
The Effect of an Optimized Wet Milling Technology on the Crystallinity, Morphology and Dissolution Properties of Micro- and Nanonized Meloxicam

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Effects of Milling Time on Particle size Distribution (PSD) and Specific Surface Area (SSA)
2.2. Characterization of the Dried Products
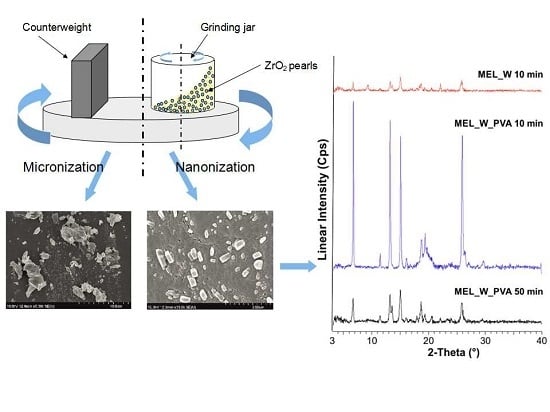
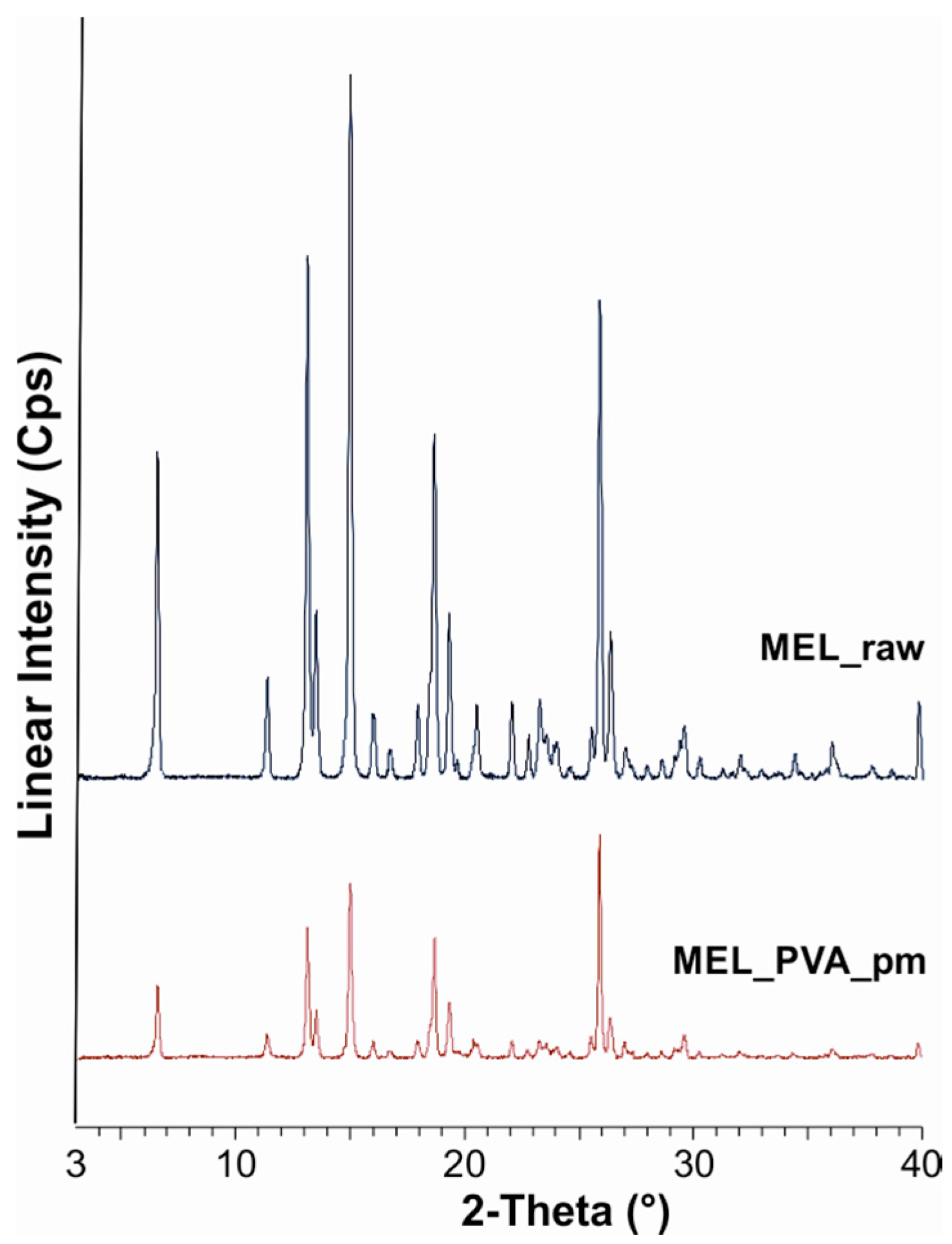
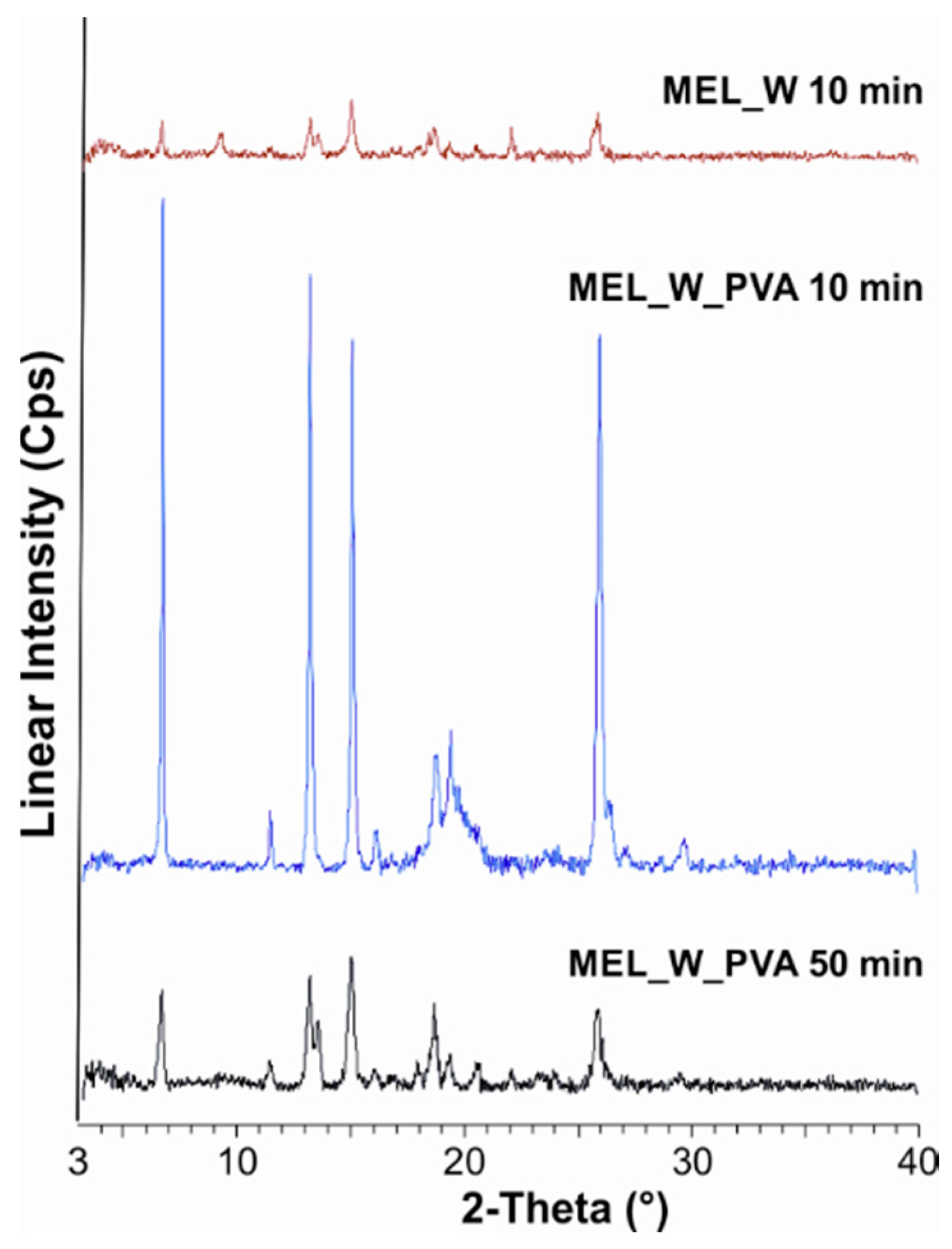
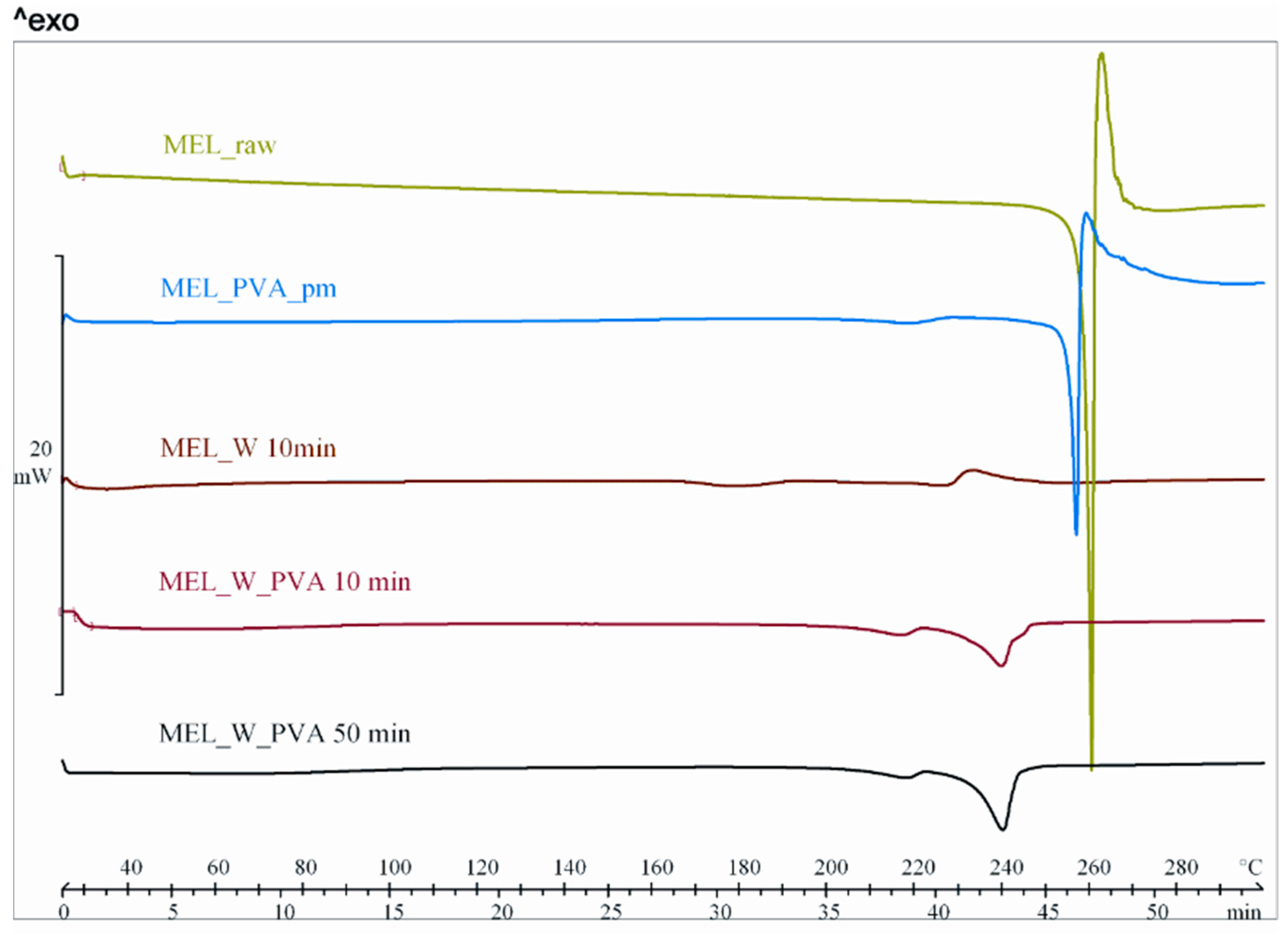
2.2.1. Physical Structure (XRPD and DSC)
2.2.2. Particle Shape and Size
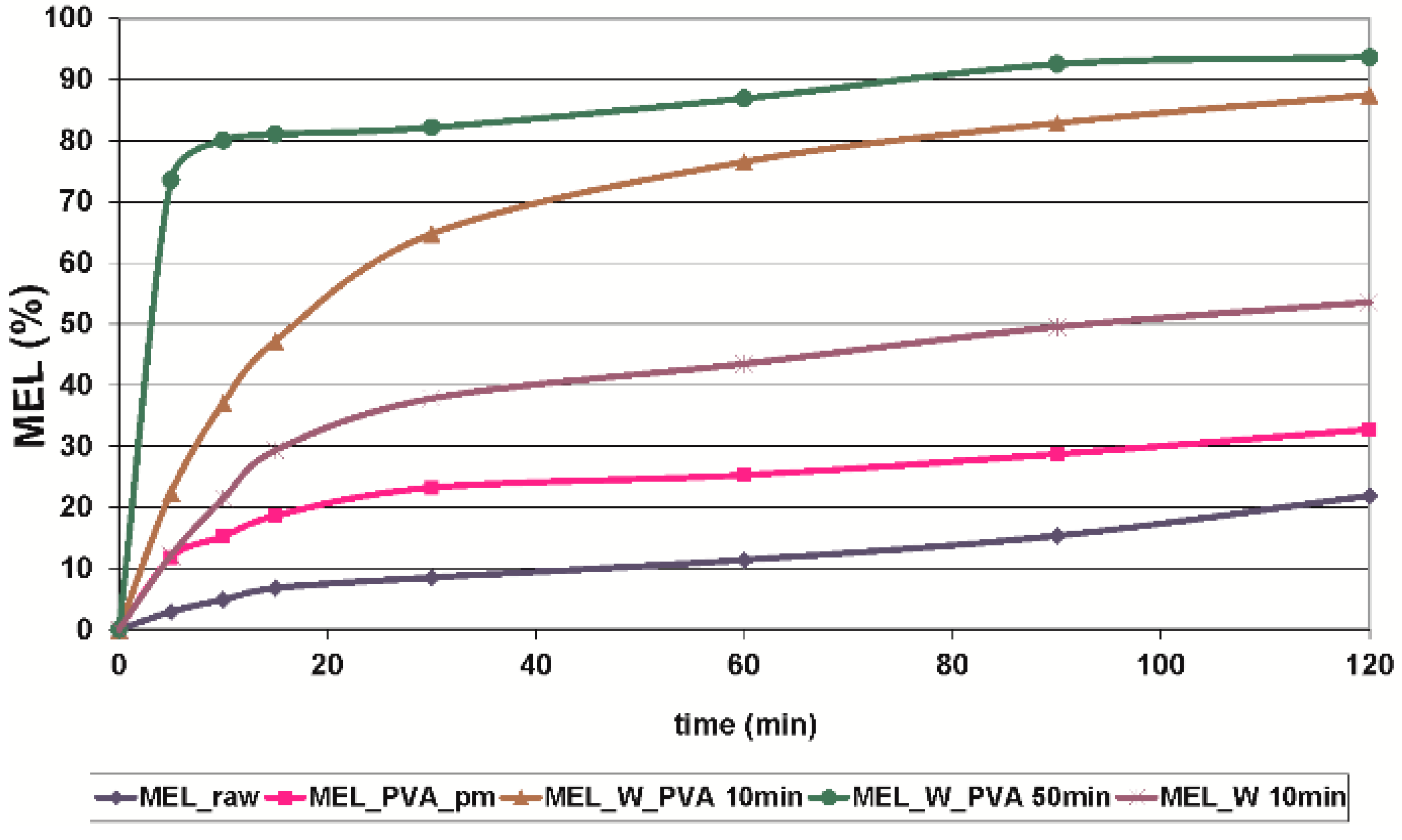
2.2.3. In Vitro Dissolution Tests
3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Combined Milling
3.2.2. Determination of Particle Size Distribution and Specific Surface Area by a Laser Diffraction Method
3.2.3. Preparation of Solid Products for Physical-Chemical Investigations of Products
3.2.4. Further Investigations of the Products
Structural Analysis
Image Analysis (SEM)
