Non-Covalent Organocatalyzed Domino Reactions Involving Oxindoles: Recent Advances





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
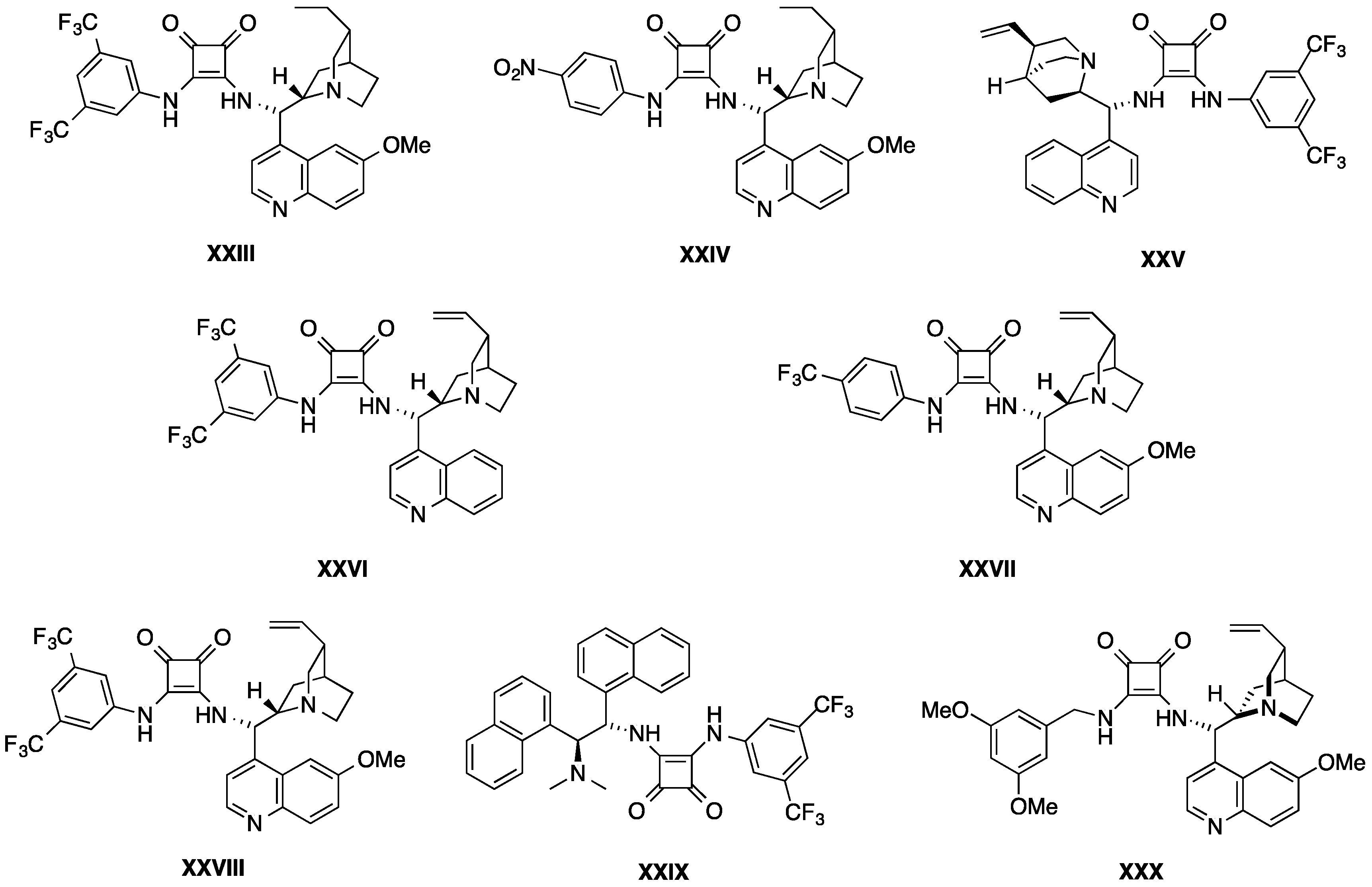{kind=link}
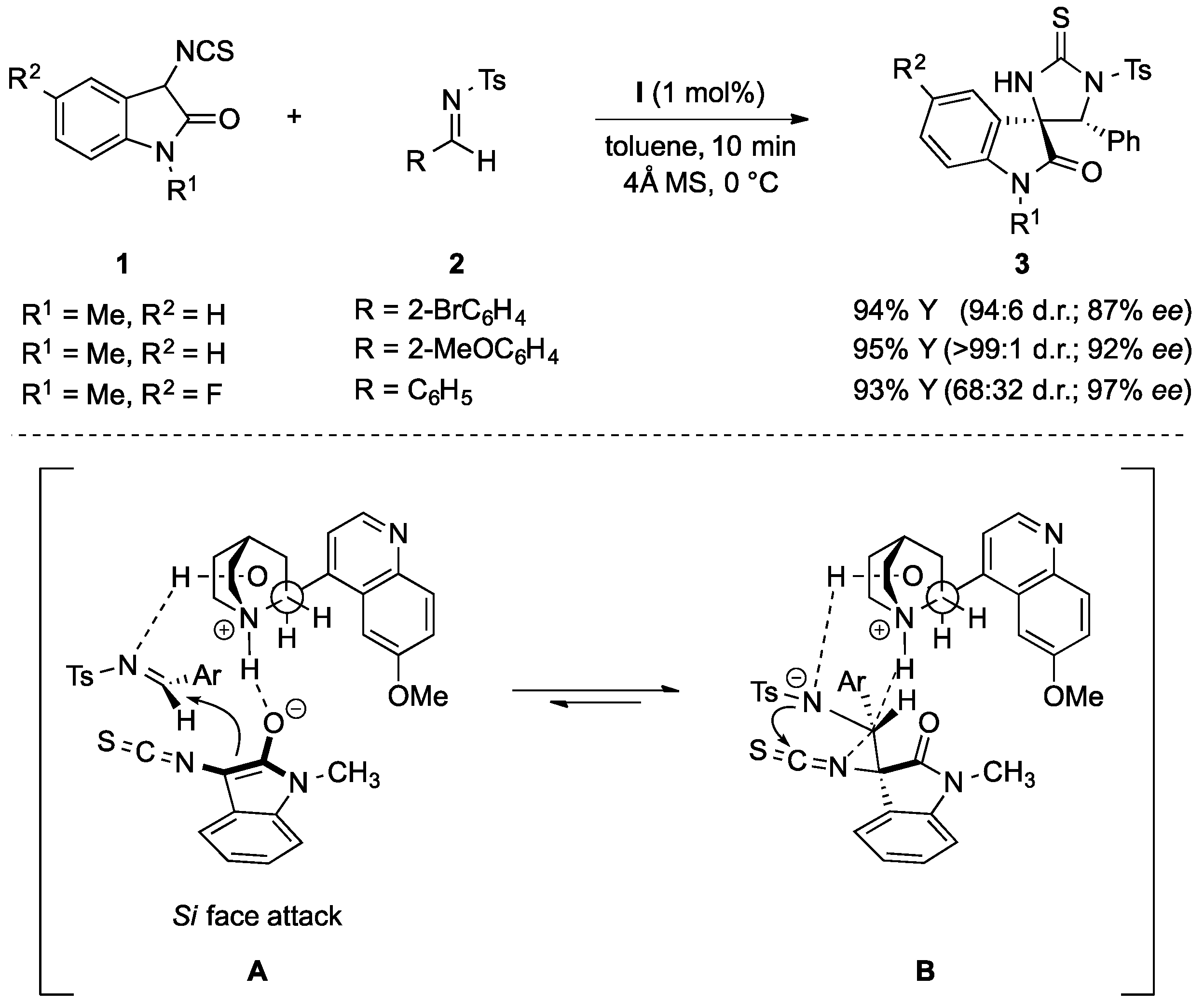{kind=link}
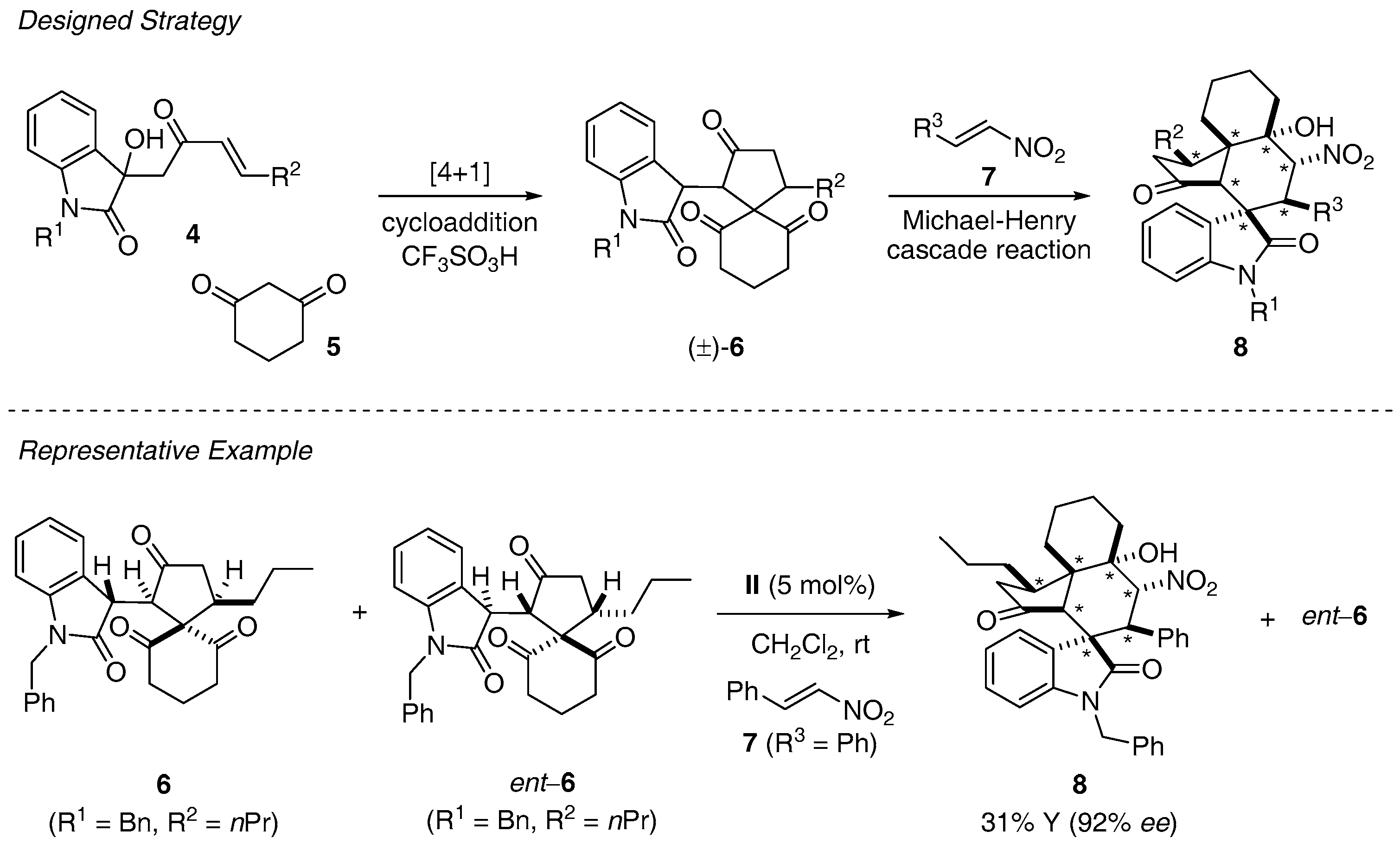{kind=link}
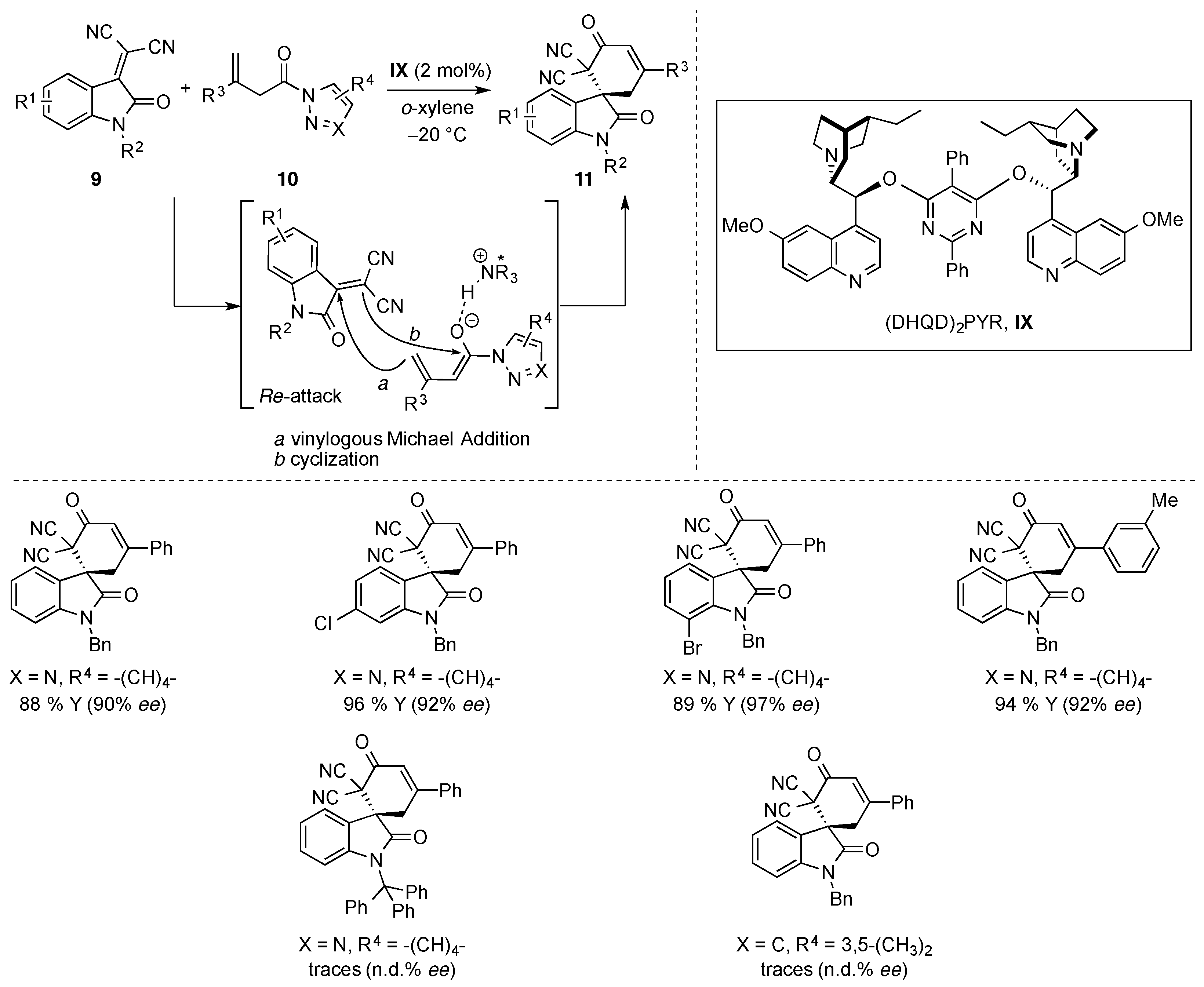{kind=link}
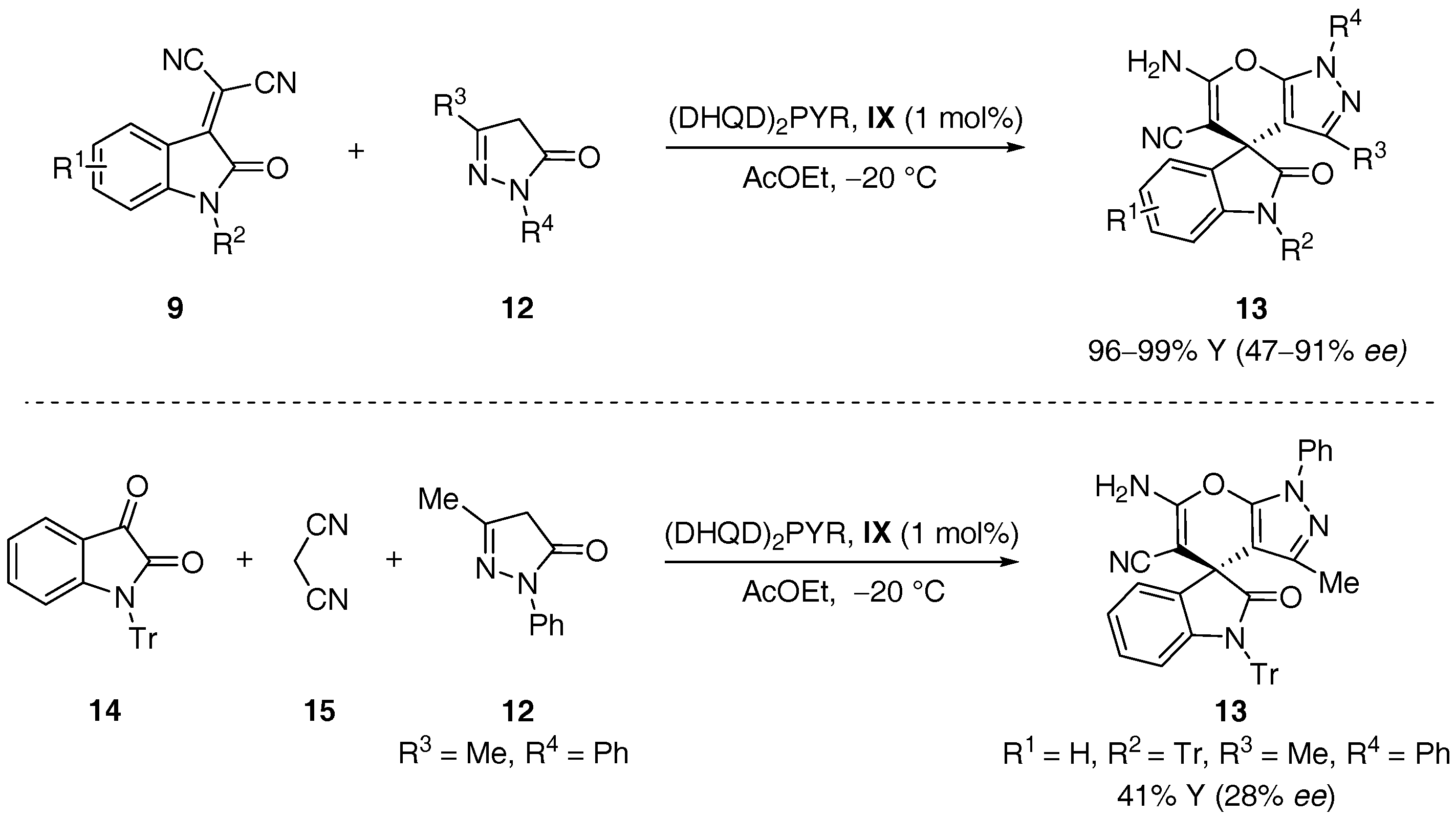{kind=link}
{kind=link}
