Abstract
A new strategy for cyclopenta[b]furan synthesis mediated by hypervalent iodine(III) has been described. The approach employs diacetoxyiodobenzene-induced initial dehydrogenation to a putative trienone intermediate and triggered sequential cycloisomerization to form the cyclo-penta[b]furan targets.
1. Introduction
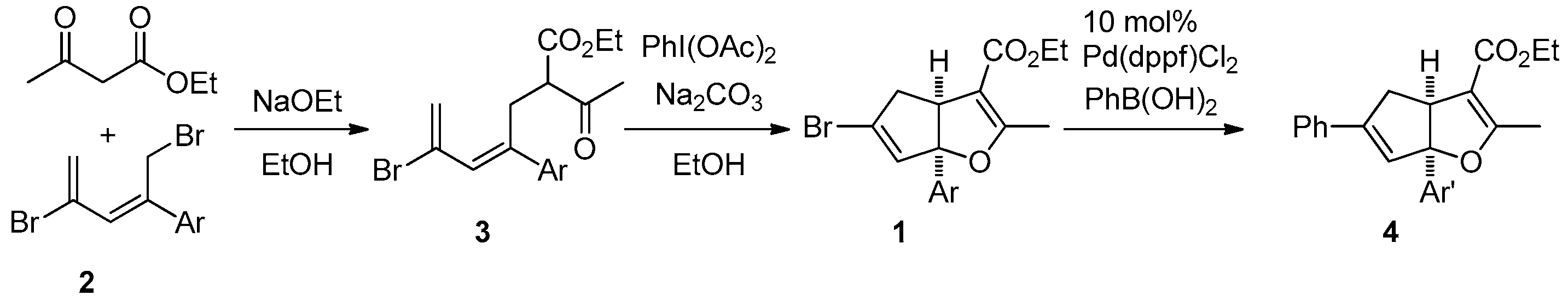
Recent medicinal chemical studies have led to the identification of the cyclopenta[b]furan ring system as a potent inhibitor of CCR2, a chemokine receptor that plays an important role in inflammation-associated diseases [1,2]. Syntheses of the cyclopenta[b]furan core component have been achieved by employing palladium(0)-catalyzed intramolecular allylations (path a, Scheme 1), [3] and phosphine-catalyzed [3 + 2] cycloadditions of allenoates with β,γ-unsaturated α-ketoesters (path b, Scheme 1) [4]. In addition, the construction of tricyclic cyclopenta[b]furan derivatives was elaborated through ethylenediammonium diacetate (EDDA)-catalyzed tandem Knoevenagel-polycyclization reactions between 1,3-dicarbonyl compounds and α,β,γ,δ-unsaturated aldehydes (path c, Scheme 1) or Lewis acid-promoted cycloisomerization of conjugated trienones (path d, Scheme 1) [5,6]. On the other hand, hypervalent hypervalent iodine species possess similar chemical properties and reactivity as heavy metal reagents but lower toxicity and an environmentally benign nature. Therefore, the utilization of hypervalent iodine in organic synthesis has been drawing intensive attention [7,8,9,10,11,12]. Among its reactions, the reactions of 1,3-dicarbonyl compounds with hypervalent iodine(III) species such as diacetoxyiodobenzene generate β-diketo iodonium ylides which behave as a 1,3-dipole, with an electrophilic center next to a carbonyl group. For example, the synthetic application of hypervalent iodine(III) in forming heterocycles involves the addition of β-diketo iodonium ylides to olefins to form fused dihydrofurans (path e, Scheme 1) [13,14]. Recent progress in the use of hypervalent iodine(III) reagents in organic synthesis has shown that diacetoxyiodobenzene-mediated dehydrogenation reactions of α-alkyl-β-dicarbonyl compounds provide an alternative route to the Knoevenagel condensation [15,16]. Herein, we have devised a route for employing hypervalent iodine(III) in the synthesis of cyclopenta[b]furan derivatives 1 (Scheme 2).
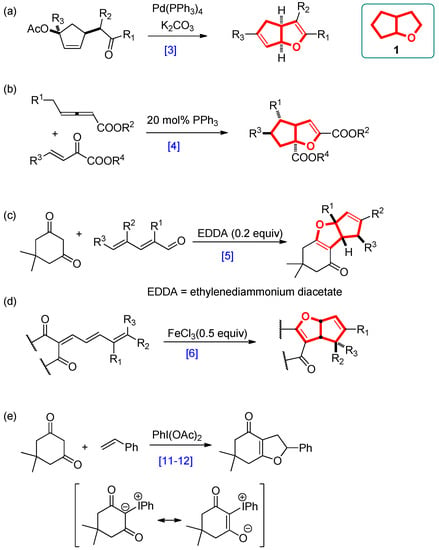
Scheme 1.
Routes for the synthesis of cyclopenta[b]furans.
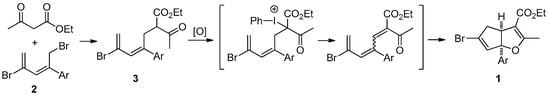
Scheme 2.
Synthetic plans for the synthesis of cyclopenta[b]furan derivatives 1.
