pyrazole. J. Chem. Soc. Perkin Trans. 1 1977, 1786–1791. [Google Scholar]
- Hüttel, R.; Büchele, F.; Jochum, P. Über Nitro-, Nitroso- und Azopyrazole. Chem. Ber. 1955, 88, 1577–1585. [Google Scholar] [CrossRef]
- Karpov, A.S.; Müller, T.J.J. New entry to a three-component pyrimidine synthesis by TMS-ynones via Sonogashira coupling. Org. Lett. 2003, 5, 3451–3454. [Google Scholar] [CrossRef]
- Bagley, M.C.; Hughes, D.D.; Sabo, H.M.; Taylor, P.H.; Xiong, X. One-pot synthesis of pyridines or pyrimidines by tandem oxidation-heteroannulation of propargylic alcohols. Synlett 2003, 1443–1446. [Google Scholar]
- Lemhadri, M.; Doucet, H.; Santelli, M. Sonogashira reaction of aryl halides with propiolaldehydediethyl acetal catalyzed by a tetraphosphine/palladium complex. Tetrahedron 2005, 61, 9839–9847. [Google Scholar]
- Cacchi, S.; Fabrizi, G.; Marinelli, F.; Moro, L.; Pace, P. Palladium-catalysed hydroarylation and hydrovinylation of 3,3-dialkoxy-1-aryl-1-propynes. An approach to 3-aryl- and 3-vinylquinolines. Tetrahedron 1996, 52, 10225–10240. [Google Scholar] [CrossRef]
- Sakamoto, T.; Shiga, F.; Yasuhara, A.; Uchiyama, D.; Kondo, Y.; Yamanaka, H. Preparation of ethyl arylpropiolates from aryl iodides by palladium-catalyzed cross-coupling reaction. Synthesis 1992, 746–748. [Google Scholar]
- Stoit, A.; Coolen, H.K.A.C.; Kruse, C.G.; Terwel, L.J.H. Heterocyclic compounds with affinity to muscarinic receptors. PCT Int. Appl. WO 2008129054 A2, 30 October 2008..
- Buchanan, J.G.; Quijano, M.L.; Wightman, R.H. C-nucleoside studies. Part 23. New and more direct synthesis of 3-(β-D-xylofuranosyl)pyrazole. J. Chem. Soc. Perkin Trans. 1 1992, 1573–1576. [Google Scholar]
- Jones, R.G.; Mann, M.J. New methods of synthesis of p-aminoethylpyrazoles. J. Am. Chem. Soc. 1953, 75, 4048–4052. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Levi, L.; Boersch, C.; Gers, C.F.; Merkul, E.; Müller, T.J.J. Consecutive Three-Component Synthesis of 3-(Hetero)Aryl-1H-pyrazoles with Propynal Diethylacetal as a Three-Carbon Building Block. Molecules 2011, 16, 9340-9356. https://doi.org/10.3390/molecules16119340
Levi L, Boersch C, Gers CF, Merkul E, Müller TJJ. Consecutive Three-Component Synthesis of 3-(Hetero)Aryl-1H-pyrazoles with Propynal Diethylacetal as a Three-Carbon Building Block. Molecules. 2011; 16(11):9340-9356. https://doi.org/10.3390/molecules16119340
Chicago/Turabian StyleLevi, Lucilla, Christina Boersch, Charlotte F. Gers, Eugen Merkul, and Thomas J. J. Müller. 2011. "Consecutive Three-Component Synthesis of 3-(Hetero)Aryl-1H-pyrazoles with Propynal Diethylacetal as a Three-Carbon Building Block" Molecules 16, no. 11: 9340-9356. https://doi.org/10.3390/molecules16119340
APA StyleLevi, L., Boersch, C., Gers, C. F., Merkul, E., & Müller, T. J. J. (2011). Consecutive Three-Component Synthesis of 3-(Hetero)Aryl-1H-pyrazoles with Propynal Diethylacetal as a Three-Carbon Building Block. Molecules, 16(11), 9340-9356. https://doi.org/10.3390/molecules16119340