3.2. Chemistry
3.2.1. 2,4-Dibromo-1-methyl-5-nitro-1H-imidazole (2)
First step: to a solution of 4(5)-nitro-1H-imidazole (20 g, 177 mmol), NaHCO3 (44.6 g, 3 equiv., 531 mmol) in water (120 mL) was added dropwise at 0 °C dibromine (27 mL, 3 equiv., 531 mmol) and the reaction mixture was heated at 65 °C for 6 h. After cooling, the solution was poured into bath of ice. A yellow precipitate appeared and was filtered, washed with water (3 × 100 mL) and dried in vacuum drying oven (dessicator cabinet). The aqueous layer was extracted with ethyle acetate (3 × 100 mL) and the organic layer was washed with brine (3 × 100 mL), dried over Na2SO4 and evaporated. Yellow solid was obtained and added to the precipitate to give 36.3 g (80%) of 2,4(5)-dibromo-5(4)-nitroimidazole.
Second step; to a solution of 2,4(5)-dibromo-5(4)-nitroimidazole (34 g, 126 mmol) in DMF (80 mL) was added dropwise at 0 °C dimethyl sulfate (13.1 mL, 1.1 equiv., 138 mmol) and the reaction mixture was heated at 100 °C for 2 h. After cooling, the solution was treated with a saturated solution of sodium hydrogen carbonate at 0 °C. A white precipitate appeared and was filtered. One part of the precipitate was solubilised in ethyl acetate and evaporated after filtration. The precipitate not dissolved was the 2,5-dibromo-1-methyl-4-nitro-1
H-imidazole (
1, 38%, 14 g), M.p. 192 °C (lit. [
26] 204–205 °C).
1H-NMR (CDCl
3): δ = 3.75 (s, 3H, NCH
3) ppm.
13C-NMR (CDCl
3): δ = 35.0 (NCH
3), 106.7 (C
Ar), 120.4 (C
Ar) ppm (CNO
2 was not visible under these conditions). The aqueous layer was extracted with ethyl acetate (3 × 100 mL) and the organic layer was washed with brine (3 × 100 mL), dried over Na
2SO
4 and evaporated. The crude product was purified by column chromatography [silica gel, petroleum ether/ethyl acetate (9/1) and the 2,4-dibromo-1-methyl-5-nitro-1
H-imidazole (
2) was obtained in 33% in yield (12.1 g). M.p. 167 °C (lit. [
42] 160–161 °C).
1H-NMR (CDCl
3): δ = 4.02 (s, 3H, NCH
3) ppm.
13C-NMR (CDCl
3): δ = 37.2 (NCH
3), 119.8 (C
Ar), 126.4 (C
Ar) ppm (CNO
2 was not visible under these conditions).
3.2.2. General Procedure for the Regioselective Suzuki-Miyaura Coupling Reaction
A solution of 2,4-dibromo-1-methyl-5-nitro-1H-imidazole (2, 0.1 g, 0.35 mmol), boronic acid (0.46 mmol, 1.3 equiv.), Pd(OAc)2 (3 mg, 0.014 mmol, 0.04 equiv.), Na2CO3 (0.11 g, 1.05 mmol, 3 equiv.), in a DME/EtOH/H2O mixture (3/1/1, 5 mL) was heated at 60 °C under microwave irradiation for 2 h. After cooling, 60 mL of water were added and the solution was extracted with dichloromethane (3 × 50 mL). The organic layer was washed with water (3 × 100 mL), dried over Na2SO4 and evaporated. The crude product was purified by column chromatography [silica gel, petroleum ether/ethyl acetate (9/1), (1/1 for 3i), cyclohexane/ethyl acetate (9/1 for 3a)] and recrystallized from propan-2-ol.
2-Bromo-1-methyl-5-nitro-4-phenyl-1H-imidazole (3a): Yield 64% (63 mg); yellow solid; M.p. 96 °C. 1H-NMR (CDCl3): δ = 4.01 (s, 3H, NCH3), 7.43–7.45 (m, 3H, 3 Ar-H), 7.73–7.77 (m, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.5 (NCH3), 126.6 (CAr), 128.1 (2 CHAr), 129.5 (2 CHAr), 129.9 (CHAr), 130.7 (CAr), 136.0 (CAr), 144.3 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C10H8BrN3O2]+: 281.9873; found 281.9874.
2-Bromo-4-(4-methoxypheny)-1-methyl-5-nitro-1H-imidazole (3b): Yield 65% (71 mg); orange solid; M.p. 125 °C. 1H-NMR (CDCl3): δ = 3.86 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 6.95 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.77 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.6 (NCH3), 55.3 (OCH3), 113.6 (2 CHAr), 123.0 (CAr), 126.8 (CAr), 131.3 (2 CHAr), 135.8 (CAr), 144.5 (CAr), 161.0 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C11H10BrN3O3]+: 311.9978; found 311.9977.
2-Bromo-1-methyl-5-nitro-4-[4-(trifluoromethyl)phenyl]-1H-imidazole (3c): Yield 62% (76 mg); yellow solid; M.p. 102 °C. 1H-NMR (CDCl3): δ = 4.03 (s, 3H, NCH3), 7.69 (d, 3JH-H = 8.2 Hz, 2H, 2 Ar-H), 8.87 (d, 3JH-H = 8.2 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.6 (NCH3), 123.9 (q, 1JC-F = 272.4 Hz, CF3), 125.1 (q, 3JC-F = 3.9 Hz, 2 CHAr), 126.9 (C), 129.9 (2 CHAr), 131.6 (q, 2JC-F = 32.5 Hz, CAr), 134.2 (CAr), 136.3 (CAr), 142.4 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C11H7BrF3N3O2]+: 349.9746; found 349.9744.
2-Bromo-1-methyl-5-nitro-4-(3-nitrophenyl)-1H-imidazole (3d): Yield 61% (70 mg); yellow solid; M.p. 166 °C. 1H-NMR (CDCl3): δ = 4.06 (s, 3H, NCH3), 7.63 (t, 3JH-H = 8.2 Hz, 1H, Ar-H), 8.10 (d, 3JH-H = 7.7 Hz, 1H, Ar-H), 8.28-8.32 (m, 1H, Ar-H), 8.66 (s, 1H, Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.8 (NCH3), 124.5 (CHAr), 124.8 (CHAr), 127.1 (CAr), 129.2 (CHAr), 132.4 (CAr), 135.4 (CHAr), 136.3 (CAr), 141.5 (CAr), 148.0 (CAr) ppm. Anal. Calcd. for C10H7BrN4O4 (327.09): C 36.72, H 2.16, N 17.13; found C 36.82, H 2.06, N 17.13.
