
Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability


Abstract
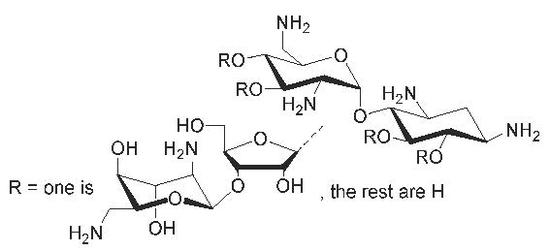
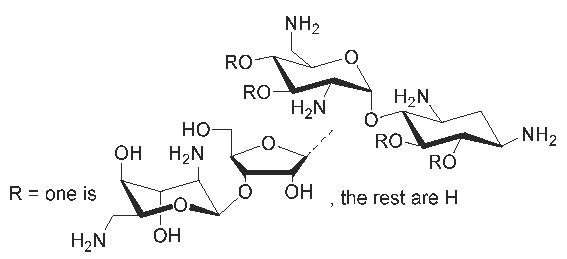
:
1. Introduction
2. Results and Discussion
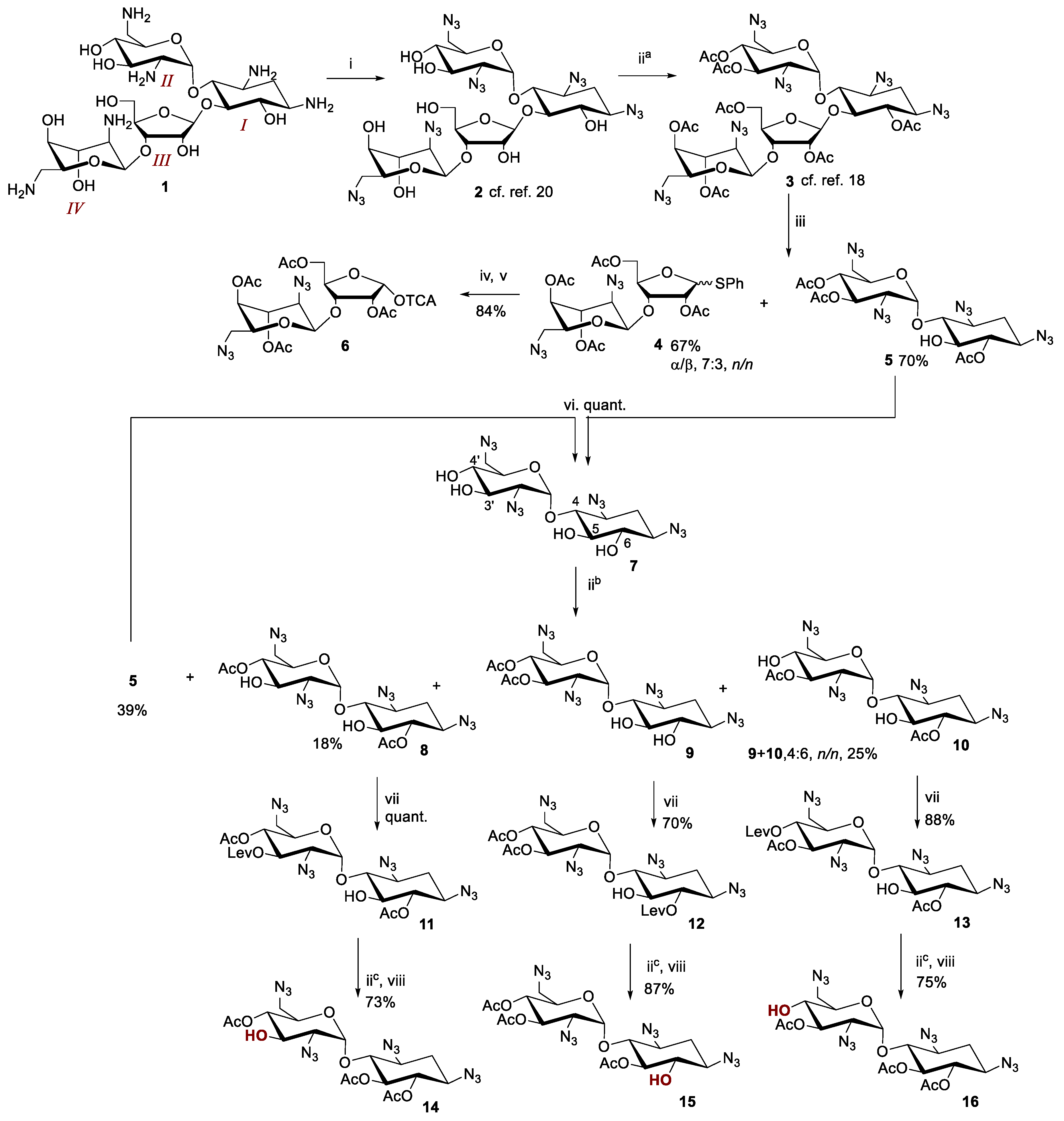
2.1. Synthesis of Glycosyl Donor 6 and Acceptors 14–16
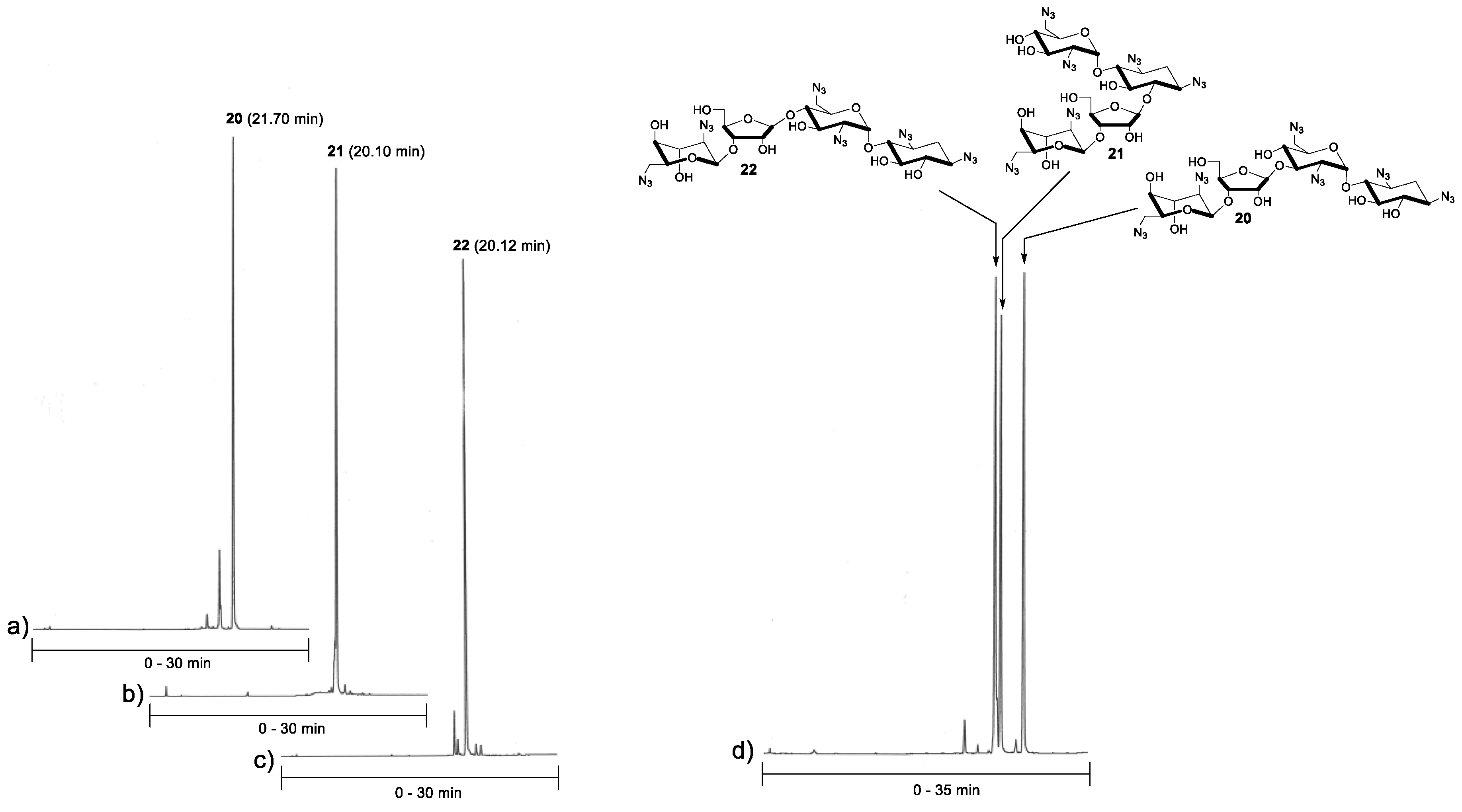
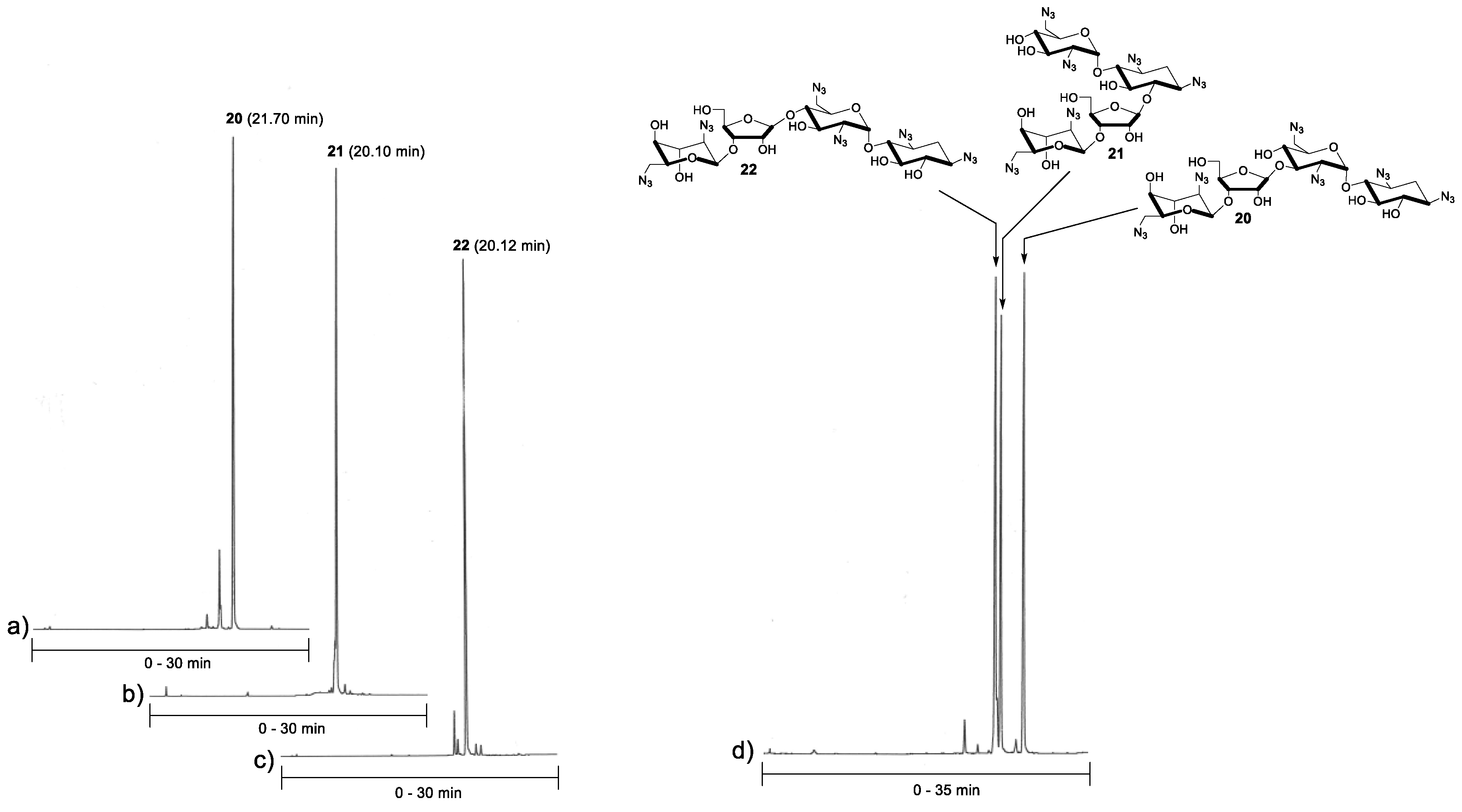
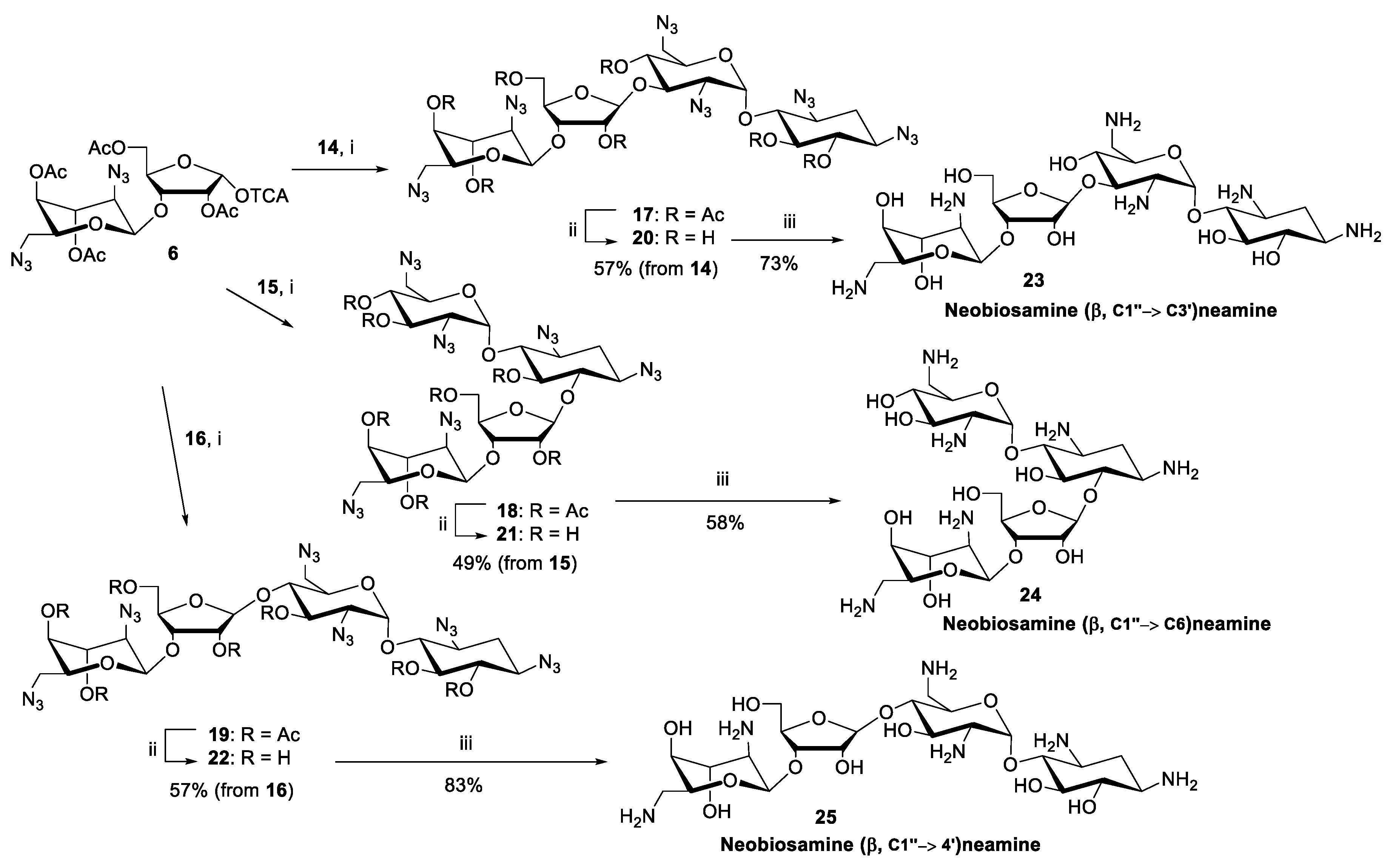
2.2. Glycosylation and Global Deprotection
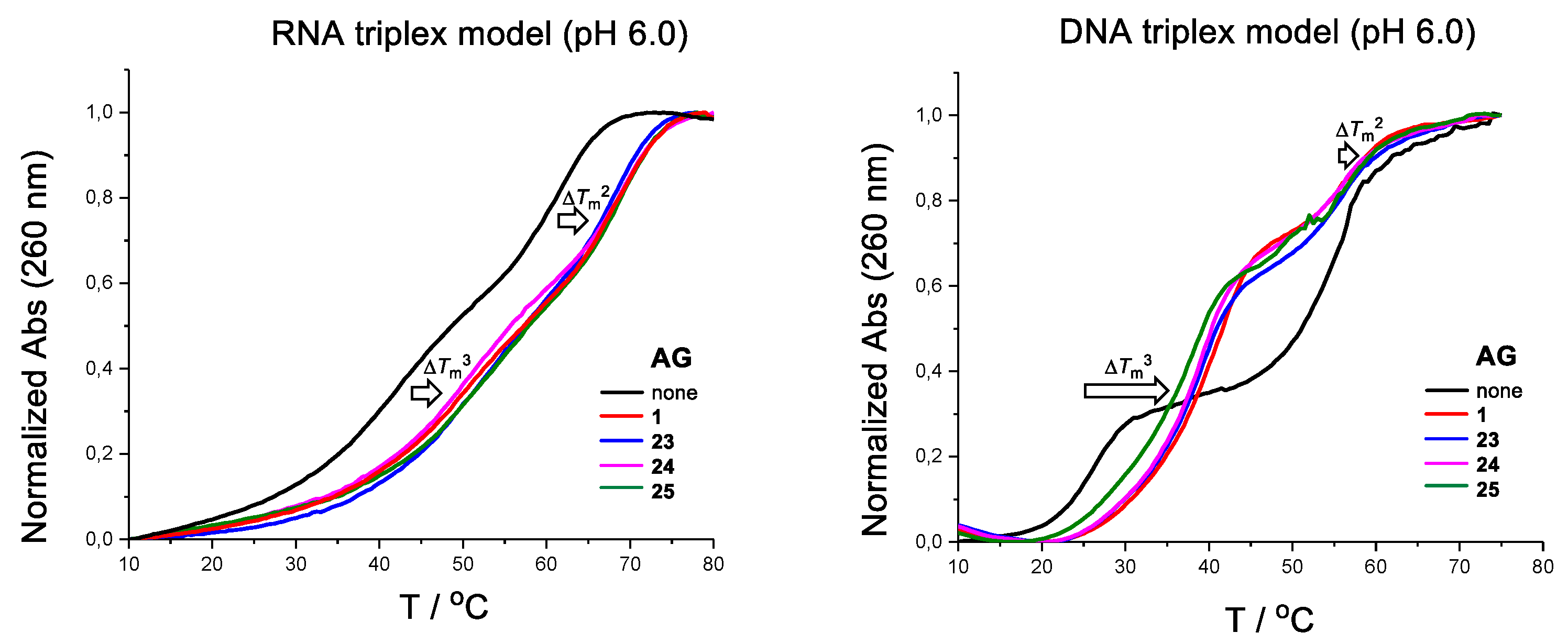
2.3. The Effect of 23–25 on DNA and RNA Triplex Stability
3. Materials and Methods
General Remarks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thomas, J.R.; Hergenrother, P.J. Targeting of RNA with Small Molecules. Chem. Rev. 2008, 108, 1171–1224. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Gorini, L.; Davis, B.D. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol. Pharmacol. 1965, 1, 93–106. [Google Scholar] [PubMed]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Von Ahsen, U.; Noller, H.F. Footprinting the sites of interaction of antibiotics with catalytic group I intron RNA. Science 1993, 260, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
- Francois, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding site oligonucleotides: Role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acid Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Chen, W.; Juskeviciene, R.; Teo, Y.; Shcherbakov, D.; Vasella, A.; Böttger, E.C.; Crich, D. Influence of 4′-O-glycoside constitution and configuration on ribosomal selectivity of paromomycin. J. Am. Chem. Soc. 2015, 137, 7706–7717. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, W.A.; Priestley, S.; Sears, P.S.; Alper, P.B.; Rosenbohm, A.; Hendrix, M.; Hung, S.-C.; Wong, C.-H. Design and synthesis of new aminoglycoside antibiotics containing neamine as an optimal core structure: Correlation of antibiotic activity with in vitro inhibition of translation. J. Am. Chem. Soc. 1999, 121, 6527–6541. [Google Scholar] [CrossRef]
- Von Ahsen, U.; Davies, J.; Schroeder, R. Antibiotic inhibition of group I ribozyme function. Nature 1991, 353, 368–370. [Google Scholar] [CrossRef]