2. Results and Discussions
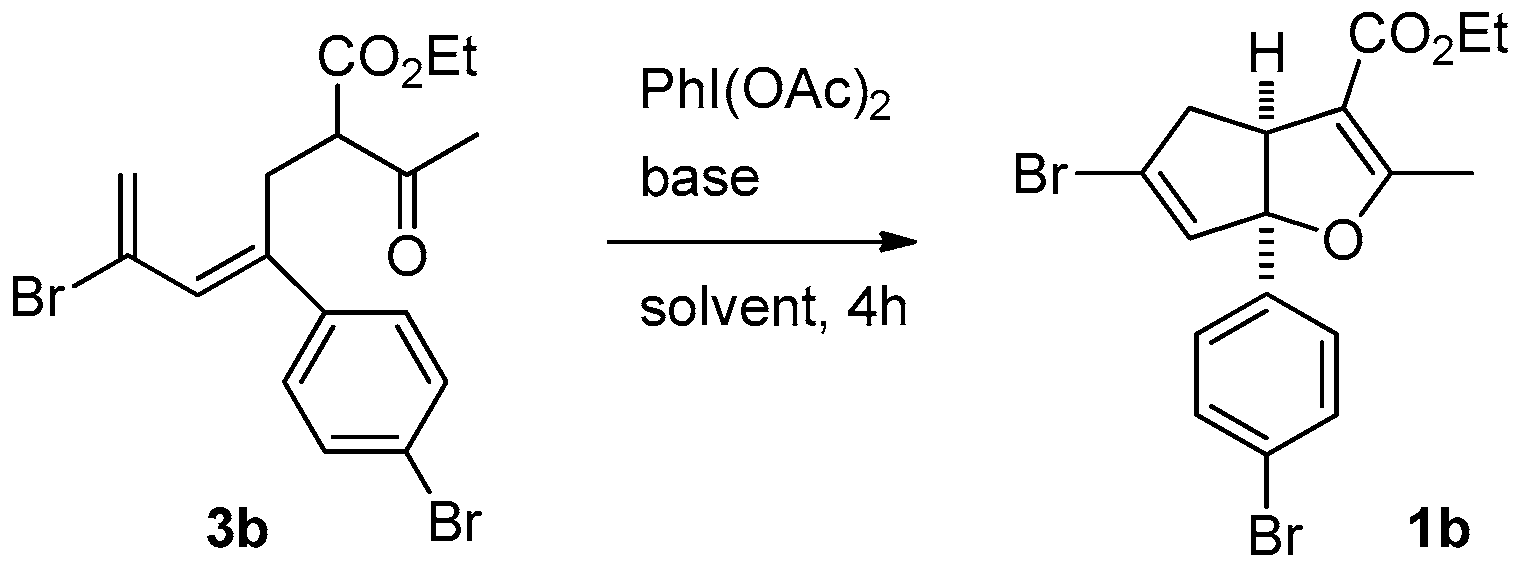
In the study described below, we have explored a method for the synthesis of cyclopenta[b]furans containing a stereogenic, quaternary, aryl-substituted bridgehead carbon. For the preparation of 2-aryl-1,4-dibromopenta-2,4-dienes 2 we used a literature procedure developed in our group [17]. The β-ketoesters linked with pentadiene tether 3 were prepared by C-alkylation of ethyl acetoacetate enolate with 2; however, the chromatographic purification of compounds 3 is difficult in most cases (Table 1). In fact, only 3h can be isolated in pure form by recrystallization after chromatographic purification. Crude analogues 3a–g as obtained after silica-gel plug treatment were carried directly into the next step, therefore, purity and exact yields of 3a–g are not given in Table 1. In order to evaluate the feasibility of the new strategy for cyclopenta[b]furan synthesis, 3b serves as effective substrate in the reaction with diacetoxyiodobenzene that takes place by a one-pot, initial dehydrogenation to give a putative trienone intermediate and sequential cycloisomerization to form the cyclopenta[b]furan (Table 2). The results show that the putative trienone produced in these reactions is not isolable using chromatography, and at ambient temperature only the formation of the trienone was observed (Table 2, entries 1 and 8). At elevated temperature, the putative trienone underwent cycloisomerization to form cyclopenta[b]furans (entry 1 vs. entry 2, entry 7 vs. entry 8), reactions in which inorganic bases gave better yields than organic bases (entry 4 vs. entry 5, entry 8 vs. entry 9). In addition, the oxidation-cycloisomerization process happened smoothly when [bis(trifluoroacetoxy)iodo]benzene was employed. The ideal conditions for this process involve the use of diacetoxyiodobenzene and sodium carbonate in refluxing ethanol (Table 2, entry 7). Next, a variety of aryl-substituted substrates 3 were examined and Suzuki reactions were pursued in order to confirm the structural determination and the relative stereochemistry at the ring junction of the cyclopenta[b]furans 1 (Table 1).

Table 1.
Synthesis of cyclopenta[b]furans.
Table 2.
Reaction Optimizations a.
The stereochemistry of the fused-bicyclic ring system in 4b was shown to be cis using X-ray crystallographic analysis (Figure 1 and Supplementary Material) [18].