4-(2-Bromo-1-methyl-5-nitro-1H-imidazol-4-yl)benzonitrile (3e): Yield 64% (69 mg); yellow solid; M.p. 155 °C. 1H-NMR (CDCl3): δ = 4.04 (s, 3H, NCH3), 7.73 (d, 3JH-H = 8.5 Hz, 2H, 2 Ar-H), 7.88 (d, 3JH-H = 8.5 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 35.8 (NCH3), 113.3 (C), 118.4 (CAr), 127.1 (CAr), 130.2 (2 CHAr), 131.9 (2 CHAr), 135.0 (CAr), 136.4 (CAr), 141.8 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C11H7BrN4O2]+: 306.9825; found 306.9824.
2-Bromo-4-(4-fluorophenyl)-1-methyl-5-nitro-1H-imidazole (3f): Yield 62% (65 mg); yellow solid; M.p. 112 °C. 1H-NMR (CDCl3): δ = 4.01 (s, 3H, NCH3), 7.08–7.15 (m, 2H, 2 Ar-H), 7.74–7.80 (m, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.6 (NCH3), 115.2 (d, 2JC-F = 21.5 Hz, 2 CHAr), 126.7 (CAr), 126.8 (CAr), 131.7 (d, 3JC-F = 8.8 Hz, 2 CHAr), 135.9 (CAr), 143.3 (CAr), 163.6 (d, 1JC-F = 250.9 Hz, CAr) ppm. Anal. Calcd. for C10H7BrFN3O2 (300.08): C 40.02, H 2.35, N 14.00; found C 39.86, H 2.23, N 13.80.
2-Bromo-1-methyl-5-nitro-4-(3,4,5-trimethoxyphenyl)-1H-imidazole (3g): Yield 55% (72 mg); yellow solid; M.p. 134 °C. 1H-NMR (CDCl3): δ = 3.89 (s, 9H, NCH3, 2 OCH3), 4.00 (s, 3H, OCH3), 7.07 (s, 2H, 2 Ar-H), ppm. 13C-NMR (CDCl3δ = 36.7 (NCH3), 56.2 (2 OCH3), 60.9 (OCH3), 107.0 (2 CHAr), 107.4 (CAr), 125.7 (CAr), 126.6 (CAr), 139.6 (CAr), 144.0 (CAr), 152.8 (2 CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C13H14BrN3O5]+: 372.0190; found 372.0187.
2-Bromo-1-methyl-5-nitro-4-p-tolyl-1H-imidazole (3h): Yield 63% (65 mg); yellow solid; M.p. 160 °C. 1H-NMR (CDCl3): δ = 2.40 (s, 3H, CH3), 4.01 (s, 3H, NCH3), 7.24 (d, 3JH-H = 8.2 Hz, 2H, 2 CH ar), 7.66 (d, 3JH-H = 8.2 Hz, 2H, 2 CH ar) ppm. 13C-NMR (CDCl3): δ = 21.4 (CH3), 36.5 (NCH3), 125.4 (CAr), 126.6 (CAr), 127.8 (CAr), 128.9 (2 CHAr), 129.4 (2 CHAr), 140.2 (CAr), 144.5 (CAr) ppm. Anal. Calcd. for C11H10BrN3O2 (296.12): C 44.62, H 3.40, N 14.19; found C 44.63, H 3.27, N 13.95.
[4-(2-Bromo-1-methyl-5-nitro-1H-imidazol-4-yl)phenyl]methanol (3i): Yield 58% (65 mg); yellow solid; M.p. 132 °C. 1H-NMR (DMSO-d6): δ = 3.91 (s, 3H, NCH3), 4.55 (d, 3JH-H = 5.8 Hz, 2H, CH2), 5.29 (t, 3JH-H = 5.7 Hz, 1H, OH), 7.40 (d, 3JH-H = 8.2 Hz, 2H, 2 Ar-H), 7.63 (d, 3JH-H = 8.2 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (DMSO-d6): δ = 36.7 (NCH3), 62.8 (CH2), 126.1 (2 CHAr), 127.4 (CAr), 129.1 (2 CHAr), 129.6 (CAr), 136.1 (CAr), 143.0 (CAr), 144.3 (CAr) ppm. Anal. Calcd. for C11H10BrN3O3 (312.12): C 42.33, H 3.23, N 13.46; found C 42.34, H 3.11, N 13.33.
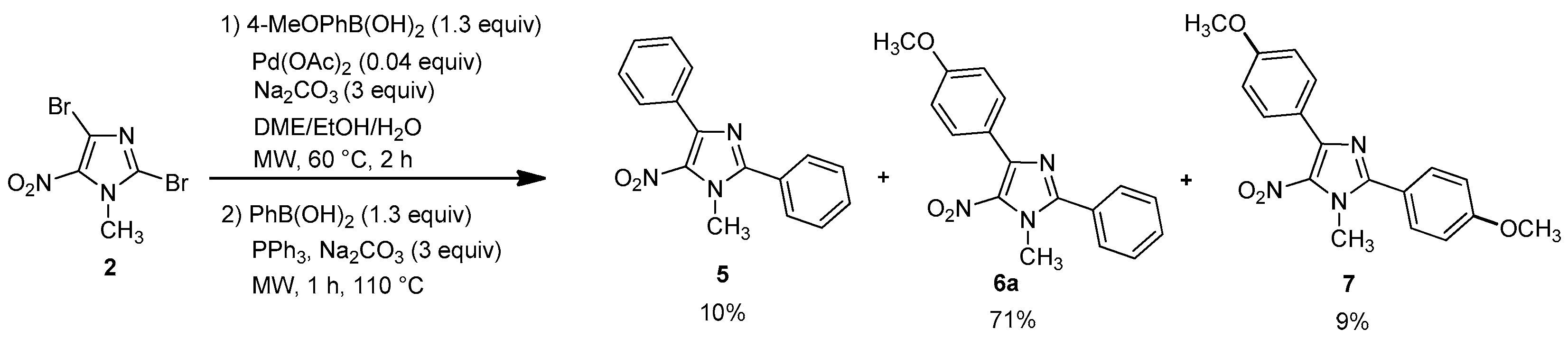
1-Methyl-5-nitro-2-4-diphenyl-1H-imidazole (5): Yield 9% using general procedure for the regioselective Suzuki-Miyaura coupling reaction (9 mg); yellow solid; M.p. 129 °C. 1H-NMR (CDCl3): δ = 4.00 (s, 3H, NCH3), 7.44–7.47 (m, 3H, 3 Ar-H), 7.54–7.56 (m, 3H, 3 Ar-H), 7.69–7.72 (m, 2H, 2 Ar-H), 7.83–7.87 (m, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.2 (NCH3), 128.1 (2 CHAr), 128.3 (CAr), 128.9 (2 CHAr), 129.5 (CHAr), 129.7 (4 CHAr), 130.7 (CHAr), 131.7 (CAr), 135.9 (CAr), 144.2 (CAr), 150.5 (CAr) ppm. Anal. Calcd. for C16H13N3O2 (279.29): C 68.71, H 4.69, N 15.05; found C 68.43, H 4.62, N 14.85.