{kind=link}
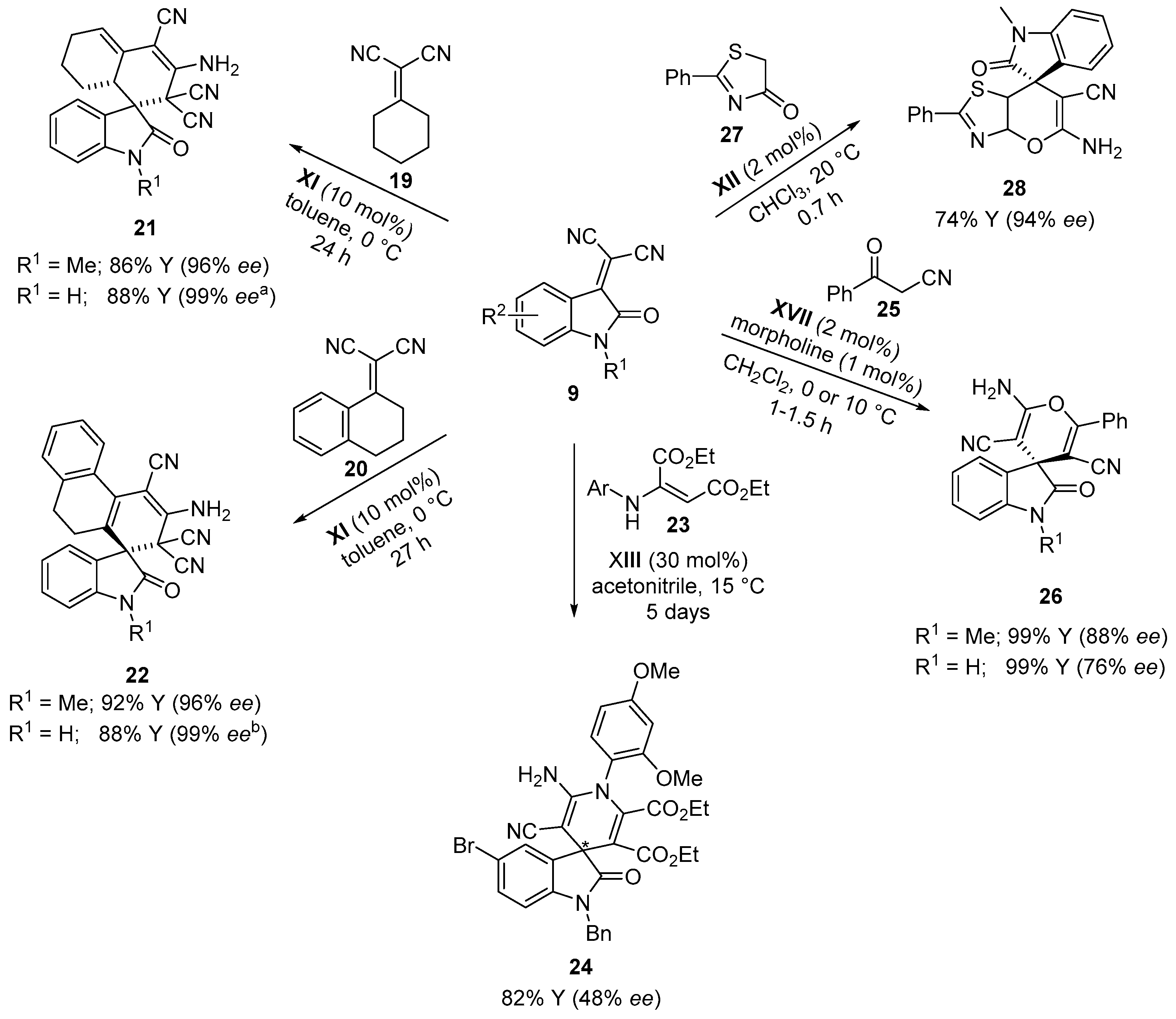
{kind=link}
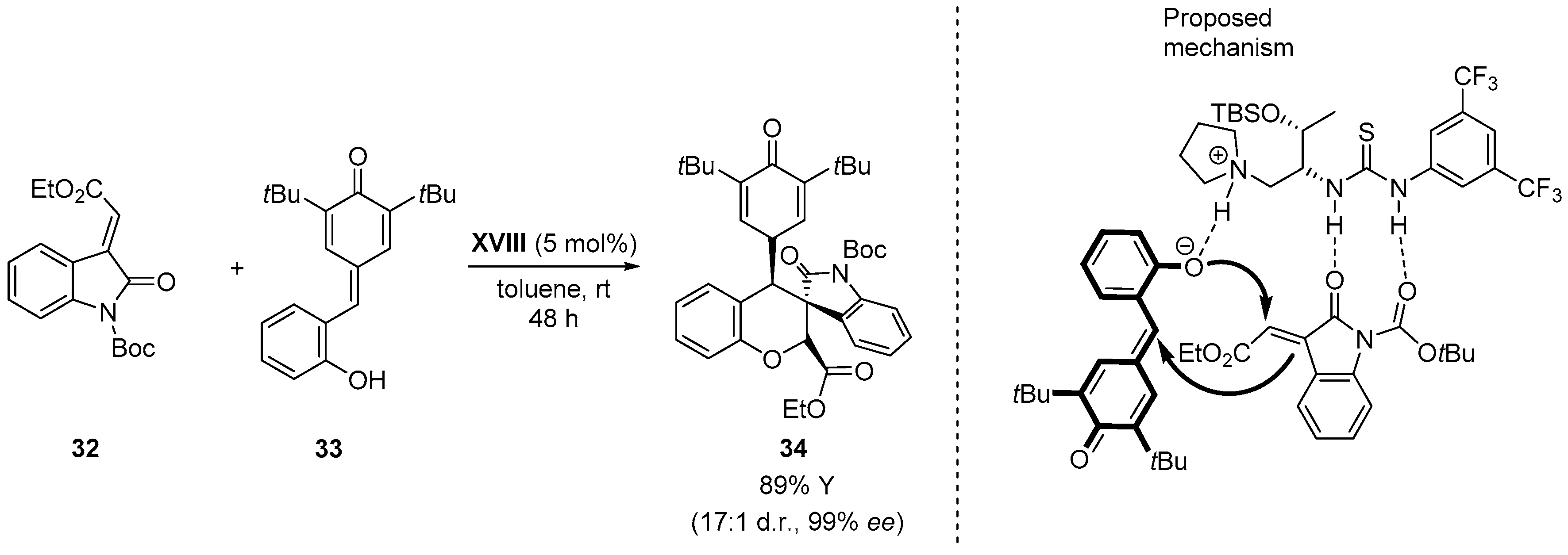{kind=link}
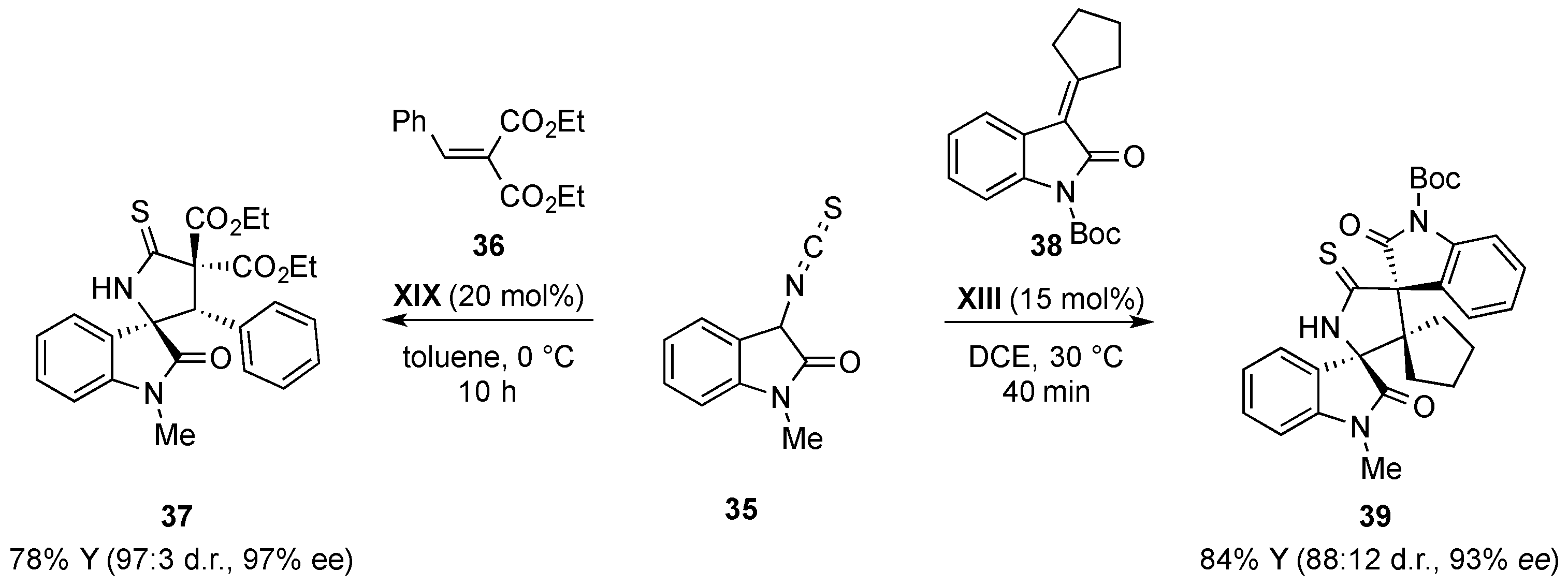{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
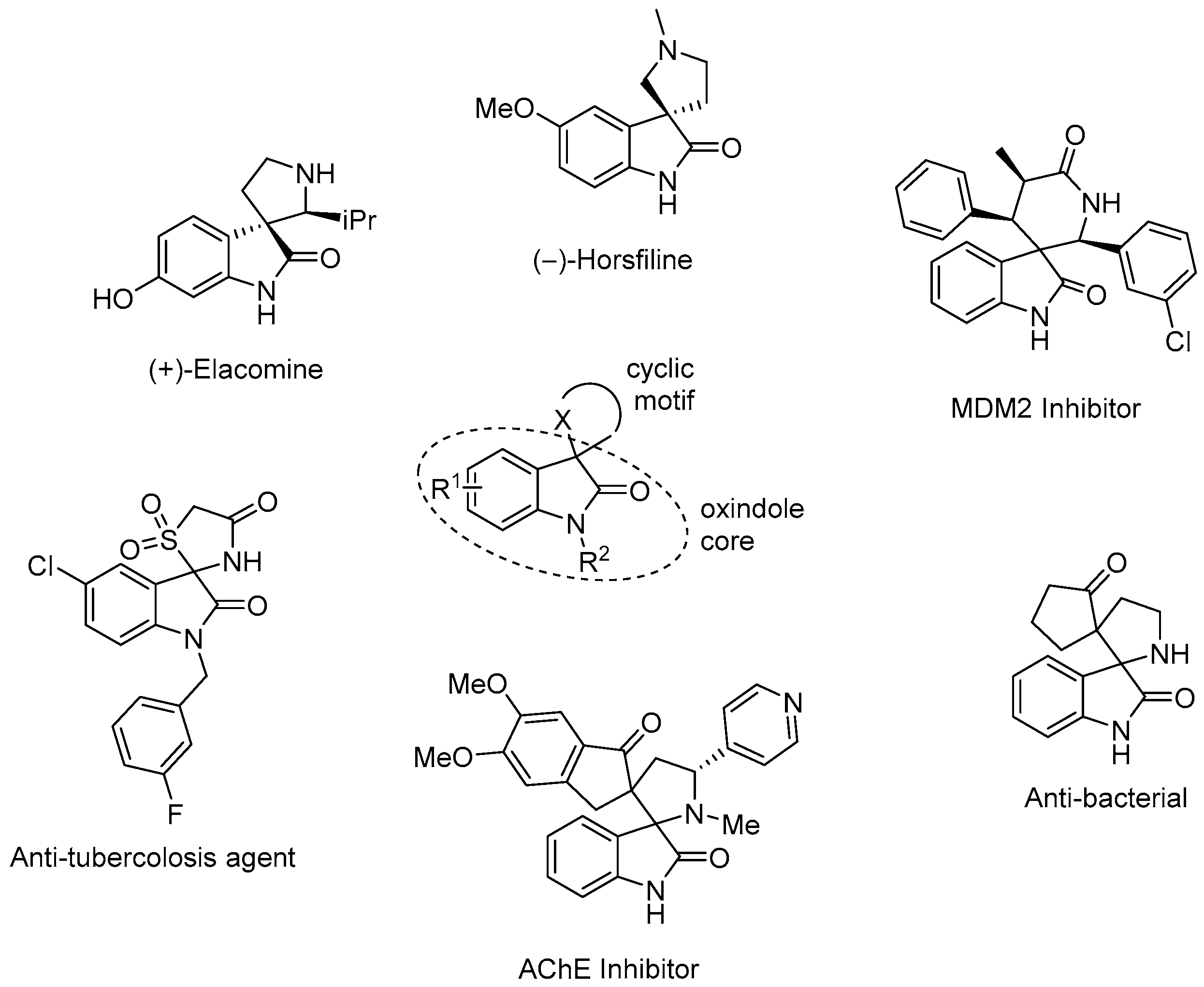
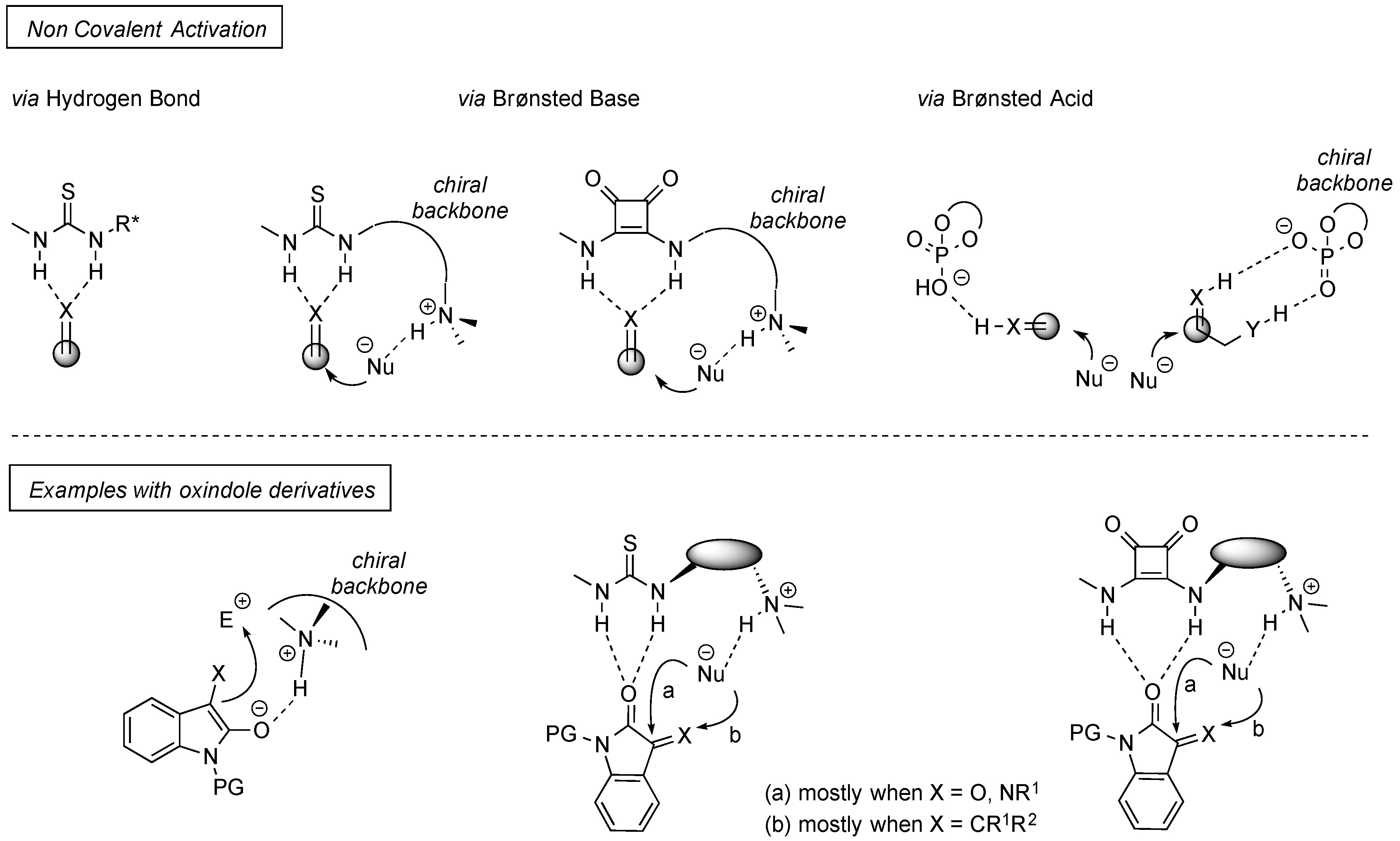
:1. Introduction
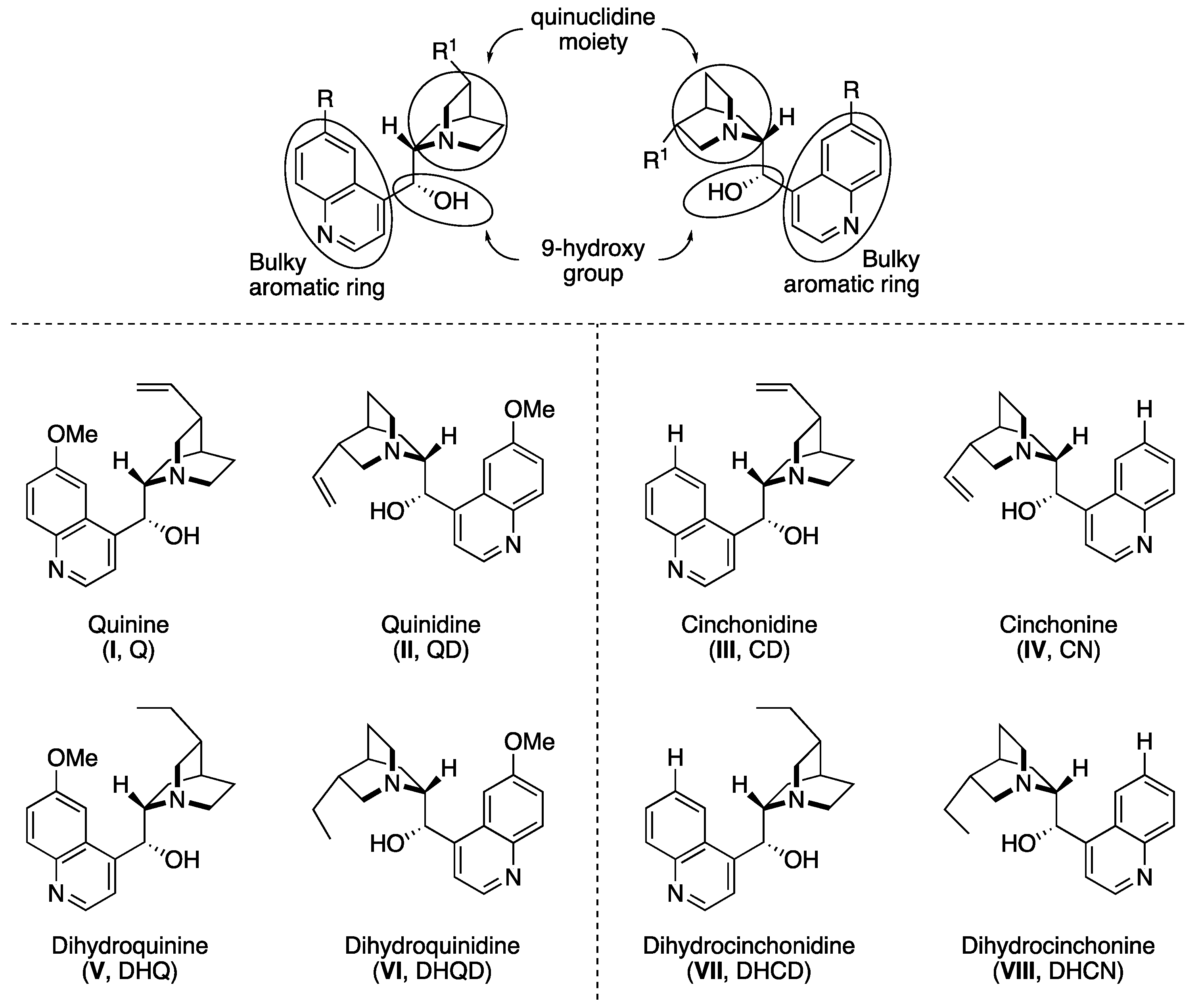
2. Cinchona Alkaloid Catalysts
- (i)
- the basicity and bulkiness of quinuclidine moiety, apt to activate a nucleophile by deprotonation as well as to stabilize the developing positive charge,
- (ii)
- the secondary 9-hydroxy group, which acts as both an acid and a H-bond donor, and is suitable for further chemical modification of the catalyst structure. Additionally, the overall catalytic action is also ascribed to the possible π–π interactions with the aromatic quinoline ring.