In Vitro Release
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Truong-Dinh Tran, T.; Tran, K.A.; Ha-Lien Tran, P. Modulation of particle size and molecular interactions by sonoprecipitation method for enhancing dissolution rate of poorly water-soluble drug. Ultrason. Sonochem. 2015, 24, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Kralj, T.P.; Morton, D.; Chen, X.D.; Woo, M.W. Squeezing out ultrafine hydrophobic and poor water-soluble drug particles with water vapour. Adv. Powder Technol. 2014, 25, 1190–1194. [Google Scholar] [CrossRef]
- Caliandro, R.; di Profio, G.; Nicolotti, O. Multivariate analysis of quaternary carbamazepine–saccharin mixtures by X-ray diffraction and infrared spectroscopy. J. Pharm. Biomed. Anal. 2013, 78–79, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Miclea, L.M.; Vlaia, L.; Vlaia, V.; D.I. Hădărugă, C. Mircioiu, Preparation and characterization of inclusion complexes of meloxicam and α-cyclodextrin and β-cyclodextrin. Farmacia 2010, 58, 583–593. [Google Scholar]
- Verma, S.; Gokhale, R.; Burgess, D.J. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int. J. Pharm. 2009, 380, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.A.; Suleiman, M.S.; Najib, N.M. Improvement of the in vitro dissolution characteristics of famotidine by inclusion in β-cyclodextrin. Int. J. Pharm. 1990, 58, 19–24. [Google Scholar] [CrossRef]
- Paulino, A.S.; Rauber, G.; Campos, C.E.M.; Maurício, M.H.P.; de Avillez, R.R.; Capobianco, G.; Cardoso, S.G.; Cuffini, S.L. Dissolution enhancement of deflazacort using hollow crystals prepared by antisolvent crystallization process. Eur. J. Pharm. Sci. 2013, 49, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.T.Y.; Ng, W.K.; Tan, R.B.H. Dissolution enhancement of indomethacin via amorphization using co-milling and supercritical co-precipitation processing. Powder Technol. 2013, 240, 79–87. [Google Scholar] [CrossRef]