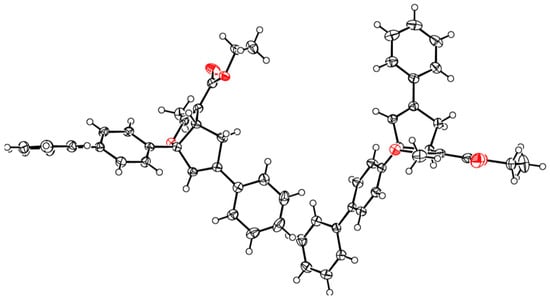
Figure 1.
ORTEP plot of X-ray crystallographic data for 4b.
A proposed plausible mechanism is shown in Scheme 3. First, the formation of putative trienone A through diacetoxyiodobenzene-mediated dehydrogenation reaction gave the newly formed C–C double bond as E/Z-form mixtures or ones where the Z-isomer predominated [15]. However, only the E-isomer possesses the proper steric alignment to form the target cyclopenta[b]furans 1. Therefore, a simple thermal cycloisomerization of the trienone to a cyclopenta[b]furan is not likely in this scenario. In the diacetoxyiodobenzene-mediated dehydrogenation process at elevated temperature, acetic acid produced during the reaction presumably plays an important role in promoting the sequential cycloisomerization from a cationic dienyl structure B to form the cyclopenta[b]furans.
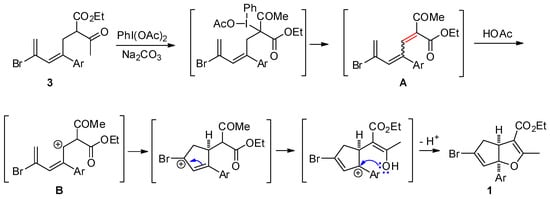
Scheme 3.
Plausible Mechanism.
3. Materials and Methods
3.1. General Information
All commercially available chemicals were used without further purification. TLC analyses were run on a TLC glass plate (silica gel 60 F254, EMD Millipore, Darmstadt, Germany) and were visualized using UV and a solution of phosphomolybdic acid in ethanol (5 wt %) or p-anisaldehyde stain. Flash chromatography was performed using silica gel (70–230 mesh, EMD Millipore). 1H-NMR spectra were recorded on a 300 MHz spectrometer (Bruker AV-300, Bruker BioSpin GmbH, Karlsruhe, Germany). 13C-NMR spectra were recorded at 75 MHz on the same instrument with complete proton decoupling (see Supplementary Material). Chemical shifts are reported relative to CHCl3 [δH 7.24, δC (central line) 77.0]. Mass spectra and high-resolution mass spectra were recorded on the Thermo/Finnigan Quest MAT Mass Spectrometer (Thermo Finnigan LLC, San Jose, CA, USA).
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Cyclopenta[b]furans 1
A mixture of bromodiene 2 (0.3 mmol), ethyl acetoacetate (0.3 mmol, 39 mg), and NaOEt (0.4 mmol, 27 mg) in EtOH (2 mL) was stirred at reflux overnight. The mixture was cooled to ambient temperature (25–28 °C) and transferred to a separatory funnel, followed by addition of Et2O (5 mL) and water (10 mL). The aqueous layer was back extracted with Et2O (2 mL × 2). The combined organic layers were washed with brine (5 mL), dried over MgSO4, and concentrated in a rotary evaporator. The residue was purified by a silica gel plug using Et2O/hexanes (10/1, 10 mL) as the eluent, followed by concentration to give the bromoester 3. To a solution of bromoester 3 (0.3 mmol, based on bromo-diene 2) in EtOH (1.2 mL) was added Na2CO3 (0.6 mmol, 64 mg) and PhI(OAc)2 (0.6 mmol, 193 mg) at ambient temperature. The resulting mixture was heated to reflux under N2 for 3 h. The reaction was then cooled room temperature and H2O (2 mL) was added. Et2O (5 mL) was added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with Et2O (5 mL × 2). The combined organic layers were washed with 2 N HCl (2 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography using Et2O/hexanes (1/10) as eluent to give the title products as yellow oils. Using this general procedure, the following title compounds were obtained:
Ethyl 5-bromo-2-methyl-6a-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1a). Yield 55 mg (72%). TLC (Et2O/hexanes (1:10)) Rf = 0.44; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.83 (dt, J = 17.4, 2.1 Hz, 1H), 3.15 (ddd, J = 17.4, 7.8, 2.1 Hz, 1H), 3.74 (dt, J = 7.8, 1.5 Hz, 1H), 4.09–4.20 (m, 2H), 5.98 (t, J = 2.1 Hz, 1H), 7.25–7.35 (m, 5H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 46.9 (CH2), 53.9 (CH), 59.6 (CH2), 100.8 (C) 105.8 (C), 124.5 (CH × 2), 128.0 (CH), 128.6 (CH × 2), 129.1 (C), 131.9 (CH), 142.0 (C), 165.5 (C), 167.0 (C); IR (neat) 2977, 1698, 1644 cm−1; EI-MS m/z (rel intensity) 350 ([M + 2]+, 10), 348 ([M]+, 10), 302 (29), 223 (100); HRMS [M]+ calcd. for C17H17BrO3: 348.0361, found 348.0366.