3. (DHQD)2 Based Catalysts
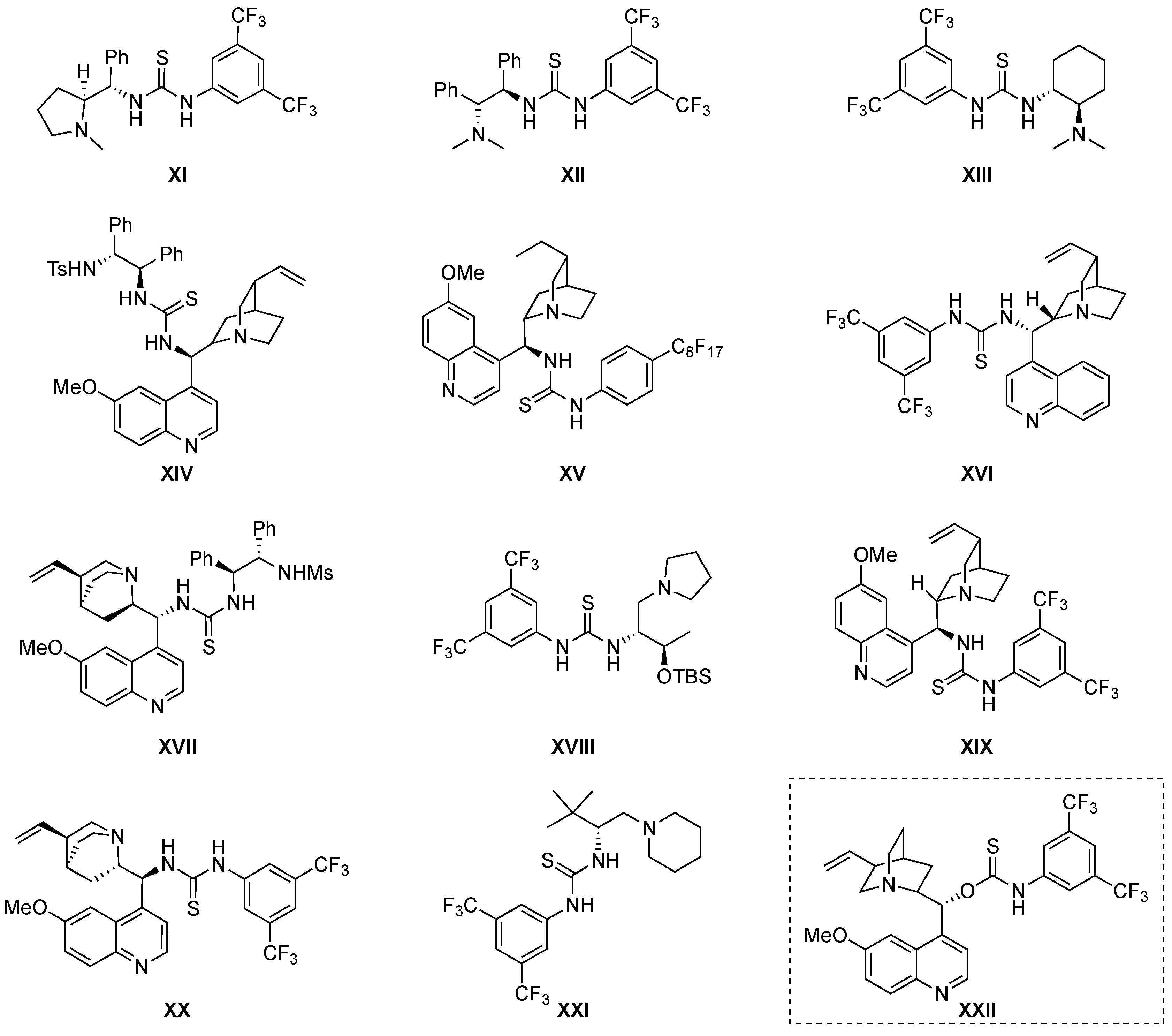
4. Thiourea-Based Catalysts
4.1. Michael Addition/Cyclization Sequence
4.2. Michael Addition/Mannich/Cyclization Sequence
4.3. Double Michael Addition Sequence
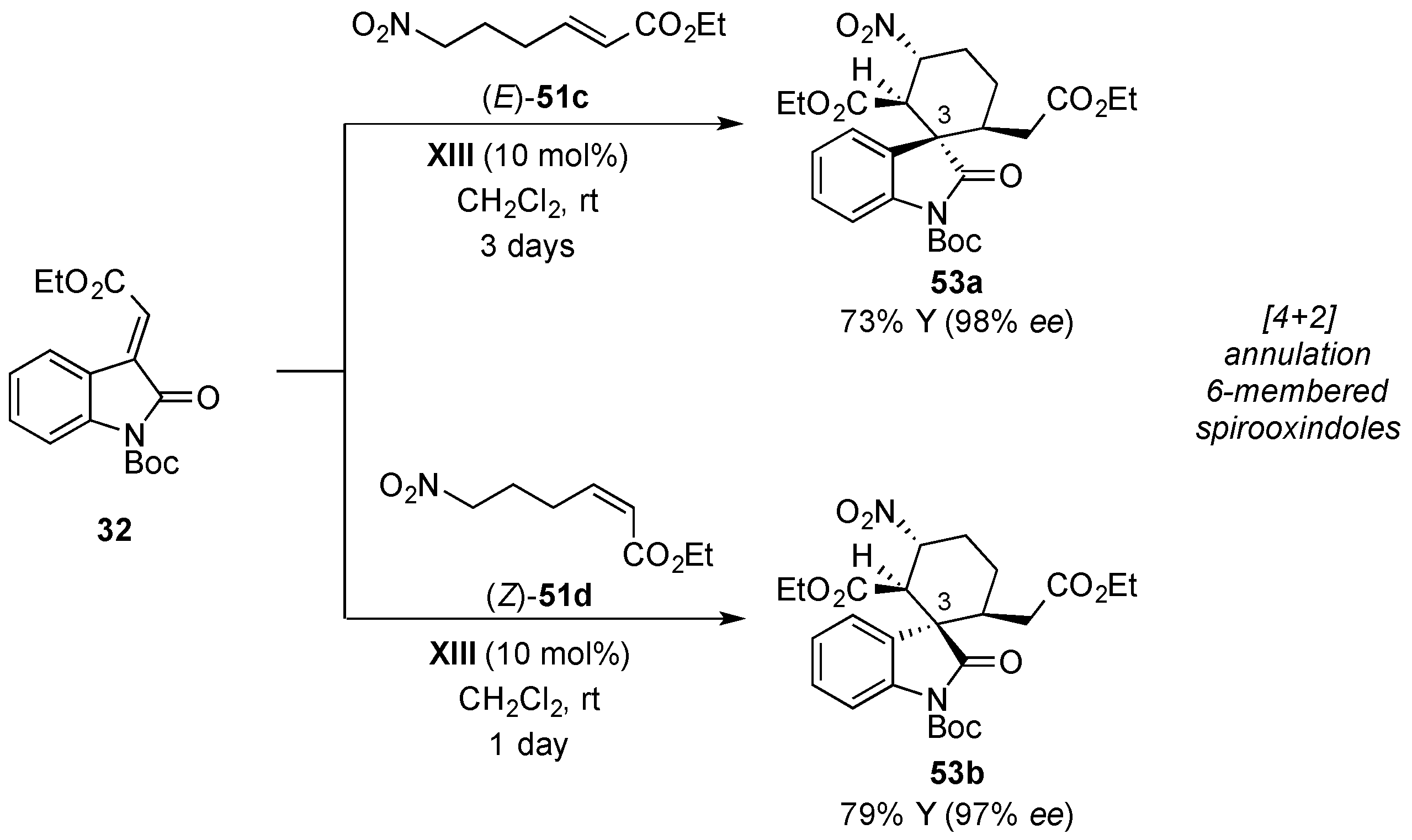
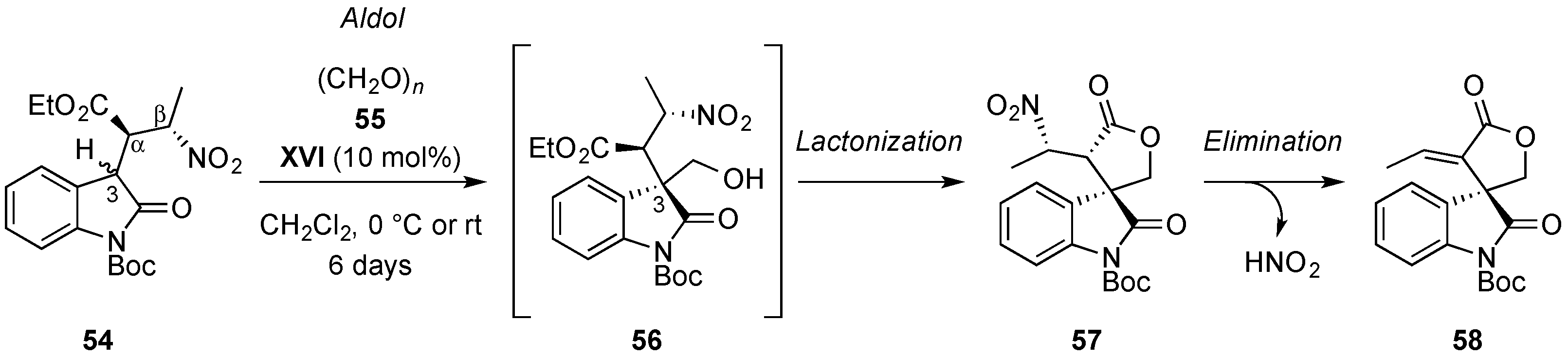
4.4. Aldol/Lactonization/Elimination Sequence
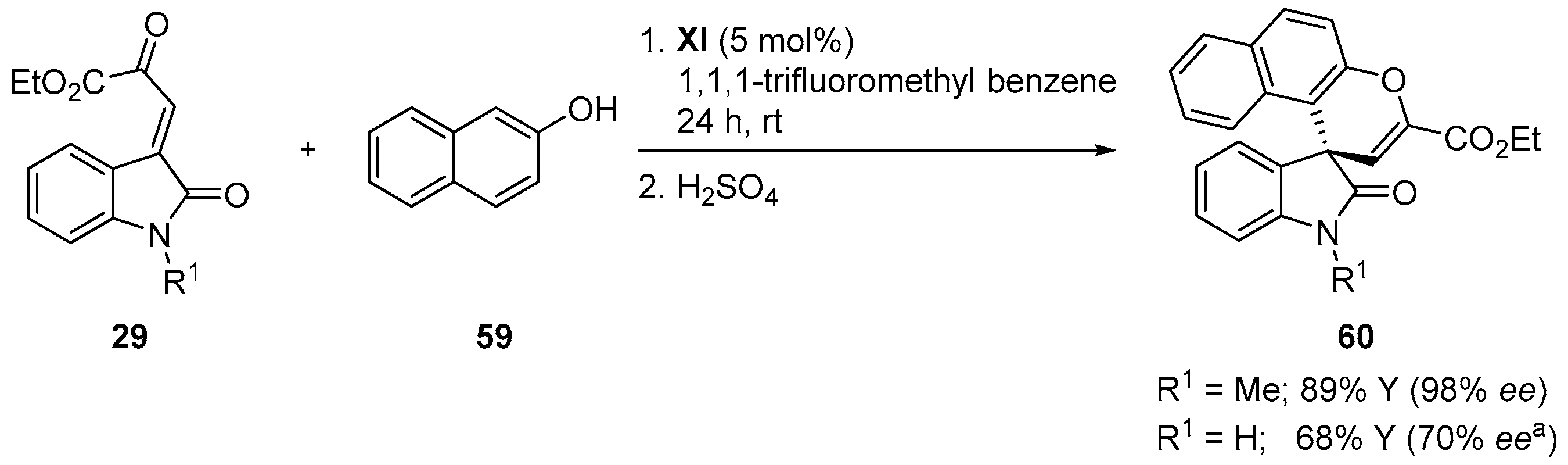
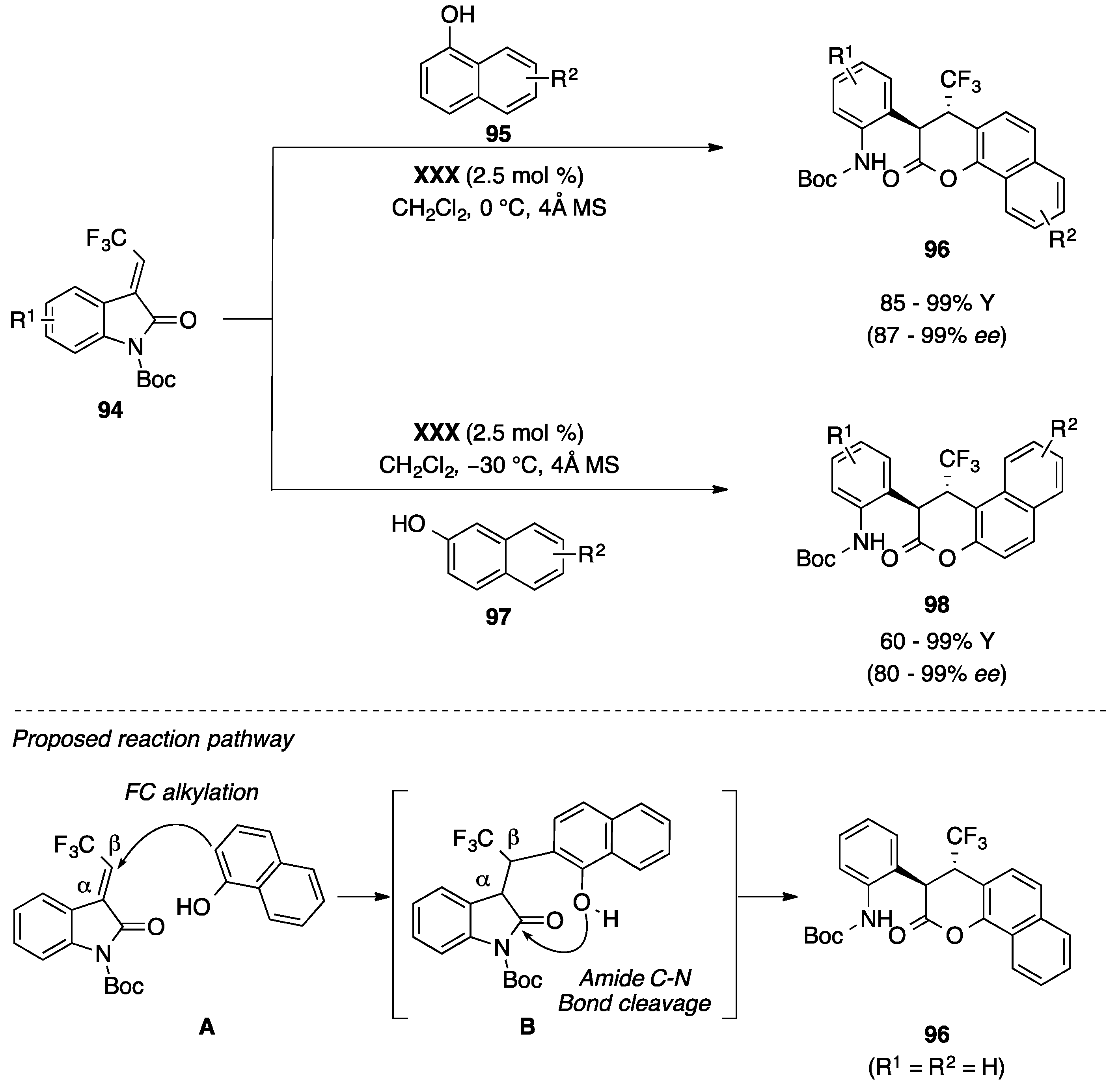
4.5. Friedel-Crafts/Hemiketalization Sequence
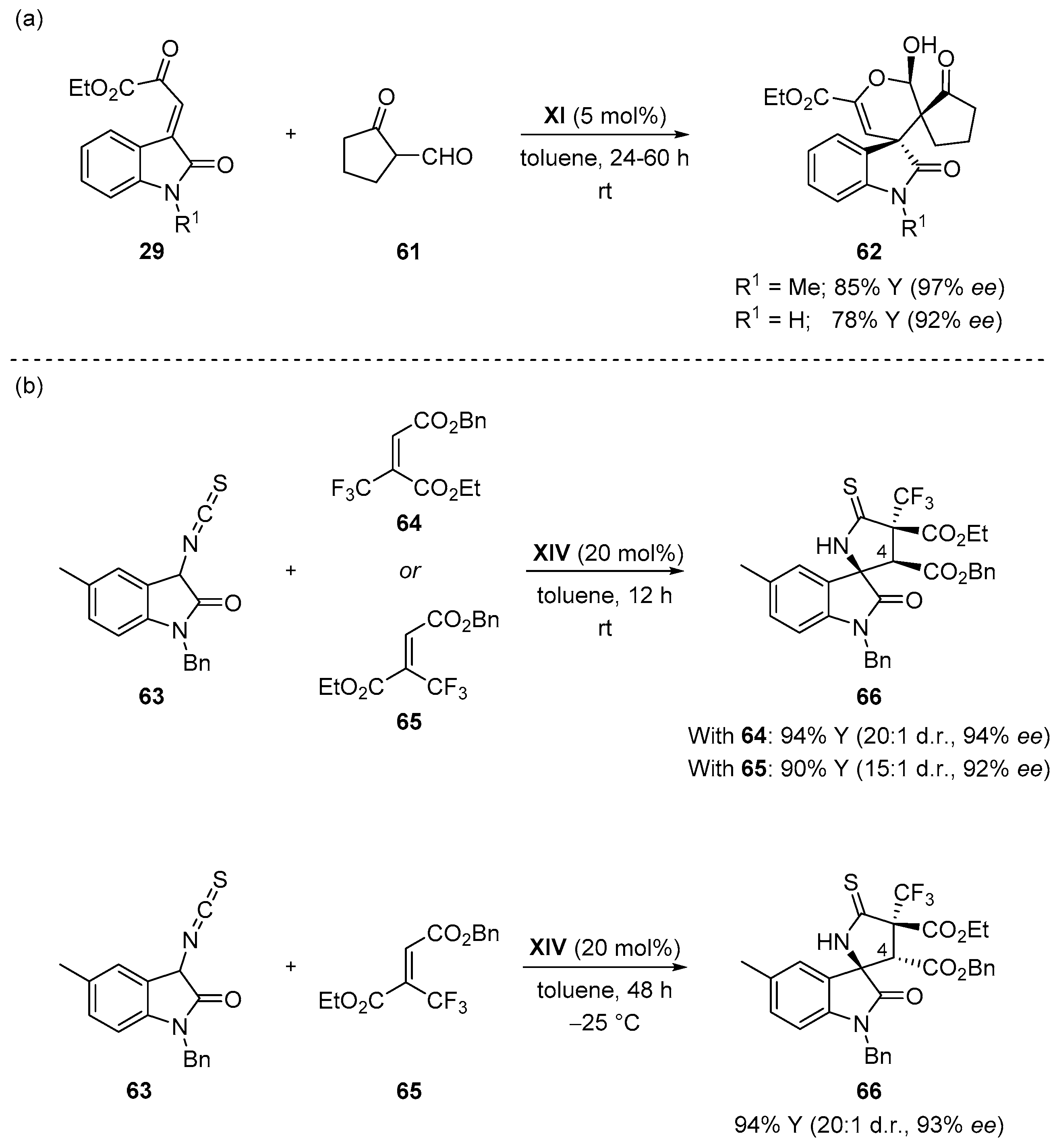
4.6. Miscellaneous
5. Squaramide Catalysts
5.1. Double Michael Addition Sequence
5.2. Michael Addition/Cyclization Reaction Sequence
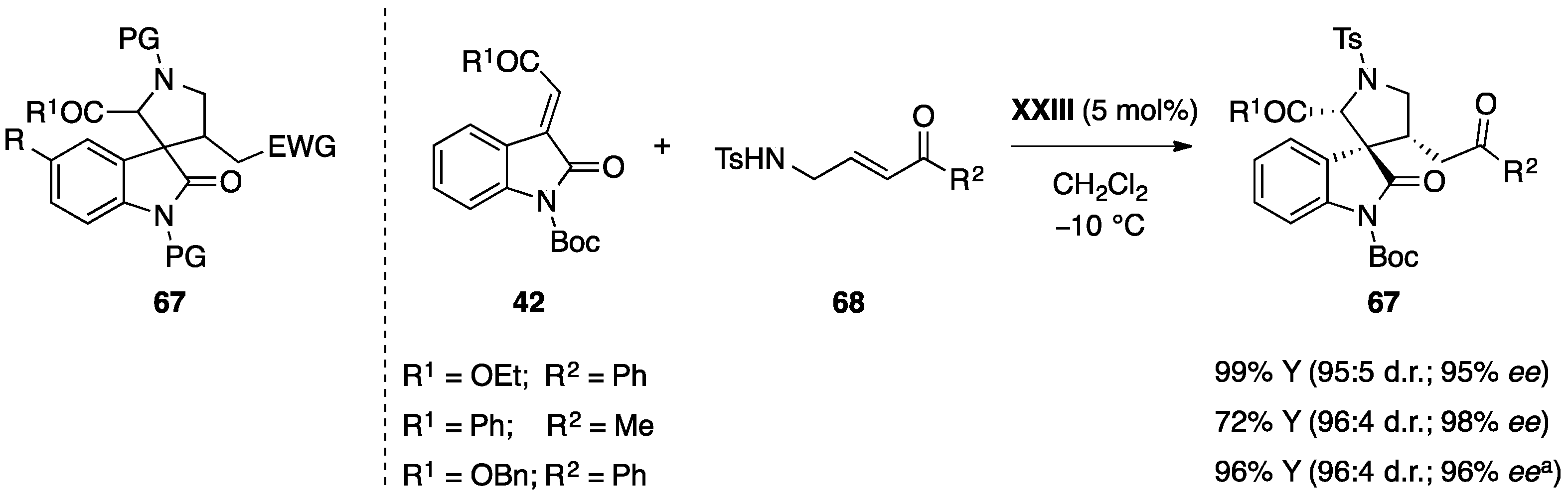
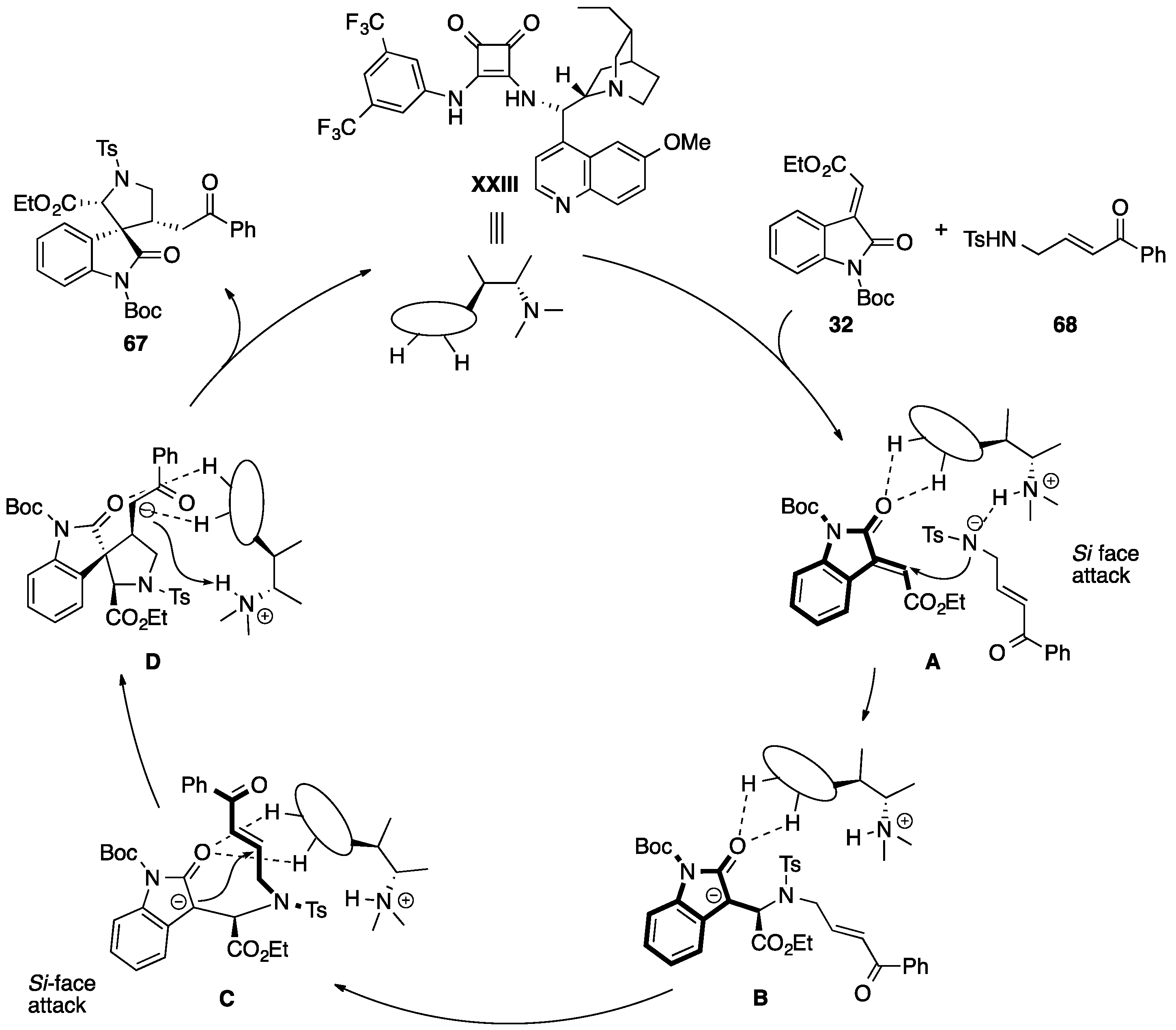
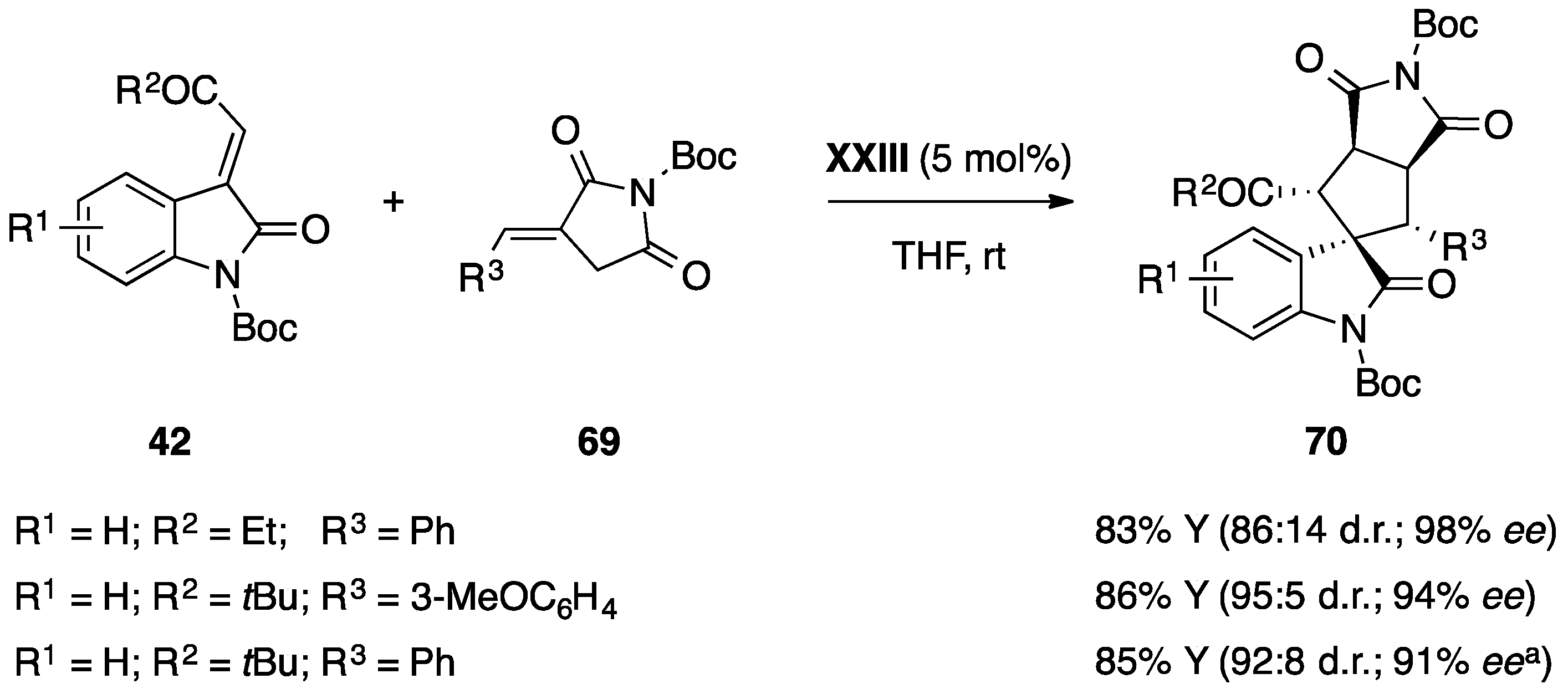
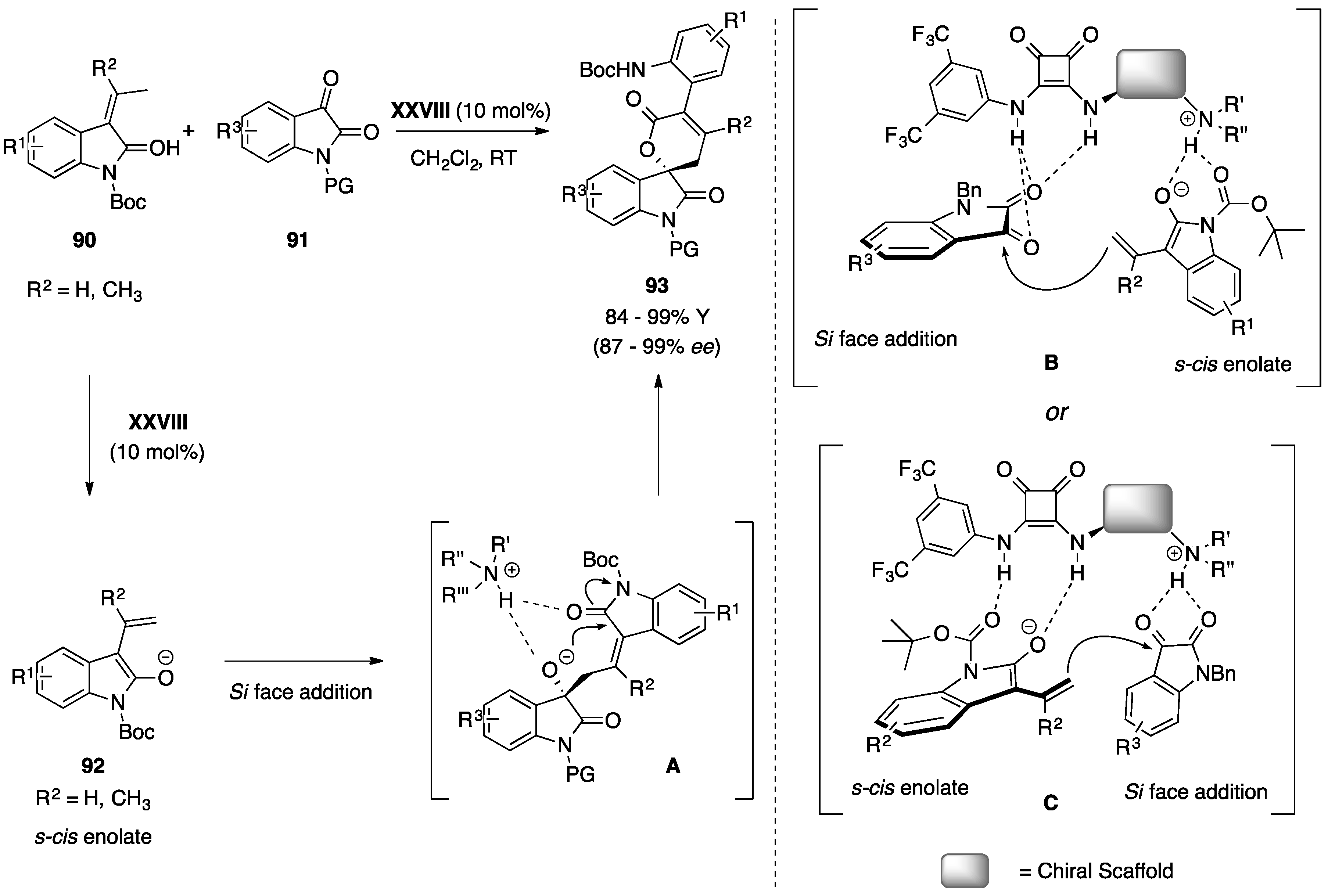
5.3. Miscellaneous
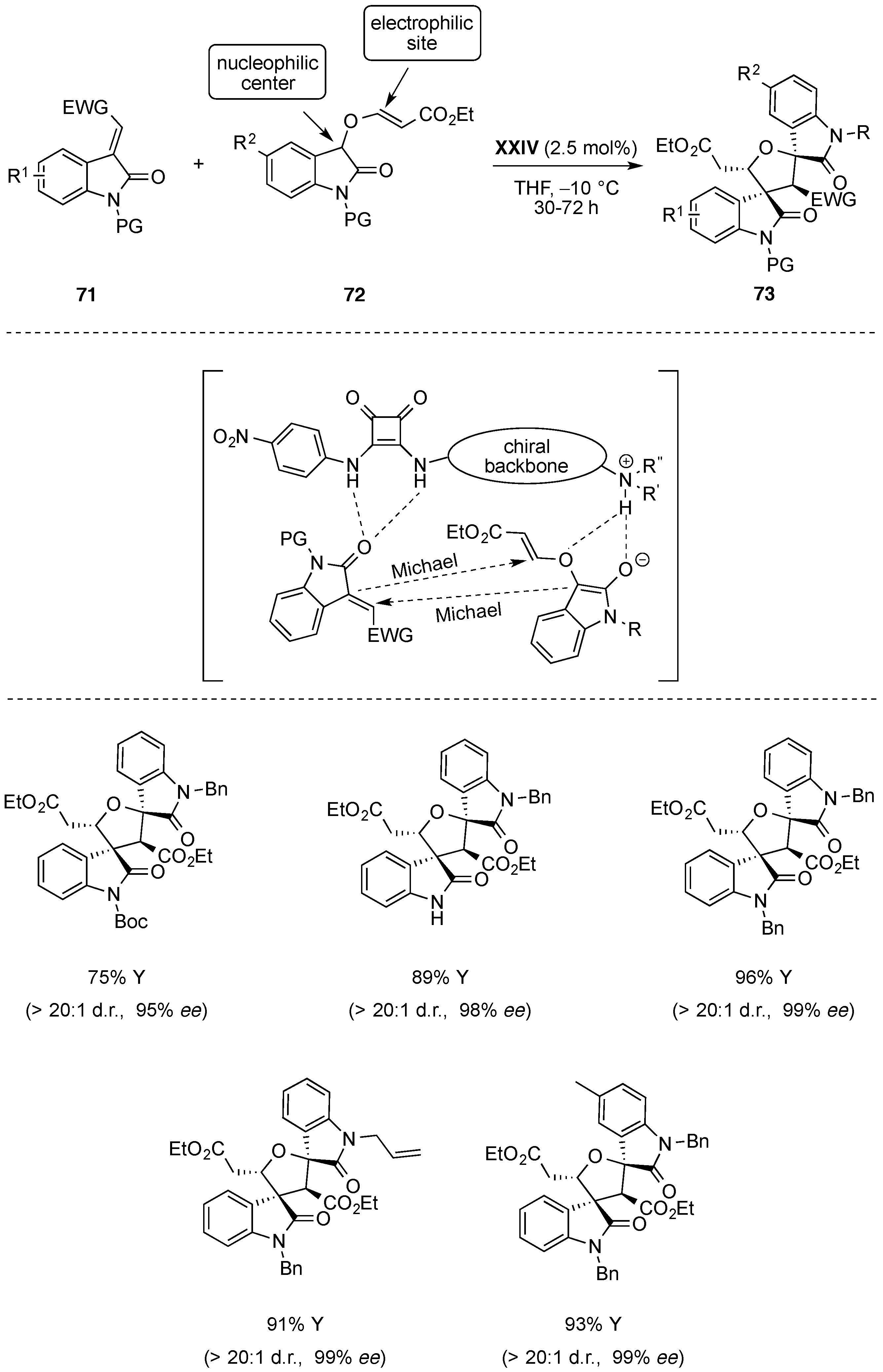
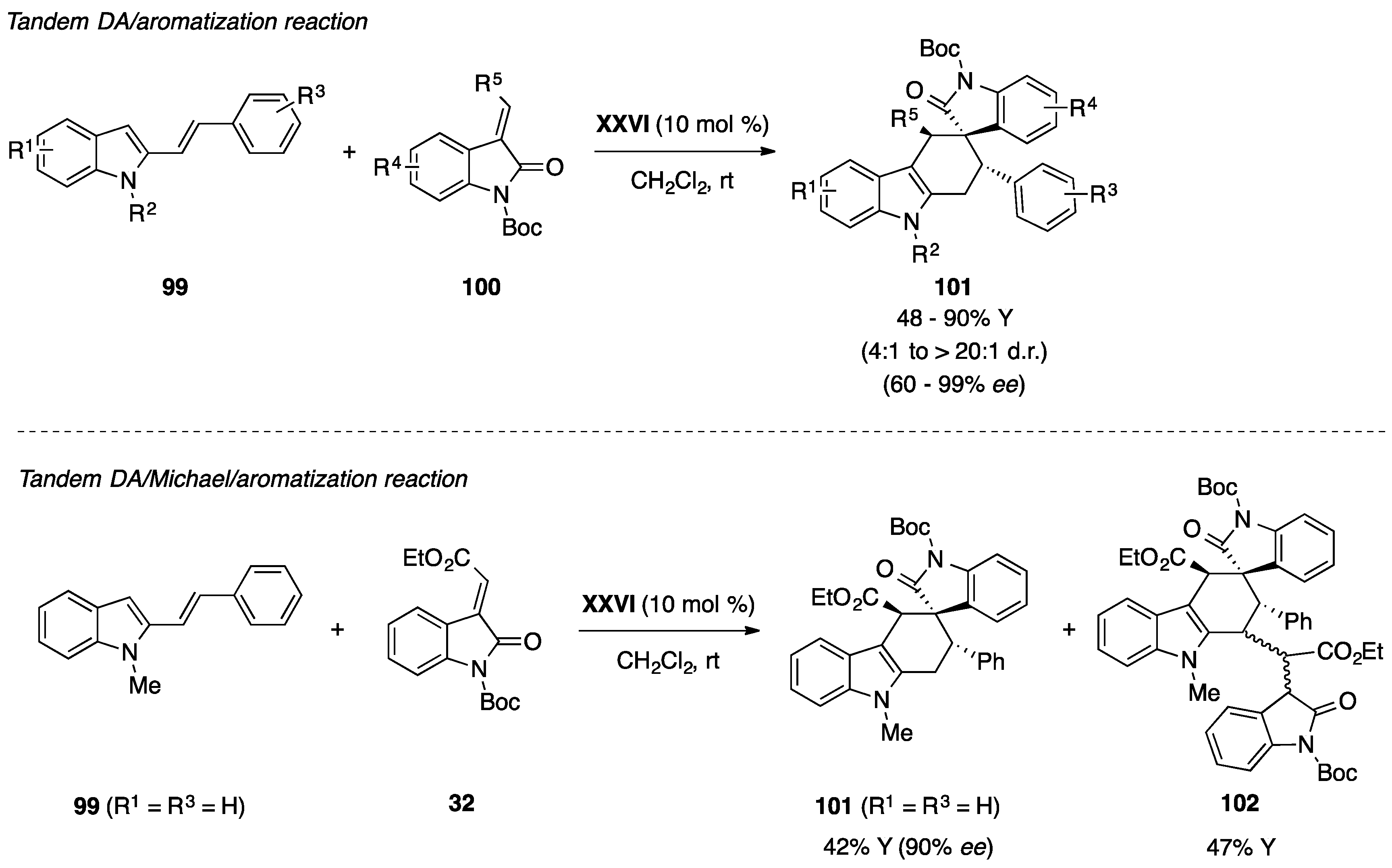
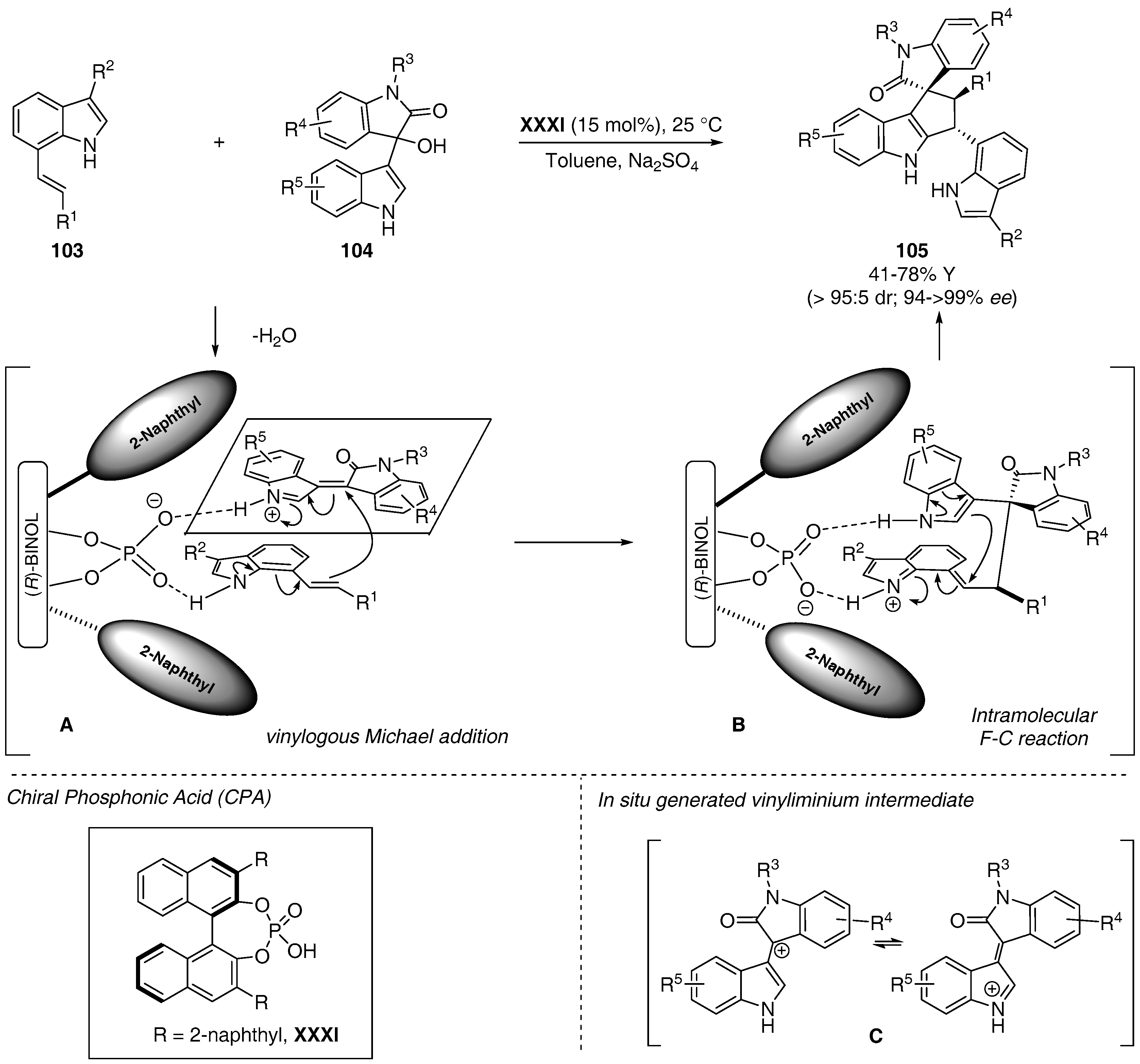
6. Miscellaneous
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Y.; Lu, H.; Xu, P.F. Asymmetric catalytic cascade reactions for constructing diverse scaffolds and complex molecules. Acc. Chem. Res. 2015, 48, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.-Y.; Zhou, J. Catalytic asymmetric synthesis of polysubstituted spirocyclopropyl oxindoles: Organocatalysis versus transition metal catalysis. Org. Chem. Front. 2015, 2, 849–858. [Google Scholar] [CrossRef]
- Trost, B.; Brennan, M. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis 2009, 18, 3003–3025. [Google Scholar] [CrossRef]
- Antonchick, A.P.; Gerding-Reimers, C.; Catarinella, M.; Schurmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2010, 2, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Xing, H.; Yu, D.Q.; Liu, H.M. Catalytic asymmetric synthesis of biologically important 3-hydroxyoxindoles: An update. Beilstein J. Org. Chem. 2016, 12, 1000–1039. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Chimni, S.S.; Mahajan, S.; Kumar, A. Stereoselective synthesis of 3-amino-2-oxindoles from isatin imines: New scaffolds for bioactivity evaluation. RSC Adv. 2015, 5, 52481–52496. [Google Scholar] [CrossRef]
- Tietze, L.F.; Brasche, G.; Gericke, K.M. Domino Reaction in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Volla, C.M.; Atodiresei, I.; Rueping, M. Catalytic c-c bond-forming multi-component cascade or domino reactions: Pushing the boundaries of complexity in asymmetric organocatalysis. Chem. Rev. 2014, 114, 2390–2431. [Google Scholar] [CrossRef] [PubMed]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Luo, Y.-C.; Hu, X.-Q.; Xu, P.-F. Recent developments in the synthesis of chiral compounds with quaternary centers by organocatalytic cascade reactions. Asian J. Org. Chem. 2016, 5, 580–607. [Google Scholar] [CrossRef]
- Gasperi, T.; Vetica, F.; de Figueiredo, R.; Orsini, M.; Tofani, D. Recent advances in organocatalytic cascade reactions toward the formation of quaternary stereocenters. Synthesis 2015, 47, 2139–2184. [Google Scholar] [CrossRef]
- Hong, L.; Wang, R. Recent advances in asymmetric organocatalytic construction of 3,3′-spirocyclic oxindoles. Adv. Synth. Catal. 2013, 355, 1023–1052. [Google Scholar] [CrossRef]
- Bencivenni, G.; Wu, L.Y.; Mazzanti, A.; Giannichi, B.; Pesciaioli, F.; Song, M.P.; Bartoli, G.; Melchiorre, P. Targeting structural and stereochemical complexity by organocascade catalysis: Construction of spirocyclic oxindoles having multiple stereocenters. Angew. Chem. Int. Ed. 2009, 48, 7200–7203. [Google Scholar] [CrossRef] [PubMed]