3.2.3. General Procedure for the One-Pot Regioselective Bis-Suzuki-Miyaura Coupling Reaction
A solution of 2,4-dibromo-1-methyl-5-nitro-1H-imidazole (2, 0.1 g, 0.35 mmol), 4-methoxy-phenylboronic acid (72 mg, 0.46 mmol, 1.3 equiv.), Pd(OAc)2 (3 mg, 0.014 mmol, 0.04 equiv.), Na2CO3 (0.11 g, 1.05 mmol, 3 equiv.), in a DME/EtOH/H2O mixture (3/1/1, 5 mL) was heated at 60 °C under microwave irradiation for 2 h. After cooling, boronic acid (0.45 mmol, 1.3 equiv.), PPh3 (9 mg, 0.035 mmol, 0.1 equiv.), Na2CO3 (0.11 g, 1.05 mmol, 3 equiv.), were introduced under argon. The mixture was heated at 110 °C for 1 h under microwave irradiation. After cooling, 60 mL of water were added and the solution was extracted with dichloromethane (3 × 50 mL). The organic layer was washed with water (3 × 100 mL), dried over Na2SO4 and evaporated. The crude product was purified by column chromatography [silica gel, petroleum ether/ethyl acetate (9/1), (7/3 for 6e), (5/5 for 6j)] and recrystallized from propan-2-ol.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-phenyl-1H-imidazole (6a): Yield 71% (77 mg); yellow solid; M.p. 120 °C. 1H-NMR (CDCl3): δ = 3.86 (s, 3H, NCH3), 3.99 (s, 3H, OCH3), 6.98 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H), 7.53–7.56 (m, 3H, 3 Ar-H), 7.68–7.72 (m, 2H, 2 Ar-H), 7.87 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.3 (NCH3), 55.3 (OCH3), 113.5 (2 CHAr), 123.9 (CAr), 128.3 (CAr), 128.9 (2 CHAr), 129.7 (2 CHAr), 130.7 (CHAr), 131.4 (2 CHAr), 135.6 (CAr), 144.3 (CAr), 150.6 (CAr), 160.7 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C17H15N3O3]+: 310.1186; found 310.1185.
2-(4-Fluorophenyl)-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (6b): Yield 70% (80 mg); yellow solid; M.p. 132 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 3.97 (s, 3H, OCH3), 6.98 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.21–7.28 (m, 2H, 2 Ar-H), 7.68–7.73 (m, 2H, 2 Ar-H), 7.85 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.3 (NCH3), 55.4 (OCH3), 113.6 (2 CHAr), 116.3 (d, 2JC-F = 21.6 Hz, 2 CHAr), 123.9 (CAr), 123.6 (CAr), 131.4 (2 CHAr), 131.9 (d, 3JC-F = 8.7 Hz, 2 CHAr), 144.3 (CAr), 148.9 (CAr), 149.6 (CAr), 160.0 (d, 1JC-F = 272.1 Hz, CAr), 160.8 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C17H14FN3O3]+: 328.1092; found 328.1093.
2-(4-Chlorophenyl)-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (6c): Yield 60% (72 mg); yellow solid; M.p. 134 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 3.98 (s, 3H, OCH3), 6.98 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.53 (d, 3JH-H = 8.7 Hz, 2H, 2 Ar-H), 7.66 (d, 3JH-H = 8.7 Hz, 2H, 2 Ar-H), 7.84 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.3 (NCH3), 55.3 (OCH3), 113.6 (2 CHAr), 123.8 (CAr), 126.8 (CAr), 129.3 (2 CHAr), 131.0 (2 CHAr), 131.4 (2 CHAr), 134.8 (CAr), 137.1 (CAr), 144.3 (CAr), 149.4 (CAr), 160.8 (CAr) ppm. Anal. Calcd. for C17H14ClN3O3 (343.76): C 59.40, H 4.10, N 12.22; found C 59.16, H 3.82, N 12.31.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-(3-nitrophenyl)-1H-imidazole (6d): Yield 65% (81 mg); yellow solid; M.p. 192 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 4.03 (s, 3H, OCH3), 6.99 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.76 (t, 3JH-H = 8.0 Hz, 1H, Ar-H), 7.85 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 8.06 (d, 3JH-H = 7.7 Hz, 1H, Ar-H), 8.42 (d, 3JH-H = 8.2 Hz, 1H, Ar-H), 8.59 (t, 4JH-H = 1.7 Hz, 1H, Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.3 (NCH3), 55.4 (OCH3), 113.6 (2 CHAr), 123.5 (CAr), 124.6 (CHAr), 125.2 (CHAr), 130.2 (CHAr), 131.3 (2 CHAr), 135.3 (CHAr), 135.9 (CAr), 144.2 (CAr), 147.7 (CAr), 148.4 (CAr), 156.1 (CAr), 160.9 (CAr) ppm. Anal. Calcd. for C17H14N4O5 (354.32): C 57.63, H 3.98, N 15.81; found C 57.38, H 3.79, N 15.59.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-(3,4,5-trimethoxyphenyl)-1H-imidazole (6e): Yield 50% (70 mg); yellow solid; M.p. 158 °C. 1H-NMR (CDCl3): δ = 3.86 (s, 3H, NCH3), 3.91 (s, 9H, 3 OCH3), 3.98 (s, 3H, OCH3), 6.87 (s, 2H, 2 Ar-H), 6.97 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H), 7.86 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.4 (NCH3), 55.3 (OCH3), 56.4 (2 OCH3), 61.0 (OCH3), 107.0 (2 CHAr), 113.5 (2 CHAr), 123.6 (CAr), 123.9 (CAr), 131.4 (2 CHAr), 135.6 (CAr), 140.2 (CAr), 144.3 (CAr), 150.7 (CAr), 153.5 (2 CAr), 160.7 (CAr) ppm. Anal. Calcd. for C20H21N3O6 (399.40): C 60.14, H 5.30, N 10.52; found C 59.67, H 5.05, N 10.67.