Ethyl 5-bromo-6a-(4-bromophenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1b). Yield 100 mg (78%). TLC (Et2O/hexanes (1:10)) Rf = 0.43; 1H-NMR (CDCl3) δ 1.26 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.80 (dt, J = 17.4, 1.8 Hz, 1H), 3.14 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.69 (dt, J = 7.8, 1.5 Hz, 1H), 4.06–4.28 (m, 2H), 5.93 (t, J = 1.5 Hz, 1H), 7.15 (d, J = 7.8 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.4 (CH3), 46.8 (CH2), 53.9 (CH), 59.7 (CH2), 100.3 (C) 105.9 (C), 122.0 (C), 126.3 (CH × 2), 129.6 (C), 131.5 (CH), 131.7 (CH × 2), 141.1 (C), 165.4 (C), 166.8 (C); IR (neat) 2976, 1703, 1488 cm−1; EI-MS m/z (rel intensity) 428 ([M + 2]+, 19), 426 ([M]+, 10), 382 (59), 301 (100); HRMS [M]+ calcd. for C17H16Br2O3: 425.9466, found 425.9462.
Ethyl 5-bromo-6a-(4-chlorophenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1c). Yield 104 mg (90%). TLC (Et2O/hexanes (1:10)) Rf = 0.43; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.83 (dt, J = 17.4, 2.1 Hz, 1H), 3.13 (ddd, J = 17.4, 7.8, 2.1 Hz, 1H), 3.68 (dt, J = 7.8, 1.5 Hz, 1H), 4.09–4.20 (m, 2H), 5.93 (t, J = 2.1 Hz, 1H), 7.20 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 8.7 Hz, 2H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.3 (CH3), 46.8 (CH2), 53.9 (CH), 59.6 (CH2), 100.2 (C) 105.9 (C), 125.9 (CH × 2), 128.7 (CH × 2), 129.5 (C), 131.5 (CH), 133.8 (C), 140.5 (C), 165.3 (C), 166.7 (C); IR (neat) 2977, 1700, 1646 cm−1; EI-MS m/z (rel intensity) 384 ([M + 2]+, 15), 382 ([M]+, 12), 338 (44), 257 (100); HRMS [M]+ calcd. for C17H16BrClO3: 381.9971, found 381.9978.
Ethyl 5-bromo-2-methyl-6a-(p-tolyl)-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1d). Yield 76 mg (70%). TLC (Et2O/hexanes (1:10)) Rf = 0.51; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.32 (s, 3H), 2.82 (dt, J = 17.4, 1.8 Hz, 1H), 3.13 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.72 (dt, J = 7.8, 1.5 Hz, 1H), 4.09–4.20 (m, 2H), 5.97 (t, J = 1.8 Hz, 1H), 7.15 (s, 4H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 21.0 (CH3), 46.8 (CH2), 53.8 (CH), 59.5 (CH2), 100.7 (C) 105.8 (C), 124.4 (CH × 2), 128.9 (C), 129.2 (CH × 2), 131.9 (CH), 137.8 (C), 139.0 (C), 165.6 (C), 167.0 (C); IR (neat) 2979, 1702, 1644 cm−1; EI-MS m/z (rel intensity) 364 ([M + 2]+, 5), 362 ([M]+, 6), 316 (19), 237 (100); HRMS [M]+ calcd. for C18H19BrO3: 362.0518, found 362.0515.
Ethyl 5-bromo-6a-(4-methoxyphenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1e). Yield 74 mg (65%). TLC (Et2O/hexanes (1:10)) Rf = 0.48; 1H-NMR (CDCl3) δ 1.25 (t, J = 7.2 Hz, 3H), 2.27 (d, J = 1.5 Hz, 3H), 2.81 (dt, J = 17.4, 1.8 Hz, 1H), 3.12 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.71 (dt, J = 7.8, 1.5 Hz, 1H), 3.78 (s, 3H), 4.06–4.22 (m, 2H), 5.97 (t, J = 1.8 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.7 Hz, 2H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 46.8 (CH2), 53.7 (CH), 55.3 (CH3), 59.5 (CH2), 100.6 (C), 105.8 (C), 113.9 (CH × 2), 125.8 (CH × 2), 128.8 (C), 131.9 (CH), 134.1 (C), 159.3 (C), 165.6 (C), 166.9 (C); IR (neat) 2981, 1698, 1643 cm−1; EI-MS m/z (rel intensity) 380 ([M + 2]+, 6), 378 ([M]+, 6), 332 (20), 253 (100); HRMS [M]+ calcd. for C18H19BrO4: 378.0467, found 378.0458.