- Hof, K.; Lippeert, K.M.; Schreiner, P.R. Science of Synthesis: Asymmetric Organocatalysis 2; Georg Thieme Verlag KG: Stuttgart, Germany, 2012. [Google Scholar]
- Knowles, R.R.; Jacobsen, E.N. Attractive non-covalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzenbach-Effers, K.; Berkessel, A. Non-covalent organocatalysis based on hydrogen bonding: Elucidation of reaction paths by computational methods. In Asymmetric Organocatalysis; List, B., Ed.; Springer: Berlin, Germany, 2009; Volume 291, pp. 1–27. [Google Scholar]
- De Figueiredo, R.M.; Mazziotta, A.; de Sant’Ana, D.P.; Palumbo, C.; Gasperi, T. Active methylene compounds in asymmetric organocatalytic synthesis of natural products and pharmaceutical scaffolds. Curr. Org. Chem. 2012, 16, 2231–2289. [Google Scholar] [CrossRef]
- Song, C.E. Cinchona Alkaloids in Synthesis and Catalysis, Ligands, Immobilization and Organocatalysis; WILEY-VCH Verlag GmbH & Co, KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Bai, M.; Cui, B.-D.; Zuo, J.; Zhao, J.-Q.; You, Y.; Chen, Y.-Z.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Quinine-catalyzed asymmetric domino Mannich-cyclization reactions of 3-isothiocyanato oxindoles with imines for the synthesis of spirocyclic oxindoles. Tetrahedron 2015, 71, 949–955. [Google Scholar] [CrossRef]
- Huang, J.R.; Sohail, M.; Taniguchi, T.; Monde, K.; Tanaka, F. Formal [4 + 1] cycloaddition and enantioselective Michael-Henry cascade reactions to synthesize spiro[4,5]decanes and spirooxindole polycycles. Angew. Chem. Int. Ed. Eng. 2017, 56, 5853–5857. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-Z.; Xie, J.; Jiang, Y.; Sha, F.; Wu, X.-Y. Enantioselective vinylogous Michael/cyclization cascade reaction of acyclic β,γ-unsaturated amides with isatylidene malononitriles: Asymmetric construction of spirocyclic oxindoles. Adv. Synth. Catal. 2015, 357, 3507–3511. [Google Scholar] [CrossRef]
- Xie, J.; Xing, X.Y.; Sha, F.; Wu, Z.Y.; Wu, X.Y. Enantioselective synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives via an organocatalytic asymmetric michael/cyclization cascade reaction. Org. Biomol. Chem. 2016, 14, 8346–8355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhi, Y.; Li, X.; Puttreddy, R.; Rissanen, K.; Enders, D. Asymmetric synthesis of 3,3′-pyrrolidinyl-dispirooxindoles via a one-pot organocatalytic Mannich/deprotection/aza-Michael sequence. Chem. Commun. 2016, 52, 2249–2252. [Google Scholar] [CrossRef] [PubMed]
- Connon, S.J. Organocatalysis mediated by (thio)urea derivatives. Chem. Eur. J. 2006, 12, 5418–5427. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y. Development of chiral thiourea catalysts and its application to asymmetric catalytic reactions. Chem. Pharm. Bull. 2010, 58, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, C.J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine-thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-L.; Wei, Y.; Shi, M. Applications of chiral thiourea-amine/phosphine organocatalysts in catalytic asymmetric reactions. ChemCatChem 2017, 9, 718–727. [Google Scholar] [CrossRef]
- Reddy Gajulapalli, V.P.; Vinayagam, P.; Kesavan, V. Enantioselective assembly of functionalized carbocyclic spirooxindoles using anl-proline derived thiourea organocatalyst. RSC Adv. 2015, 5, 7370–7379. [Google Scholar] [CrossRef]