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-[3-(trifluoromethyl)-phenyl]-1H-imidazole (6f): Yield 58% (76 mg); yellow solid; M.p. 117 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 6.99 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.69 (t, 3JH-H = 7.7 Hz, 1H, Ar-H), 7.81–7.90 (m, 4H, 4 Ar-H), 8.00 (s, 1H, Ar-H). 13C-NMR (CDCl3): δ = 36.2 (NCH3), 55.3 (OCH3), 113.6 (2 CHAr), 123.5 (q, 1JC-F = 272.9 Hz, CF3), 123.6 (CAr), 126.7 (q, 3JC-F = 3.7 Hz, CHAr), 127.4 (q, 3JC-F = 3.7 Hz, CHAr), 129.3 (CAr), 129.6 (CHAr), 131.3 (2 CHAr), 131.7 (q, 2JC-F = 33.0 Hz, CAr), 132.8 (CHAr), 135.8 (CAr), 144.2 (CAr), 148.8 (CAr), 160.9 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C18H14F3N3O3]+: 378.1060; found 378.1058.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-p-tolyl-1H-imidazole (6g): Yield 56% (64 mg); yellow solid; M.p. 136 °C. 1H-NMR (CDCl3): δ = 2.45 (s, 3H, CH3), 3.86 (s, 3H, NCH3), 3.97 (s, 3H, OCH3), 6.98 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.35 (d, 3JH-H = 7.9 Hz, 2H, 2 Ar-H), 7.58 (d, 3JH-H = 8.2 Hz, 2H, 2 Ar-H), 7.86 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 21.5 (CH3), 36.3 (NCH3), 55.3 (OCH3), 113.5 (2 CHAr), 124.1 (CAr), 125.5 (CAr), 129.5 (2 CHAr), 129.6 (2 CHAr), 131.4 (2 CHAr), 135.6 (CAr), 141.1 (CAr), 144.5 (CAr), 150.8 (CAr), 160.7 (CAr) ppm. Anal. Calcd. for C18H17N3O3 (323.35): C 66.86, H 5.30, N 13.00; found C 66.74, H 5.22, N 12.81.
4-(4-Methoxyphenyl)-1-methyl-2-(naphthalen-2-yl)-5-nitro-1H-imidazole (6h): Yield 61% (77 mg); yellow solid; M.p. 143 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 4.05 (s, 3H, OCH3), 6.99 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.56–7.64 (m, 2H, 2 Ar-H), 7.77 (dd, 4JH-H = 1.6 Hz, 3JH-H = 8.5 Hz, 1H, Ar-H), 7.90 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.92–7.96 (m, 2H, 2 Ar-H), 8.01 (d, 3JH-H = 8.5 Hz, 1H, Ar-H), 8.22 (s, 1H, Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.4 (NCH3), 55.3 (OCH3), 113.6 (2 CHAr), 124.0 (CAr), 125.7 (CAr), 125.9 (CHAr), 127.1 (CHAr), 127.8 (CHAr), 127.9 (CHAr), 128.6 (CHAr), 128.8 (CHAr), 130.2 (CHAr), 131.4 (2 CHAr), 132.8 (CAr), 134.0 (CAr), 135.8 (CAr), 144.4 (CAr), 150.7 (CAr), 160.8 (CAr) ppm. Anal. Calcd. for C21H17N3O3 (359.38): C 70.18, H 4.77, N 11.69; found C 70.23, H 4.69, N 11.69.
4-(4-Methoxyphenyl)-1-methyl-2-(5-methylthiophen-2-yl)-5-nitro-1H-imidazole (6i): Yield 52% (60 mg); yellow solid; M.p. 151 °C. 1H-NMR (CDCl3): δ = 2.57 (s, 3H, CH3), 3.86 (s, 3H, NCH3), 4.09 (s, 3H, OCH3), 6.86-6.88 (m, 1H, Ar-H), 6.97 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.37 (d, 4JH-H = 3.6 Hz, 1H, Ar-H), 7.85 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 15.4 (CH3), 36.0 (NCH3), 55.3 (OCH3), 113.5 (2 CHAr), 124.0 (CAr), 126.4 (CHAr), 127.7 (CAr), 129.9 (CHAr), 131.5 (2 CHAr), 135.5 (CAr), 136.8 (CAr), 145.0 (CAr), 145.3 (CAr), 160.8 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C16H15N3O3S]+: 330.0907; found 330.0907.
3-[4-(4-Methoxyphenyl)-1-methyl-5-nitro-1H-imidazol-2-yl]pyridine (6j): Yield 69% (75 mg); yellow solid; M.p. 152 °C. 1H-NMR (CDCl3): δ = 3.86 (s, 3H, NCH3), 4.01 (s, 3H, OCH3), 6.98 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.48–7.53 (m, 1H, Ar-H), 8.84 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 8.04–8.08 (m, 1H, Ar-H), 8.79–8.80 (m, 1H, Ar-H), 8.96 (s, 1H, Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.2 (NCH3), 55.3 (OCH3), 113.6 (2 CHAr), 123.7 (CHAr), 125.0 (CAr), 131.3 (2 CHAr), 135.9 (CAr), 137.2 (CHAr), 144.4 (CAr), 147.5 (CAr), 149.9 (CHAr), 151.4 (CHAr), 160.8 (2 CAr). HRMS (ESI) m/z [M + H]+ calcd. for [C16H14N4O3]+: 311.1139; found 311.1136.
2-(4-Methoxyphenyl)-1-methyl-5-nitro-4-phenyl-1H-imidazole (6): Yield 64% (69 mg); yellow solid; M.p. 128 °C. 1H-NMR (CDCl3): δ = 3.88 (s, 3H, NCH3), 3.99 (s, 3H, OCH3), 7.04 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.43–7.48 (m, 3H, 3 Ar-H), 7.65 (d, 3JH-H = 8.7 Hz, 2H, 2 Ar-H), 7.82–7.86 (m, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.1 (NCH3), 55.5 (OCH3), 114.6 (2 CHAr), 121.0 (CAr), 128.0 (2 CHAr), 129.4 (CHAr), 129.7 (2 CHAr), 131.3 (2 CHAr), 132.1 (CAr), 136.1 (CAr), 144.2 (CAr), 150.6 (CAr), 161.7 (CAr) ppm. Anal. Calcd. for C17H15N3O3 (309.32): C 66.01, H 4.89, N 13.58; found C 65.98, H 4.75, N 13.42.