Ethyl 5-bromo-6a-(3-methoxyphenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1f). Yield 72 mg (63%). TLC (Et2O/hexanes (1:10)) Rf = 0.45; 1H-NMR (CDCl3) δ 1.24 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.82 (dt, J = 17.4, 1.8 Hz, 1H), 3.14 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.73 (dt, J = 7.8, 1.8 Hz, 1H), 3.79 (s, 3H), 4.03–4.24 (m, 2H), 5.95 (t, J = 1.8 Hz, 1H), 6.79–6.86 (m, 3H), 7.24–7.29 (m, 1H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.3 (CH3), 46.8 (CH2), 53.8 (CH), 55.2 (CH3), 59.5 (CH2), 100.6 (C) 105.8 (C), 110.7 (CH), 112.7 (CH), 116.7 (CH), 129.0 (C), 129.7 (CH), 131.8 (CH), 143.6 (C), 159.7 (C), 165.5 (C), 166.9 (C); IR (neat) 2981, 1702, 1644 cm−1; EI-MS m/z (rel intensity) 380 ([M + 2]+, 10), 378 ([M]+, 10), 332 (21), 253 (100); HRMS [M]+ calcd. for C18H19BrO4: 378.0467, found 378.0461.
Ethyl 5-bromo-6a-(3-bromophenyl)-2-methyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1g). Yield 112 mg (87%). TLC (Et2O/hexanes (1:10)) Rf = 0.53; 1H-NMR (CDCl3) δ 1.24 (t, J = 7.2 Hz, 3H), 2.28 (d, J = 1.5 Hz, 3H), 2.83 (dt, J = 17.7, 2.1 Hz, 1H), 3.15 (ddd, J = 17.7, 7.8, 2.1 Hz, 1H), 3.70 (dt, J = 7.8, 2.1 Hz, 1H), 4.06–4.21 (m, 2H), 5.95 (t, J = 2.1 Hz, 1H), 7.15–7.20 (m, 2H), 7.37–7.41 (m, 2H); 13C-NMR (CDCl3) δ 14.2 (CH3), 14.3 (CH3), 46.7 (CH2), 53.9 (CH), 59.6 (CH2), 99.9 (C) 105.8 (C), 122.7 (C), 123.7 (CH), 127.6 (CH), 129.6 (CH), 130.1 (CH), 131.0 (CH), 131.3 (CH), 144.2 (C), 165.2 (C), 166.6 (C); IR (neat) 2979, 1704, 1643 cm−1; EI-MS m/z (rel intensity) 428 ([M + 2]+, 19), 426 ([M]+, 10), 382 (59), 301 (100); HRMS [M]+ calcd. for C17H16Br2O3: 425.9466, found 425.9460.
Ethyl 5-bromo-2-methyl-6a-(naphthalen-2-yl)-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (1h). Yield 77 mg (64%). TLC (Et2O/hexanes (1:10)) Rf = 0.48; 1H-NMR (CDCl3) δ 1.26 (t, J = 7.2 Hz, 3H), 2.35 (d, J = 1.5 Hz, 3H), 2.90 (dt, J = 17.4, 1.8 Hz, 1H), 3.22 (ddd, J = 17.4, 7.8, 1.8 Hz, 1H), 3.84 (dt, J = 7.8, 1.8 Hz, 1H), 4.11–4.23 (m, 2H), 6.09 (t, J = 1.8 Hz, 1H), 7.35 (dd, J = 8.4, 1.8 Hz, 1H), 7.44–7.51 (m, 2H), 7.74 (s, 1H), 7.80–7.85 (m, 3H); 13C-NMR (CDCl3) δ 14.3 (CH3), 14.4 (CH3), 46.9 (CH2), 53.7 (CH), 59.6 (CH2), 100.9 (C) 105.9 (C), 112.6 (CH), 123.1 (CH), 126.2 (CH), 126.4 (CH), 127.6 (CH), 128.1 (CH), 128.7 (CH), 129.3 (C), 131.8 (CH), 132.8 (C), 132.9 (C), 139.0 (C), 165.5 (C), 167.0 (C); IR (neat) 3058, 1700, 1637 cm−1; EI-MS m/z (rel intensity) 400 ([M + 2]+, 11), 398 ([M]+, 11), 273 (99), 202 (100); HRMS [M]+ calcd. for C21H19BrO3: 398.0518, found 398.0516.
3.2.2. General Procedure for the Suzuki Reactions of Cyclopenta[b]furans 1
To a solution of cyclopenta[b]furan 1 (0.5 mmol) in dioxane/H2O (5 mL/1 mL) was added Cs2CO3 (1.5 mmol, 489 mg), PhB(OH)2 (0.75 mmol, 91 mg), and Pd(dppf)Cl2 (0.05 mmol, 37 mg) at ambient temperature. The resulting mixture was vacuumed and purged with N2 for 3 times. The reaction was heated to 100 °C under N2 for 2 h. The reaction was then cooled room temperature and concentrated in a rotary evaporator. EtOAc (5 mL) and H2O (1 mL) were added and the mixture was transferred to a separatory funnel. The aqueous layer was back extracted with EtOAc (5 mL × 2). The combined organic layers were dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The residue was purified by silica gel chromatography to give the title product. The following compounds were prepared using this method:
Ethyl 2-methyl-5,6a-diphenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4a). Yield 149 mg (86%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.50; 1H-NMR (CDCl3) δ 1.29 (t, J = 7.2 Hz, 3H), 2.31 (s, 3H), 3.02 (dt, J = 17.1, 2.1 Hz, 1H), 3.29 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.88 (dt, J = 7.8, 1.5 Hz, 1H), 4.11–4.26 (m, 2H), 6.23 (t, J = 2.1 Hz, 1H), 7.26–7.39 (m, 8H), 7.54 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 40.2, 52.8, 59.3, 102.3, 106.4, 124.6, 125.5, 126.4, 127.5, 128.4, 128.5, 134.7, 143.1, 146.5, 165.9, 166.6; IR (neat) 2977, 1698, 1644 cm−1; EI-MS m/z (rel intensity) 346 ([M]+, 12), 300 (100), 257 (30), 229 (47); HRMS [M]+ calcd. for C23H22O3: 346.1569, found 346.1561.