- Auria-Luna, F.; Marques-Lopez, E.; Mohammadi, S.; Heiran, R.; Herrera, R.P. New organocatalytic asymmetric synthesis of highly substituted chiral 2-oxospiro-[indole-3,4′-(1′,4′-dihydropyridine)] derivatives. Molecules 2015, 20, 15807–15826. [Google Scholar] [CrossRef] [PubMed]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xing, W.-L.; Sha, F.; Wu, X.-Y. Enantioselective cascade reaction of α-cyano ketones and isatylidene malononitriles: Asymmetric construction of spiro[4h-pyran-oxindoles]. Eur. J. Org. Chem. 2016, 2016, 3983–3992. [Google Scholar] [CrossRef]
- Cui, L.Y.; Wang, Y.M.; Zhou, Z.H. Enantioselective construction of novel chiral spirooxindoles incorporating a thiazole nucleus. RSC Adv. 2016, 6, 64474–64481. [Google Scholar] [CrossRef]
- Pratap Reddy Gajulapalli, V.; Lokesh, K.; Vishwanath, M.; Kesavan, V. Organocatalytic construction of spirooxindole naphthoquinones through Michael/hemiketalization using L-proline derived bifunctional thiourea. RSC Adv. 2016, 6, 12180–12184. [Google Scholar] [CrossRef]
- Yin, S.-J.; Zhang, S.-Y.; Zhang, J.-Q.; Sun, B.-B.; Fan, W.-T.; Wu, B.; Wang, X.-W. Organocatalytic tandem enantioselective Michael-cyclization of isatin-derived β,γ-unsaturated α-ketoesters with 3-hydroxy-4h-chromen-4-one or 2-hydroxy-1,4-naphthoquinone derivatives. RSC Adv. 2016, 6, 84248–84254. [Google Scholar] [CrossRef]
- Zhao, K.; Zhi, Y.; Shu, T.; Valkonen, A.; Rissanen, K.; Enders, D. Organocatalytic domino oxa-Michael/1,6-addition reactions: Asymmetric synthesis of chromans bearing oxindole scaffolds. Angew. Chem. Int. Ed. 2016, 55, 12104–12108. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Kumar, M.; Ghosh, S.K. Organocatalyzed enantioselective Michael addition/cyclization cascade reaction of 3-isothiocyanato oxindoles with arylidene malonates. Org. Biomol. Chem. 2016, 14, 11250–11260. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jing, L.; Qin, D.; Yin, M.; He, Q. Organocatalytic asymmetric synthesis of trans-configured trispirooxindoles through a cascade Michael-cyclization reaction. Tetrahedron Lett. 2016, 57, 2857–2860. [Google Scholar] [CrossRef]
- Zhao, J.Q.; Zhou, M.Q.; Wu, Z.J.; Wang, Z.H.; Yue, D.F.; Xu, X.Y.; Zhang, X.M.; Yuan, W.C. Asymmetric Michael/cyclization cascade reaction of 3-isothiocyanato oxindoles and 3-nitroindoles with amino-thiocarbamate catalysts: Enantioselective synthesis of polycyclic spirooxindoles. Org. Lett. 2015, 17, 2238–2241. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.-B.; Zhang, Z.; Huang, X.; Tanner, A.; Cai, C.; Zhang, W. One-pot fluorination and asymmetric Michael addition promoted by recyclable fluorous organocatalysts. RSC Adv. 2013, 3, 18267–18270. [Google Scholar] [CrossRef]
- Huang, X.; Pham, K.; Yi, W.; Zhang, X.; Clamens, C.; Hyatt, J.H.; Jasinsk, J.P.; Tayvah, U.; Zhang, W. Recyclable organocatalyst-promoted one-pot asymmetric synthesis of spirooxindoles bearing multiple stereogenic centers. Adv. Synth. Catal. 2015, 357, 3820–3824. [Google Scholar] [CrossRef]
- Huang, X.; Liu, M.; Pham, K.; Zhang, X.; Yi, W.B.; Jasinski, J.P.; Zhang, W. Organocatalytic one-pot asymmetric synthesis of thiolated spiro-γ-lactam oxindoles bearing three stereocenters. J. Org. Chem. 2016, 81, 5362–5369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Curran, D.P. Synthetic applications of fluorous solid-phase extraction (f-spe). Tetrahedron 2006, 62, 11837–11865. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Zhi, Y.; Zhao, K.; von Essen, C.; Rissanen, K. Thiourea-catalyzed domino Michael–Mannich [3 + 2] cycloadditions: A strategy for the asymmetric synthesis of 3,3′-pyrrolidinyl-dispirooxindoles. Synlett 2017. [Google Scholar] [CrossRef]