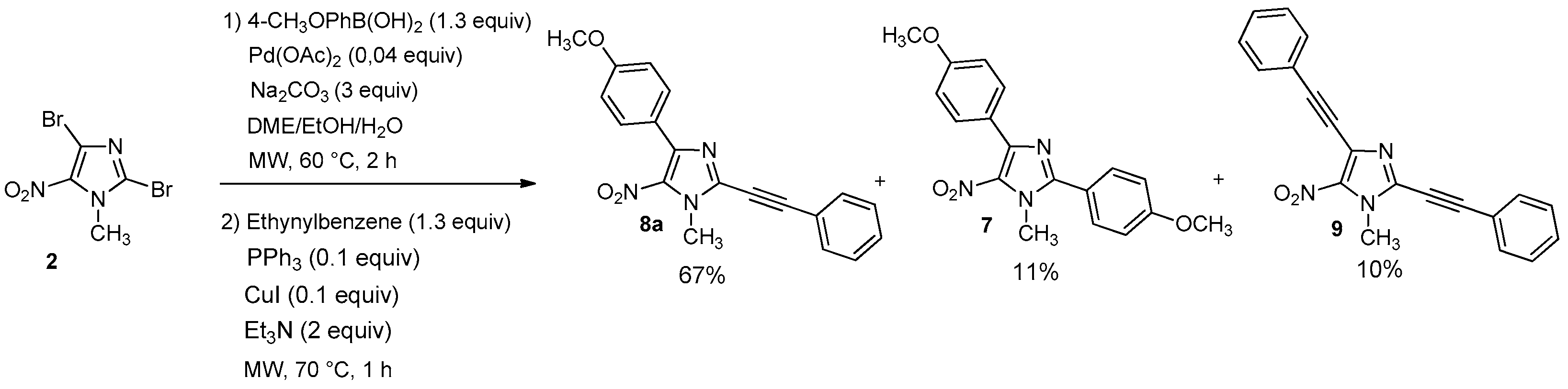
2,4-bis(4-methoxypheny)-1-methyl-5-nitro-1H-imidazole (7): Yield 9% using general procedure for the one-pot regioselective bis-Suzuki-Miyaura coupling reaction (11 mg); yellow solid; M.p. 121 °C. 1H-NMR (CDCl324 °C): δ = 3.86 (s, 3H, NCH3), 3.89 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 6.98 (d, 3JH-H = 7.6 Hz, 2H, 2 Ar-H), 7.02 (d, 3JH-H = 8.1 Hz, 2H, 2 Ar-H), 7.65 (d, 3JH-H = 7.9 Hz, 2H, 2 Ar-H), 7.86 (d, 3JH-H = 7.6 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 36.5 (NCH3), 55.3 (OCH3), 55.5 (OCH3), 113.6 (2 CHAr), 114.5 (2 CHAr), 120.5 (CAr), 124.0 (CAr), 131.3 (2 CHAr), 131.5 (2 CHAr), 135.6 (CAr), 144.5 (CAr), 150.8 (CAr), 160.8 (CAr), 161.6 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C18H17N3O4] 340.1292; found 340.1292.
3.2.4. General Procedure for the One-Pot Regioselective Suzuki-Miyaura/Sonogashira Coupling Reaction
A solution of 2,4-dibromo-1-methyl-5-nitro-1H-imidazole (2, 0.1 g, 0.35 mmol), 4-methoxy-phenylboronic acid (72 mg, 0.46 mmol, 1.3 equiv.), Pd(OAc)2 (3 mg, 0.014 mmol, 0.04 equiv.), Na2CO3 (0.11 g, 1.05 mmol, 3 equiv.), in a DME/EtOH/H2O mixture (3/1/1, 5 mL) was heated at 60 °C under microwave irradiation for 2 h. After cooling, terminal alkynes (0.45 mmol, 1.3 equiv.), PPh3 (9 mg, 0.035 mmol, 0.1 equiv.), CuI (7 mg, 0.035 mmol, 0.1 equiv.), Et3N (0.1 mL, 0.7 mmol, 2 equiv.), were introduced under argon. The mixture was heated at 70 °C for 1 h under microwave irradiation. After cooling, 60 mL of water were added and the solution was extracted with dichloromethane (3 × 50 mL). The organic layer was washed with water (3 × 100 mL), dried over Na2SO4 and evaporated. The crude product was purified by column chromatography [silica gel, petroleum ether/ethyl acetate (10%), (50% for 8g)] and recrystallized from propan-2-ol.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-(phenylethynyl)-1H-imidazole (8a): Yield 67% (78 mg); yellow solid; M.p. 156 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 4.14 (s, 3H, OCH3), 6.97 (d, 3JH-H = 8.0 Hz, 2H, 2 Ar-H), 7.39–7.51 (m, 3H, 3 Ar-H), 7.64 (d, 3JH-H = 6.6 Hz, 2H, 2 Ar-H), 8.82 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 35.8 (NCH3), 55.3 (OCH3), 77.2 (C), 96.5 (C), 113.5 (2 CHAr), 120.3 (CAr), 123.6 (CAr), 128.7 (2 CHAr), 130.3 (CHAr), 131.2 (2 CHAr), 132.2 (2 CHAr), 134.1 (CAr), 134.4 (CAr), 144.7 (CAr), 160.8 (CAr) ppm. Anal. Calcd. for C19H15N3O3 (333.34): C 68.46, H 4.54, N 12.61; found C 68.39, H 4.44, N 12.53.
4-(4-Methoxyphenyl)-1-methyl-5-nitro-2-(m-tolylethynyl)-1H-imidazole (8b): Yield 74% (90 mg); yellow solid; M.p. 138 °C. 1H-NMR (CDCl3): δ = 2.56 (s, 3H, CH3), 3.86 (s, 3H, NCH3), 4.14 (s, 3H, OCH3), 6.97 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.20–7.39 (m, 3H, 3 Ar-H), 7.60 (d, 3JH-H = 8.4 Hz, 1H, Ar-H), 8.81 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 20.9 (CH3), 35.7 (NCH3), 55.3 (OCH3), 81.0 (C), 95.6 (C), 113.5 (2 CHAr), 120.2 (CAr), 123.6 (CAr), 126.0 (CHAr), 129.8 (CHAr), 130.3 (CHAr), 131.2 (2 CHAr), 132.8 (CHAr), 134.1 (CAr), 134.6 (CAr), 141.1 (CAr), 144.7 (CAr), 160.8 (CAr) ppm. Anal. Calcd. for C20H17N3O3 (347.37): C 69.15, H 4.93, N 12.10; found C 68.94, H 4.79, N 11.89.