- Liu, P.; Rong, X.; Laru, J.; van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Laaksonen, T.; Peltonen, L. Nanosuspensions of poorly soluble drugs: Preparation and development by wet milling. Int. J. Pharm. 2011, 411, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Manfredini & Schianchi. MS DRYTECH: Continuous Evolution in the Dry Preparation of Raw Materials. Available online: http://www.manfredinieschianchi.com/406-2EN-advantages-of-dry-grindiing.htm (accessed on 20 March 2016).
- Swarbrick, J. Encyclopedia of Pharmaceutical Technology, 3rd ed.; Informa Healthcare: New York, NY, USA, 2013; Volume 6. [Google Scholar]
- Merisko-Liversidge, E.; Liversidge, G.G. Nanosizing for oral and parenteral drug delivery: A perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv. Drug. Deliv. Rev. 2011, 63, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Blagden, N.; Matas, M.; Gavan, P.T.; York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 2007, 59, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Bund, R.K.; Pandit, A.B. Sonocrystallization: Effect on lactose recovery and crystal habit. Ultrason. Sonochem. 2007, 14, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bakar, M.R.A.; Nagy, Z.K.; Saleemi, A.N.; Rielly, C.D. The impact of direct nucleation control on crystal size distribution in pharmaceutical crystallization processes. Cryst. Growth Des. 2009, 9, 1378–1384. [Google Scholar] [CrossRef]
- Salazar, J.; Ghanem, A.; Müller, R.H.; Möschwitzer, J.P. Nanocrystals: Comparison of the size reduction effectiveness of a novel combinative method with conventional top-down approaches. Eur. J. Pharm. Biopharm. 2012, 81, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Möschwitzer, J.P. Drug nanocrystals in the commercial pharmaceutical development process. Int. J. Pharm. 2013, 453, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Direct Industry, Retsch®: All Retsch Catalogues and Technical Brochures. Available online: http://pdf.directindustry.com/pdf/retsch/the-sample-high-energy-ball-mills/19308-518973.html (accessed on 20 March 2016).