- Monari, M.; Montroni, E.; Nitti, A.; Lombardo, M.; Trombini, C.; Quintavalla, A. Highly stereoselective [4 + 2] and [3 + 2] spiroannulations of 2-(2-oxoindolin-3-ylidene)acetic esters catalyzed by bifunctional thioureas. Chem. Eur. J. 2015, 21, 11038–11049. [Google Scholar] [CrossRef] [PubMed]
- Cerisoli, L.; Lombardo, M.; Trombini, C.; Quintavalla, A. The first enantioselective organocatalytic synthesis of 3-spiro-α-alkylidene-gamma-butyrolactone oxindoles. Chem. Eur. J. 2016, 22, 3865–3872. [Google Scholar] [CrossRef] [PubMed]
- Quintavalla, A.; Lanza, F.; Montroni, E.; Lombardo, M.; Trombini, C. Organocatalytic conjugate addition of nitroalkanes to 3-ylidene oxindoles: A stereocontrolled diversity oriented route to oxindole derivatives. J. Org. Chem. 2013, 78, 12049–12064. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, S.; Prakash, M.; Ramakrishnan, C.; Gromiha, M.M.; Kesavan, V. Organocatalytic enantioselective assembly of spirooxindole-naphthopyrans through tandem Friedel-Crafts type/hemiketalization. ChemCatChem 2016, 8, 1708–1712. [Google Scholar] [CrossRef]
- Vishwanath, M.; Vinayagam, P.; Gajulapalli, V.P.R.; Kesavan, V. Asymmetric organocatalytic assembly of oxindoles fused with spiro-3,4-dihydropyrans with three contiguous stereocenters consisting of vicinal quaternary centers. Asian J. Org. Chem. 2016, 5, 613–616. [Google Scholar] [CrossRef]
- Du, D.; Jiang, Y.; Xu, Q.; Tang, X.-Y.; Shi, M. Enantioselective [3 + 2] cyclization of 3-isothiocyanato oxindoles with trifluoromethylated 2-butenedioic acid diesters. ChemCatChem 2015, 7, 1366–1371. [Google Scholar] [CrossRef]
- Cook, A.G.; Voges, A.B.; Kammarath, A.E. Aminal-catalyzed isomerization of and addition to dimethyl maleate. Tetrahedron Lett. 2001, 42, 7349–7352. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalyst. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.-L.; Du, D.-M. Organocatalytic enantioselective cascade aza-Michael/Michael addition sequence for asymmetric synthesis of chiral spiro[pyrrolidine-3,3′-oxindole]s. Asian J. Org. Chem. 2015, 4, 1120–1126. [Google Scholar] [CrossRef]
- Zhao, B.L.; Du, D.M. Organocatalytic cascade Michael/Michael reaction for the asymmetric synthesis of spirooxindoles containing five contiguous stereocenters. Chem. Commun. 2016, 52, 6162–6165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.-L.; Du, D.-M. Squaramide-catalyzed enantioselective cascade approach to bispirooxindoles with multiple stereocenters. Adv. Synth. Catal. 2016, 358, 3992–3998. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Z.J.; Zhang, M.L.; Yue, D.F.; Zhang, X.M.; Xu, X.Y.; Yuan, W.C. Organocatalytic asymmetric Michael/cyclization cascade reactions of 3-hydroxyoxindoles/3-aminooxindoles with α,β-unsaturated acyl phosphonates for the construction of spirocyclic oxindole-γ-lactones/lactams. J. Org. Chem. 2015, 80, 12668–12675. [Google Scholar] [CrossRef] [PubMed]
- Ming, S.; Zhao, B.L.; Du, D.M. Chiral squaramide-catalysed enantioselective Michael/cyclization cascade reaction of 3-hydroxyoxindoles with α,β-unsaturated N-acylated succinimides. Org. Biomol. Chem. 2017, 15, 6205–6213. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, L.; Du, D.-M. Squaramide-catalyzed asymmetric Michael/cyclization cascade reaction of 3-isothiocyanato oxindoles with chalcones for synthesis of pyrrolidinyl spirooxindoles. Org. Chem. Front. 2017, 4, 1229–1238. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, B.-L.; Du, D.-M. Organocatalytic asymmetric Michael/cyclization cascade reaction of 3-isothiocyanato oxindoles with maleimides for the efficient construction of pyrrolidonyl spirooxindoles. Eur. J. Org. Chem. 2016, 2016, 4711–4718. [Google Scholar] [CrossRef]