2-[(3-Fluorophenyl)ethynyl]-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8c): Yield 59% (73 mg); yellow solid; M.p. 148 °C. 1H-NMR (CDCl3): δ = 3.86 (s, 3H, NCH3), 4.13 (s, 3H, OCH3), 6.97 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H), 7.13–7.21 (m, 1H, Ar-H), 7.30–7.42 (m, 3H, 3 Ar-H), 8.80 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 35.9 (NCH3), 55.3 (OCH3), 77.9 (C), 94.8 (d, 4JC-F = 3.2 Hz, C), 113.5 (2 CHAr), 117.7 (d, 2JC-F = 21.1 Hz, CHAr), 119.0 (d, 2JC-F = 23.9 Hz, CHAr), 122.1 (d, 3JC-F = 9.7 Hz, CAr), 123.4 (CAr), 128.1 (d, 4JC-F = 3.2 Hz, CHAr), 130.4 (d, 3JC-F = 8.7 Hz, CHAr), 131.2 (2 CHAr), 133.9 (CAr), 134.2 (CAr), 144.6 (CAr), 160.8 (CAr), 162.2 (d, 1JC-F = 248.2 Hz, CAr) ppm. Anal. Calcd. for C19H14FN3O3 (351.33): C 64.95, H 4.02, N 11.96; found C 64.67, H 3.84, N 11.79.
2-[(4-Methoxy-2-methylphenyl)ethynyl]-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8d): Yield 53% (70 mg); yellow solid; M.p. 182 °C. 1H-NMR (CDCl3): δ = 2.53 (s, 3H, CH3), 3.84 (s, 3H, NCH3), 3.86 (s, 3H, OCH3), 4.13 (s, 3H, OCH3), 6.74–6.81 (m, 2H, 2 Ar-H), 6.97 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.53 (d, 3JH-H = 8.4 Hz, 1H, Ar-H), 7.81 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 21.1 (CH3), 35.7 (NCH3), 55.3 (OCH3), 55.4 (OCH3), 80.1 (C), 96.2 (C), 111.8 (CHAr), 112.3 (CAr), 113.5 (2 CHAr), 115.5 (CHAr), 123.7 (CAr), 131.3 (2 CHAr), 134.0 (CAr), 134.4 (CHAr), 135.1 (CAr), 143.2 (CAr), 144.8 (CAr), 160.7 (CAr), 161.1 (CAr) ppm. Anal. Calcd. for C21H19N3O4 (377.39): C 66.83, H 5.07, N 11.13; found C 66.56, H 4.91, N 11.10.
2-(Cyclopentylethynyl)-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8e): Yield 60% (69 mg); yellow solid; M.p. 98 °C. 1H-NMR (CDCl3): δ = 1.59–1.68 (m, 2H, CH2), 1.73–1.82 (m, 4H, 2 CH2), 2.00–2.10 (m, 2H, CH2), 2.92 (q, 3JH-H = 7.5 Hz, 1H, CH), 3.84 (s, 3H, NCH3), 4.01 (s, 3H, OCH3), 6.94 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H), 7.77 (d, 3JH-H = 9.0 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 25.2 (2 CH2), 30.5 (CH), 33.4 (2 CH2), 35.5 (NCH3), 55.3 (OCH3), 68.9 (C), 103.5 (C), 113.4 (2 CHAr), 123.7 (CAr), 131.2 (2 CHAr), 133.6 (CAr), 134.9 (CAr), 144.4 (CAr), 160.7 (CAr) ppm. Anal. Calcd. for C18H19N3O3 (325.36): C 66.45, H 5.89, N 12.91; found C 66.21, H 5.76, N 12.88.
2-[(4-tert-Butylphenyl)ethynyl]-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8f): Yield 63% (86 mg); yellow oil. 1H-NMR (CDCl3): δ = 1.34 (s, 9H, 3 CH3), 3.86 (s, 3H, NCH3), 4.12 (s, 3H, OCH3), 6.96 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H), 7.43 (d, 3JH-H = 8.4 Hz, 2H, 2 Ar-H), 7.56 (d, 3JH-H = 8.4 Hz, 2H, 2 Ar-H), 7.81 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 31.0 (3 CH3), 35.0 (C), 35.7 (NCH3), 55.3 (OCH3), 76.8 (C), 96.9 (C), 113.5 (2 CHAr), 117.2 (CAr), 123.6 (CAr), 125.7 (2 CHAr), 131.2 (2 CHAr), 132.0 (2 CHAr), 134.0 (CAr), 134.6 (CAr), 144.7 (CAr), 153.9 (CAr), 160.7 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C23H23N3O3]+: 390.1812; found 390.1813.
3-[4-(4-Methoxyphenyl)-1-methyl-5-nitro-1H-imidazol-2-yl]prop-2-yn-1-ol (8g): Yield 51% (51 mg); yellow solid; M.p. 177 °C. 1H-NMR (DMSO-d6): δ = 3.81 (s, 3H, NCH3), 3.97 (s, 3H, OCH3), 4.43 (d, 3JH-H = 5.7 Hz, 2H, CH2), 5.67 (t, 3JH-H = 5.8 Hz, 1H, OH), 7.01 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H), 7.67 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H). 13C-NMR (DMSO-d6): δ = 35.9 (NCH3), 49.8 (CH2), 55.7 (OCH3), 72.7 (C), 97.5 (C), 113.9 (2 CHAr), 124.1 (CAr), 131.2 (2 CHAr), 133.6 (CAr), 134.5 (CAr), 143.2 (CAr), 160.6 (CAr) ppm. Anal. Calcd. for C14H13N3O4 (287.27): C 58.53, H 4.56, N 14.63; found C 58.64, H 4.51, N 14.40.
2-[(3-Chlorophenyl)ethynyl]-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8h): Yield 61% (79 mg); yellow solid; M.p. 139 °C. 1H-NMR (CDCl3): δ = 3.86 (s, 3H, NCH3), 4.13 (s, 3H, OCH3), 6.97 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H), 7.32–7.38 (m, 1H, Ar-H), 7.42–7.47 (m, 1H, Ar-H), 7.51 (dt, 4JH-H = 1.4 Hz, 3JH-H = 7.4 Hz, 1H, Ar-H), 7.61 (t, 4JH-H = 1.4 Hz, 1H, Ar-H), 8.80 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 35.8 (NCH3), 55.3 (OCH3), 78.2 (C), 94.7 (C), 113.6 (2 CHAr), 122.0 (CAr), 123.4 (CAr), 130.0 (CHAr), 130.3 (CHAr), 130.6 (CHAr), 131.2 (2 CHAr), 131.9 (CHAr), 133.8 (CAr), 134.2 (CAr), 134.6 (CAr), 144.6 (CAr), 160.8 (CAr). Anal. Calcd. for C19H14ClN3O3 (367.79): C 62.05, H 3.84, N 11.43; found C 61.90, H 3.77, N 11.35.