Ethyl 6a-([1,1′-biphenyl]-4-yl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4b). Yield 180 mg (85%). A colorless solid, m.p. 118–120 °C; TLC (EtOAc/hexanes (1:10)) Rf = 0.35; 1H-NMR (CDCl3) δ 1.28 (t, J = 7.2 Hz, 3H), 2.31 (s, 3H), 3.02 (dt, J = 17.1, 1.8 Hz, 1H), 3.31 (ddd, J = 17.1, 6.0, 2.1 Hz, 1H), 3.91 (dt, J = 6.0, 1.8 Hz, 1H), 4.10–4.25 (m, 2H), 6.25 (t, J = 1.8 Hz, 1H), 7.30–7.45 (m, 8H), 7.53–7.55 (m, 6H); 13C-NMR (CDCl3) δ 14.3, 40.1, 52.8, 59.3, 102.2, 106.4, 125.0, 125.4, 126.3, 126.9, 127.1, 127.2, 128.3, 128.4, 128.6, 134.6, 140.4, 140.5, 142.1, 146.5, 165.8, 166.5; IR (neat) 3031, 1700, 1637 cm−1; EI-MS m/z (rel intensity) 422 ([M]+, 10), 376 (100), 333 (13), 305 (23); HRMS [M]+ calcd. for C29H26O3: 422.1882, found 422.1878.
Ethyl 6a-(4-chlorophenyl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4c). Yield 164 mg (86%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.43; 1H-NMR (CDCl3) δ 1.31 (t, J = 7.2 Hz, 3H), 2.32 (s, 3H), 3.04 (dt, J = 17.1, 2.1 Hz, 1H), 3.30 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.84 (dt, J = 7.8, 1.8 Hz, 1H), 4.13–4.29 (m, 2H), 6.19 (t, J = 1.8 Hz, 1H), 7.27–7.40 (m, 7H), 7.54 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 40.1, 52.9, 59.4, 101.8, 106.4, 124.6, 125.0, 126.1, 128.4, 128.5, 128.6, 133.3, 134.5, 141.7, 146.9, 165.7, 166.4; IR (neat) 2979, 1698, 1643 cm−1; EI-MS m/z (rel intensity) 382 ([M + 2]+, 4), 380 ([M]+, 10), 334 (100), 228 (36); HRMS [M]+ calcd. for C23H21ClO3: 380.1179, found 380.1184.
Ethyl 2-methyl-5-phenyl-6a-(p-tolyl)-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4d). Yield 151 mg (84%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.60; 1H-NMR (CDCl3) δ 1.31 (t, J = 7.2 Hz, 3H), 2.32 (s, 3H), 2.36 (s, 3H), 3.03 (dt, J = 17.1, 2.1 Hz, 1H), 3.30 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.88 (dt, J = 7.8, 1.8 Hz, 1H), 4.17–4.25 (m, 2H), 6.24 (t, J = 1.8 Hz, 1H), 7.18 (d, J = 8.1 Hz, 2H), 7.25–7.40 (m, 5H), 7.56 (d, J = 8.1 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 14.5, 21.0, 40.2, 52.8, 59.3, 102.4, 106.4, 124.6, 125.6, 126.4, 128.4, 128.5, 129.1, 134.8, 137.3, 140.2, 146.3, 166.0, 166.6; IR (neat) 2977, 2923, 1693 cm−1; EI-MS m/z (rel intensity) 360 ([M]+, 11), 314 (100), 243 (27), 228 (20); HRMS [M]+ calcd. for C24H24O3: 360.1725, found 360.1721.
Ethyl 6a-(4-methoxyphenyl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4e). Yield 154 mg (82%). A red oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.50; 1H-NMR (CDCl3) δ 1.29 (t, J = 7.2 Hz, 3H), 2.29 (s, 3H), 2.99 (dt, J = 17.1, 1.8 Hz, 1H), 3.26 (ddd, J = 17.1, 6.6, 2.1 Hz, 1H), 3.79 (s, 3H), 3.83 (dt, J = 6.6, 1.8 Hz, 1H), 4.13–4.24 (m, 2H), 6.22 (t, J = 1.8 Hz, 1H), 6.88 (d, J = 6.6 Hz, 2H), 7.25–7.38 (m, 5H), 7.53 (d, J = 7.8 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 14.5, 40.1, 52.7, 55.2, 59.4, 102.2, 106.4, 113.7, 125.5, 125.9, 126.4, 128.4, 128.5, 134.8, 135.3, 146.3, 159.0, 166.0, 166.6; IR (neat) 2931, 1696, 1644 cm−1; EI-MS m/z (rel intensity) 376 ([M]+, 18), 330 (100), 287 (19), 259 (33); HRMS [M]+ calcd. for C24H24O4: 376.1675, found 376.1672.