- Lou, Q.; Ding, Y.; Xu, D.; Liu, G.; Zhao, J. Organocatalytic enantioselective synthesis of dihydropyrano- indole derivatives bearing trifluoromethylated all-carbon-substituted stereocenters. Adv. Synth. Catal. 2017, 359, 2557–2563. [Google Scholar] [CrossRef]
- Curti, C.; Rassu, G.; Zambrano, V.; Pinna, L.; Pelosi, G.; Sartori, A.; Battistini, L.; Zanardi, F.; Casiraghi, G. Bifunctional cinchona alkaloid/thiourea catalyzes direct and enantioselective vinylogous Michael addition of 3-alkylidene oxindoles to nitroolefins. Angew. Chem. Int. Ed. Engl. 2012, 51, 6200–6204. [Google Scholar] [CrossRef] [PubMed]
- Rassu, G.; Zambrano, V.; Pinna, L.; Curti, C.; Battistini, L.; Sartori, A.; Pelosi, G.; Zanardi, F.; Casiraghi, G. Direct regio-, diastereo-, and enantioselective vinylogous Michael addition of prochiral 3-alkylideneoxindoles to nitroolefins. Adv. Synth. Catal. 2013, 355, 1881–1886. [Google Scholar] [CrossRef]
- Rassu, G.; Zambrano, V.; Tanca, R.; Sartori, A.; Battistini, L.; Zanardi, F.; Curti, C.; Casiraghi, G. 3-alkenyl-2-silyloxyindoles: An enabling, yet understated progeny of vinylogous carbon nucleophiles. Eur. J. Org. Chem. 2012, 2012, 466–470. [Google Scholar] [CrossRef]
- Han, J.L.; Chang, C.H. An asymmetric assembly of spirooxindole dihydropyranones through a direct enantioselective organocatalytic vinylogous aldol-cyclization cascade reaction of 3-alkylidene oxindoles with isatins. Chem. Commun. 2016, 52, 2322–2325. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Lou, Q.X.; Wang, L.S.; Hu, W.H.; Zhao, J.L. Organocatalytic Friedel-Crafts alkylation/lactonization reaction of naphthols with 3-trifluoroethylidene oxindoles: The asymmetric synthesis of dihydrocoumarins. Angew. Chem. Int. Ed. Eng. 2017, 56, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-J.; Weng, J.; Wang, S.; Lu, G. Organocatalytic diels-alder reaction of 2-vinylindoles with methyleneindolinones: An efficient approach to functionalized carbazolespirooxindoles. Adv. Synth. Catal. 2015, 357, 993–1003. [Google Scholar] [CrossRef]
- Shi, F.; Zhang, H.H.; Sun, X.X.; Liang, J.; Fan, T.; Tu, S.J. Organocatalytic asymmetric cascade reactions of 7-vinylindoles: Ddiastereo- and enantioselective synthesis of C7-functionalized indoles. Chem. Eur. J. 2015, 21, 3465–3471. [Google Scholar] [CrossRef] [PubMed]




































© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasperi, T.; Miceli, M.; Campagne, J.-M.; Marcia de Figueiredo, R. Non-Covalent Organocatalyzed Domino Reactions Involving Oxindoles: Recent Advances. Molecules 2017, 22, 1636. https://doi.org/10.3390/molecules22101636
Gasperi T, Miceli M, Campagne J-M, Marcia de Figueiredo R. Non-Covalent Organocatalyzed Domino Reactions Involving Oxindoles: Recent Advances. Molecules. 2017; 22(10):1636. https://doi.org/10.3390/molecules22101636
Chicago/Turabian StyleGasperi, Tecla, Martina Miceli, Jean-Marc Campagne, and Renata Marcia de Figueiredo. 2017. "Non-Covalent Organocatalyzed Domino Reactions Involving Oxindoles: Recent Advances" Molecules 22, no. 10: 1636. https://doi.org/10.3390/molecules22101636
APA StyleGasperi, T., Miceli, M., Campagne, J.-M., & Marcia de Figueiredo, R. (2017). Non-Covalent Organocatalyzed Domino Reactions Involving Oxindoles: Recent Advances. Molecules, 22(10), 1636. https://doi.org/10.3390/molecules22101636