2-[(2-Chlorophenyl)ethynyl]-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8i): Yield 52% (67 mg); yellow solid; M.p. 171 °C. 1H-NMR (CDCl3): δ = 3.87 (s, 3H, NCH3), 4.19 (s, 3H, OCH3), 6.98 (d, 3JH-H = 8.5 Hz, 2H, 2 Ar-H), 7.29–7.42 (m, 2H, 2 Ar-H), 7.49 (d, 3JH-H = 7.9 Hz, 1H, Ar-H), 7.67 (d, 3JH-H = 7.4 Hz, 1H, Ar-H), 7.80 (d, 3JH-H = 8.7 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 35.8 (NCH3), 55.3 (OCH3), 82.1 (C), 92.8 (C), 113.6 (2 CHAr), 120.7 (CAr), 123.6 (CAr), 126.9 (CHAr), 129.6 (CHAr), 131.2 (CHAr), 131.3 (2 CHAr), 133.9 (CHAr), 134.0 (CAr), 134.1 (CAr), 136.6 (CAr), 144.6 (CAr), 160.9 (CAr) ppm. Anal. Calcd. for C19H14ClN3O3 (367.79): C 62.05, H 3.84, N 11.43; found C 61.86, H 3.66, N 11.31.
2-(Cyclopropylethynyl)-4-(4-methoxyphenyl)-1-methyl-5-nitro-1H-imidazole (8j): Yield 63% (66 mg); yellow solid; M.p. 124 °C. 1H-NMR (CDCl3): δ = 0.95–1.03 (m, 4H, 2 CH2), 1.50–1.60 (m, 1H, CH), 3.85 (s, 3H, NCH3), 4.02 (s, 3H, OCH3), 6.94 (d, 3JH-H = 8.8 Hz, 2H, 2 Ar-H), 7.77 (d, 3JH-H = 8.9 Hz, 2H, 2 Ar-H) ppm. 13C-NMR (CDCl3): δ = 0.2 (CH), 9.3 (2 CH2), 35.5 (NCH3), 55.3 (OCH3), 64.4 (C), 77.2 (C), 102.4 (CAr), 113.5 (2 CHAr), 123.7 (CAr), 131.3 (2 CHAr), 134.7 (CAr), 144.3 (CAr), 160.8 (CAr) ppm. Anal. Calcd. for C16H15N3O3 (297.31): C 64.64, H 5.09, N 14.13; found C 64.46, H 4.92, N 14.22.