- Paltonen, L.; Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: Critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 2010, 62, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Hussain, A.; Bukhari, N.I.; Ermolina, I. Quantification of residual crystallinity in ball milled commercially sourced lactose monohydrate by thermo-analytical techniques and terahertz spectroscopy. Eur. J. Pharm. Biopharm. 2015, 92, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Mártha, C.; Kürti, L.; Farkas, G.; Jójárt-Laczkovich, O.; Szalontai, B.; Glässer, E.; Deli, M.A.; Szabó-Révész, P. Effects of polymers on the crystallinity of nanonized meloxicam during a co-grinding process. Eur. Polym. J. 2013, 49, 2426–2432. [Google Scholar] [CrossRef]
- Pan, X.; Julian, T.; Augsburger, L. Increasing the dissolution rate of a low-solubility drug through a crystalline-amorphous transition: A case study with indomethacin. Drug Dev. Ind. Pharm. 2008, 34, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, R.; Kocbek, P.; Kristl, J.; Šibanc, R.; Rajkó, R.; Szabó-Révész, P. Investigation of preparation parameters to improve the dissolution of poorly water-soluble meloxicam. Int. J. Pharm. 2009, 381, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hughey, J.R.; Keen, J.M.; Brough, C.; Saeger, S.; McGinity, J.W. Thermal processing of a poorly water-soluble drug substance exhibiting a high melting point: The utility of KinetiSol® Dispersing. Int. J. Pharm. 2011, 419, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Milling Time (min) | D10 (µm) | D50 (µm) | D90 (µm) | SSA (m2/g) |
|---|---|---|---|---|
| raw MEL | 11.40 | 34.26 | 73.59 | 0.332 |
| 10 | 1.76 | 3.55 | 8.69 | 1.89 |
| 20 | 2.63 | 5.74 | 14.21 | 1.22 |
| 30 | 2.62 | 5.99 | 18.97 | 1.18 |
| 40 | 2.77 | 6.84 | 24.54 | 1.07 |
| 50 | 2.72 | 6.13 | 19.56 | 1.15 |
| 60 | 2.57 | 6.12 | 20.87 | 1.18 |
| 70 | 2.77 | 7.13 | 26.26 | 1.05 |
| 80 | 2.58 | 5.94 | 22.54 | 1.19 |
| 90 | 2.51 | 5.55 | 18.88 | 1.25 |
| Milling Time (min) | D10 (µm) | D50 (µm) | D90 (µm) | SSA (m2/g) |
|---|---|---|---|---|
| raw MEL | 11.40 | 34.26 | 73.59 | 0.332 |
| 10 | 0.24 | 2.96 | 29.40 | 7.71 |
| 20 | 0.084 | 0.169 | 2.863 | 34.6 |
| 30 | 0.090 | 0.337 | 4.180 | 27.2 |
| 40 | 0.074 | 0.134 | 1.358 | 46 |
| 50 | 0.074 | 0.126 | 0.253 | 49.5 |
| 60 | 0.072 | 0.122 | 0.219 | 52 |
| 70 | 0.070 | 0.121 | 0.225 | 52.6 |
| 80 | 0.072 | 0.119 | 0.209 | 53.2 |
| 90 | 0.064 | 0.116 | 0.219 | 57.9 |
| Sample | Crystallinity (%) |
|---|---|
| MEL_raw | 100 |
| MEL_PVA_pm | 80 |
| MEL_W 10 min | 3 |
| MEL_W 50 min | 5 |
| MEL_W 60 min | 8 |
| MEL_W 90 min | 9 |
| MEL_W_PVA 10 min | 48 |
| MEL_W_PVA 50 min | 12 |
| MEL_W_PVA 60 min | 5 |
| MEL_W_PVA 90 min | 2 |
| Parameters | Raw MEL | pm | Dried MEL_W Samples | Dried MEL_W_PVA Samples | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Milling time (min) | - | - | 10 | 50 | 60 | 90 | 10 | 50 | 60 | 90 |