Ethyl 6a-(3-methoxyphenyl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4f). Yield 152 mg (81%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.44; 1H-NMR (CDCl3) δ 1.30 (t, J = 7.2 Hz, 3H), 2.32 (s, 3H), 3.02 (dt, J = 17.1, 2.1 Hz, 1H), 3.30 (ddd, J = 17.1, 7.8, 2.1 Hz, 1H), 3.80 (s, 3H), 3.88 (dt, J = 7.8, 1.8 Hz, 1H), 4.16–4.24 (m, 2H), 6.22 (t, J = 1.8 Hz, 1H), 6.80–6.84 (m, 1H), 6.92–6.95 (m, 2H), 7.25–7.39 (m, 4H), 7.55 (d, J = 8.1 Hz, 2H); 13C-NMR (CDCl3) δ 14.4, 40.2, 52.8, 55.2, 59.4, 102.2, 106.5, 110.7, 112.4, 116.9, 125.4, 126.4, 128.4, 128.5, 129.5, 134.7, 144.8, 146.5, 159.7, 165.9, 166.5; IR (neat) 3056, 2979, 1693 cm−1; EI-MS m/z (rel intensity) 376 ([M]+, 18), 330 (100), 287 (48), 259 (41); HRMS [M]+ calcd. for C24H24O4: 376.1675, found 376.1680.
Ethyl 6a-([1,1′-biphenyl]-3-yl)-2-methyl-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4g). Yield 180 mg (85%). A red oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.53; 1H-NMR (CDCl3) δ 1.33 (t, J = 7.2 Hz, 3H), 2.38 (s, 3H), 3.10 (dt, J = 17.1, 1.8 Hz, 1H), 3.38 (ddd, J = 17.1, 6.0, 2.1 Hz, 1H), 3.99 (dt, J = 6.0, 1.8 Hz, 1H), 4.20–4.28 (m, 2H), 6.32 (t, J = 1.8 Hz, 1H), 7.34–7.49 (m, 8H), 7.53–7.63 (m, 6H); 13C-NMR (CDCl3) δ 14.4, 14.5, 40.3, 52.9, 59.4, 102.4, 106.5, 123.4, 123.6, 125.5, 126.4, 126.5, 127.2, 127.3, 128.4, 128.5, 128.7, 128.9, 134.7, 141.0, 141.5, 143.7, 146.6, 165.9, 166.6; IR (neat) 3060, 2981, 1706 cm−1; EI-MS m/z (rel intensity) 422 ([M]+, 13), 376 (100), 333 (23), 305 (27); HRMS [M]+ calcd. for C29H26O3: 422.1882, found 422.1884.
Ethyl 2-methyl-6a-(naphthalen-2-yl)-5-phenyl-4,6a-dihydro-3aH-cyclopenta[b]furan-3-carboxylate (4h). Yield 167 mg (84%). A yellow oil; TLC (EtOAc/hexanes (1:10)) Rf = 0.38; 1H-NMR (MHz, CDCl3) δ 1.31 (t, J = 7.2 Hz, 3H), 2.38 (s, 3H), 3.09 (dt, J = 17.1, 1.8 Hz, 1H), 3.37 (ddd, J = 17.1, 7.8, 1.8 Hz, 1H), 3.98 (d, J = 7.8 Hz, 1H), 4.14–4.29 (m, 2H), 6.34 (t, J = 1.8 Hz, 1H), 7.33–7.49 (m, 6H), 7.58–7.61 (m, 2H), 7.75–7.85 (m, 4H); 13C-NMR (CDCl3) δ 14.5, 14.6, 40.4, 52.9, 59.5, 102.6, 106.7, 123.2, 125.5, 126.1, 126.3, 126.5, 127.6, 128.2, 128.5, 128.6, 132.8, 133.1, 134.8, 140.4, 146.9, 166.1, 166.8; IR (neat) 3056, 2977, 1698 cm−1; EI-MS m/z (rel intensity) 396 ([M]+, 22), 350 (100), 307 (23), 279 (53); HRMS [M]+ calcd. for C27H24O3: 396.1725, found 396.1733.
4. Conclusions
In summary, the synthetic application of hypervalent iodine(III) in the formation of cyclopenta[b]furans is described. This method provides an alternative route to access the target for those cases where the corresponding diene-aldehyde precursor is not readily available or its use limited by intrinsic potential instability.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/12/1713/s1.
Acknowledgments
Financial support from the Ministry of Science and Technology of the Republic of China, Taiwan (MOST 105-2113-M-018-004) is gratefully acknowledged. We also thank the assistance from Chang-Chih Hsieh for the X-ray structure determination.