1-Methyl-5-nitro-2,4-bis(phenylethynyl)-1H-imidazole (9): Yield 10% using general procedure for the one-pot regioselective Suzuki-Miyaura/Sonogashira coupling reaction (12 mg); yellow solid; M.p. 166 °C. 1H-NMR (CDCl3): δ = 4.14 (s, 3H, NCH3), 7.38–7.48 (m, 6H, 6 Ar-H), 7.61–7.66 (m, 4H, 4 Ar-H) ppm. 13C-NMR (CDCl3): δ = 35.6 (NCH3), 76.7 (C), 81.0 (C), 96.8 (C), 97.1 (C), 120.0 (CAr), 121.7 (CAr), 127.7 (CAr), 128.5 (2 CHAr), 128.7 (2 CHAr), 129.6 (CHAr), 130.5 (CHAr), 132.2 (2 CHAr), 132.3 (2 CHAr), 133.5 (CAr), 135.1 (CAr) ppm. HRMS (ESI) m/z [M + H]+ calcd. for [C20H13N3O2]+: 328.1081; found 328.1081.
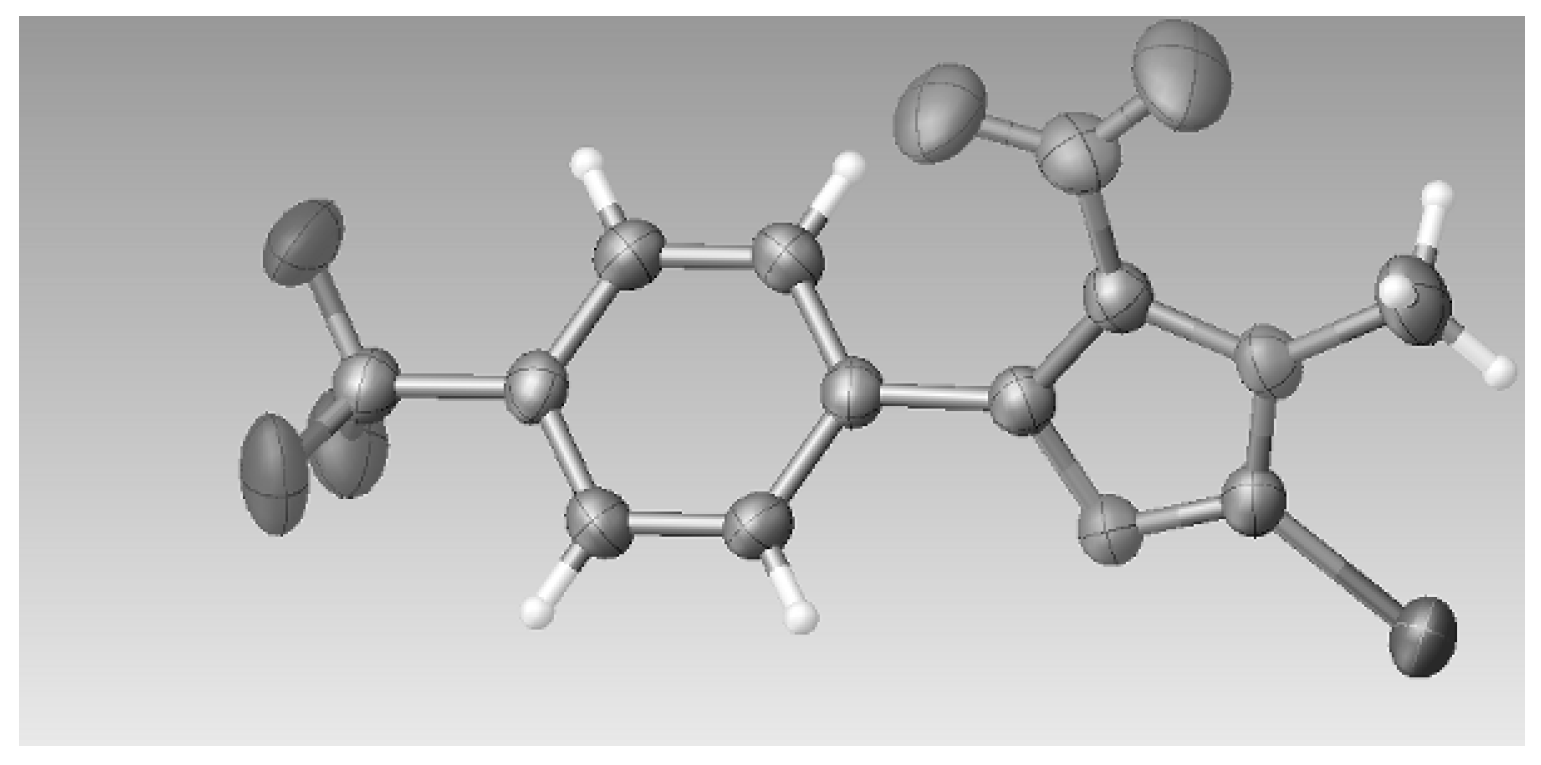
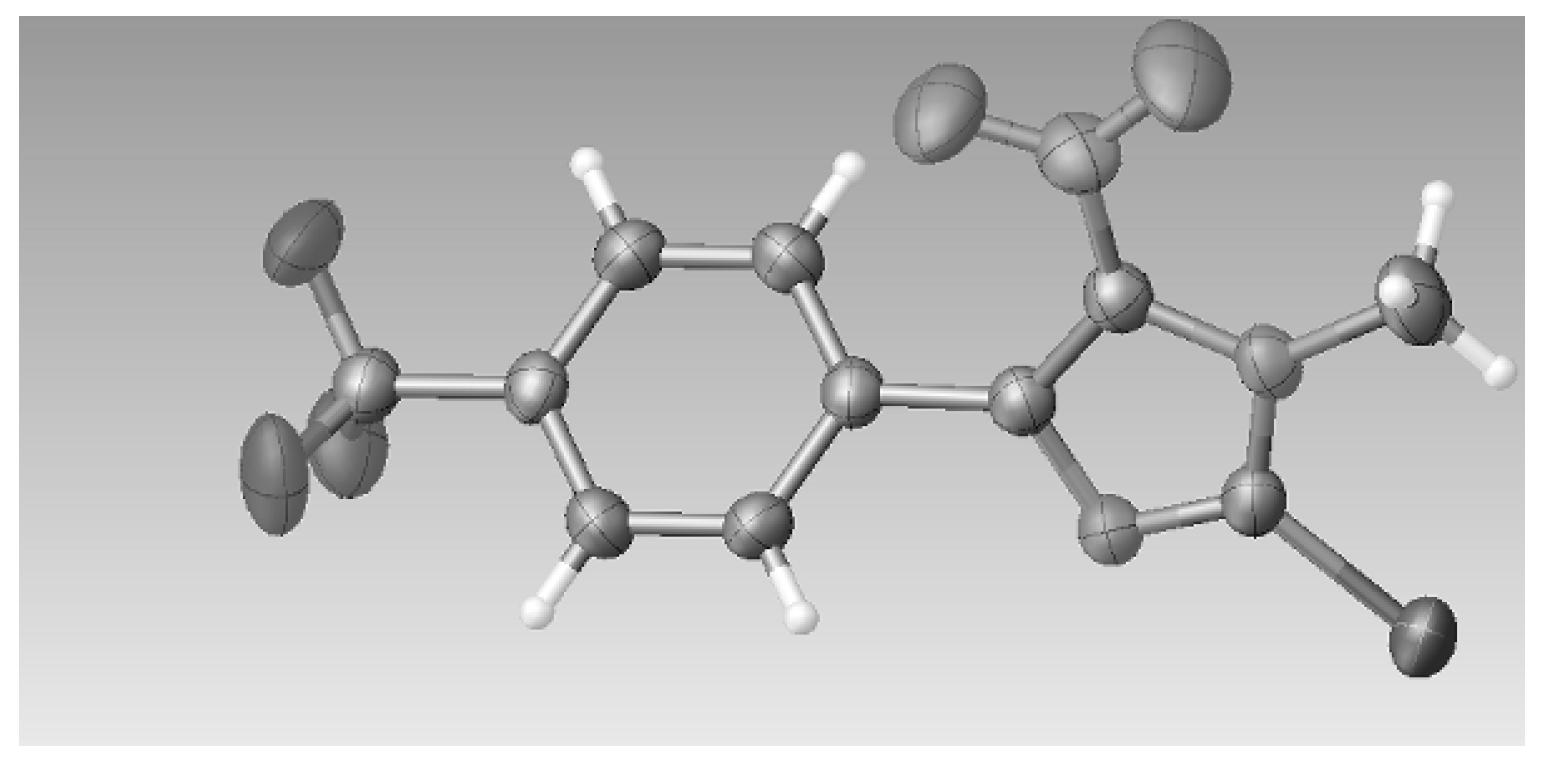
3.2.5. Crystal Data for Compound 3c
2(C
11H
7BrF
3N
3O
2),
M = 700.21,
a = 8.4376(4) Å,
b = 12.5416(6) Å,
c = 12.6319(5) Å,
α = 82.406(4)°,
β = 73.324(4)°,
γ = 80.384(4)°,
V = 1257.48(10) Å
3,
T = 293 K, space group
P Z = 2, 14503 reflections measured, 5058 independent reflections (
Rint = 0.0184). The final
R1 values were 0.0320 (
I > 2
σ(
I)). The final
wR(
F2) values were 0.0689 (
I > 2
σ(
I)). The final
R1 values were 0.0469 (all data). The final
wR(
F2) values were 0.0752 (all data). The goodness of fit on
F2 was 1.007. CCDC 1542022 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at
www.cdcc.cam.ac.uk/data_request/cif of from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; Fax: + 44 (1223) 336033; email:
deposit@ccdc.cam.ac.uk.
Figures S1–S66:
1H- and
13C-NMR of all compounds
3a–
i,
5,
6a–
j,
6,
7,
8a–
j and
9.





{kind=link}
{kind=link}
{kind=link}
{kind=link}