| ∆H * (J/g) | −148.5 | −29.9 | −7.5 | −40.1 | −64.4 | −40.8 | −60.3 | −70.9 | −32.4 | −46.4 |
| Onset (°C) | 258.4 | 235.9 | 217.9 | 233.5 | 248.5 | 247.1 | 231.6 | 232.9 | 207.1 | 206.2 |
| Peak (°C) | 259.1 | 255.9 | 227.0 | 238.7 | 250.7 | 250.2 | 239.8 | 240.1 | 216.7 | 217.4 |
| Endset (°C) | 261.0 | 255.9 | 230.0 | 240.4 | 250.8 | 251.6 | 243.1 | 243.2 | 222.3 | 222.3 |
| Sample Name | MEL (%) | PVA (%) | Description |
|---|---|---|---|
| MEL_raw | 100 | - | untreated MEL |
| MEL_PVA_pm | 80 | 20 | physical mixture of MEL and PVA |
| MEL_W 10 min | 100 | - | dried MEL, milled for 10 min in water |
| MEL_W 50 min | 100 | - | dried MEL, milled for 50 min in water |
| MEL_W 60 min | 100 | - | dried MEL, milled for 60 min in water |
| MEL_W 90 min | 100 | - | dried MEL, milled for 90 min in water |
| MEL_W_PVA 10 min | 80 | 20 | dried product of milling for 10 min in PVA-solution |
| MEL_W_PVA 50 min | 80 | 20 | dried product of milling for 50 min in PVA-solution |
| MEL_W_PVA 60 min | 80 | 20 | dried product of milling for 60 min in PVA-solution |
| MEL_W_PVA 90 min | 80 | 20 | dried product of milling for 90 min in PVA-solution |
| Sample | D50 (µm) | Crystallinity (%) | Dissolved MEL at 30 min (%) |
|---|---|---|---|
| MEL_raw | 34.26 | 100 | 8.53 |
| MEL_PVA_pm | 20.15 | 80 | 29.22 |
| MEL_W 10 min | 3.55 | 3 | 37.82 |
| MEL_W_PVA 10 min | 2.96 | 48 | 64.73 |
| MEL_W_PVA 50 min | 0.126 | 12 | 82.16 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartos, C.; Szabó-Révész, P.; Bartos, C.; Katona, G.; Jójárt-Laczkovich, O.; Ambrus, R. The Effect of an Optimized Wet Milling Technology on the Crystallinity, Morphology and Dissolution Properties of Micro- and Nanonized Meloxicam. Molecules 2016, 21, 507. https://doi.org/10.3390/molecules21040507
Bartos C, Szabó-Révész P, Bartos C, Katona G, Jójárt-Laczkovich O, Ambrus R. The Effect of an Optimized Wet Milling Technology on the Crystallinity, Morphology and Dissolution Properties of Micro- and Nanonized Meloxicam. Molecules. 2016; 21(4):507. https://doi.org/10.3390/molecules21040507
Chicago/Turabian StyleBartos, Csilla, Piroska Szabó-Révész, Csaba Bartos, Gábor Katona, Orsolya Jójárt-Laczkovich, and Rita Ambrus. 2016. "The Effect of an Optimized Wet Milling Technology on the Crystallinity, Morphology and Dissolution Properties of Micro- and Nanonized Meloxicam" Molecules 21, no. 4: 507. https://doi.org/10.3390/molecules21040507
APA StyleBartos, C., Szabó-Révész, P., Bartos, C., Katona, G., Jójárt-Laczkovich, O., & Ambrus, R. (2016). The Effect of an Optimized Wet Milling Technology on the Crystallinity, Morphology and Dissolution Properties of Micro- and Nanonized Meloxicam. Molecules, 21(4), 507. https://doi.org/10.3390/molecules21040507