Author Contributions
M.-H.L. conceived and designed the experiments; Y.-C.C. and S.-H.C. performed the experiments. Y.-F.C. performed the X-ray analysis. T.-H.C. gave insight suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Winters, M.P.; Teleha, C.A.; Kang, F.-A.; McComsey, D.; O’Neill, J.C.; Hou, C.; Kirchner, T.; Wang, P.; Johnson, D.; Sui, Z. The discovery and SAR of cyclopenta[b]furans as inhibitors of CCR2. Bioorg. Med. Chem. Lett. 2014, 24, 2137–2140. [Google Scholar] [CrossRef] [PubMed]
- Teleha, C.A.; Branum, S.; Zhang, Y.; Reuman, M.E.; van der Steen, L.; Verbeek, M.; Fawzy, N.; Leo, G.C.; Winters, M.P.; Kang, F.-A.; et al. Lab-scale preparation of a novel cyclopenta[b]furan chemokine receptor antagonist. Org. Process Res. Dev. 2014, 18, 1630–1640. [Google Scholar] [CrossRef]
- Khan, P.M.; Wu, R.; Bisht, K.S. Pd(0) catalyzed intramolecular alkylation: Stereoselective synthesis of furan and isoxazoline-2-oxide analogs. Tetrahedron 2007, 63, 1116–1126. [Google Scholar] [CrossRef]
- Li, E.; Huang, Y. Phosphine-catalyzed domino reactions: A route to functionalized bicyclic skeletons. Chem. Eur. J. 2014, 20, 3520–3527. [Google Scholar] [CrossRef] [PubMed]
- Riveira, M.J.; Mischne, M.P. One-pot organocatalytic tandem Aldol/polycyclization reactions between 1,3-dicarbonyl compounds and α,β,γ,δ-unsaturated aldehydes for the straightforward assembly of cyclopenta[b]furan-type derivatives: New insight into the Knoevenagel reaction. Chem. Eur. J. 2012, 18, 2382–2388. [Google Scholar] [CrossRef] [PubMed]
- Riveira, M.J.; Mischne, M.P. An interrupted vinylogous iso-Nazarov reaction: Cycloisomerization of conjugated trienones to cyclopenta[b]furan derivatives. J. Org. Chem. 2014, 79, 8244–8254. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. (Ed.) Topics in Current Chemistry. Hypervalent Iodine Chemistry; Springer: Berlin, Germany, 2016; Volume 373, pp. 1–309.
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: Chichester, UK, 2014. [Google Scholar]
- Wirth, T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. Engl. 2005, 44, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, X.; Ren, C.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Synthesis of oxazoles from enamides via phenyliodine diacetate-mediated intramolecular oxidative cyclization. J. Org. Chem. 2012, 77, 10353–10361. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang-Negrerie, D.; Deng, J.; Du, Y.; Zhao, K. Metal-free synthesis of 2-oxindoles via PhI(OAc)2-mediated oxidative C–C bond formation. J. Org. Chem. 2014, 79, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-Q.; Li, Y.-M. Regioselective (diacetoxyiodo)benzene-promoted halocyclization of unfunctionalized olefins. J. Org. Chem. 2014, 79, 10094–10109. [Google Scholar] [CrossRef] [PubMed]
- Karade, N.N.; Shirodkar, S.G.; Patil, M.N.; Potrekar, R.A.; Karade, H.N. Diacetoxyiodobenzene-mediated oxidative addition of 1,3-dicarbonyl compounds to olefins: An efficient one-pot synthesis of 2,3-dihydrofuran derivatives. Tetrahedron Lett. 2003, 44, 6729–6731. [Google Scholar] [CrossRef]
- Kalpogiannaki, D.; Martini, C.-I.; Nikopoulou, A.; Nyxas, J.A.; Pantazi, V.; Hadjiarapoglou, L.P. Fused dihydrofurans from the one-pot, three-component reaction of 1,3-cyclohexanedione, iodobenzene diacetate and alkenes. Tetrahedron 2013, 69, 1566–1575. [Google Scholar] [CrossRef]
- Fan, L.; Chen, W.; Tang, K.; Wu, D. Stereoselective synthesis of α-ylidene-β-dicarbonyl compounds: A mild PhI(OAc)2-mediated dehydrogenation process. Chem. Lett. 2012, 41, 940–942. [Google Scholar] [CrossRef]
- Duan, Y.-N.; Cui, L.-Q.; Zuo, L.-H.; Zhang, C. Recyclable hypervalent-iodine-mediated dehydrogenative α,β′-bifunctionalization of β-keto esters under metal-free conditions. Chem. Eur. J. 2015, 21, 13052–13057. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-H.; Li, Y.-S.; Kuo, C.-K.; Chen, C.-H.; Huang, Y.-C.; Liang, K.-Y.; Chen, Y.-C.; Tsai, C.-H.; Chuang, T.-H. Synthesis of hexahydro-1H-isoindole derivatives from arylacyl bromides via homoallenic bromohydrins. J. Org. Chem. 2015, 80, 2462–2466. [Google Scholar] [CrossRef] [PubMed]
- The structure has been deposited with the Cambridge Crystallographic Data Centre (4b: CCDC 1517150). These Data Can be Obtained Free of Charge from The Cambridge Crystallographic Data Centre via. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 14 November 2016).
- Sample Availability: Samples of the compounds 1a–h, 4a–h are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).