- Herman, T. Drugs targeting the ribosome. Curr. Opin. Struct. Biol. 2005, 15, 355–366. [Google Scholar] [CrossRef]
- Mei, H.-Y.; Galan, A.A.; Halim, N.S.; Mack, D.P.; Morelan, D.W.; Sanders, K.B.; Truong, H.N.; Czarnik, A.W. Inhibition of an HIV-1 Tat-derived peptide binding to TAR RNA by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 1995, 5, 2755–2760. [Google Scholar] [CrossRef]
- Zapp, M.L.; Stern, S.; Green, M.R. Small molecules that selectively block RNA bindingo f HIV-1 rev protein inhibit rev function and viral production. Cell 1993, 74, 969–978. [Google Scholar] [CrossRef]
- Tam, V.K.; Kwong, D.; Tor, Y. Fluorescent HIV-1 dimerization initiation site: Design, properties, and use for ligand discovery. J. Am. Chem. Soc. 2007, 129, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Ennifar, E.; Paillart, J.-C.; Bodlenner, A.; Walter, P.; Weibel, J.-M.; Aubertin, A.-M.; Pale, P.; Dumas, P.; Marquet, R. Targeting the dimerization initiation site of HIV-1 RNA with aminoglycosides: From crystal to cell. Nucleic Acid Res. 2006, 34, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Coffee, R.L., Jr. DNA triple helix stabilization by aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 2000, 10, 1897–1899. [Google Scholar] [CrossRef]
- Arya, D.P.; Micovic, L.; Charles, I.; Lane Coffee, R., Jr.; Willis, B.; Xue, L. Neomycin Binding to Watson-Hoogsteen (W-H) DNA triplex groove: A model. J. Am. Chem. Soc. 2003, 125, 3733–3744. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Xue, L.; Tennant, P. Combining the best in triplex recognition: Synthesis and nucleic acid binding of a BQQ-neomycin conjugate. J. Am. Chem. Soc. 2003, 125, 8070–8071. [Google Scholar] [CrossRef] [PubMed]
- Arya, D.P.; Coffee, R.L., Jr.; Charles, I. Neomycin induced hybrid triplex formation. J. Am. Chem. Soc. 2001, 123, 11093–11094. [Google Scholar] [CrossRef]
- Arya, D.P. New approaches towards recognition of nucleic acid triple helices. Acc. Chem. Res. 2011, 44, 134–146. [Google Scholar] [CrossRef]
- Wu, B.; Yang, J.; He, Y.; Swayze, E.E. Reexamination of neomycin B degradation: Efficient preparation of its CD and D rings as protected glycosyl donors. Org. Lett. 2002, 4, 3455–3458. [Google Scholar] [CrossRef]
- Van den Broek, S.A.M.W.; Gruijters, B.W.T.; Rutjes, F.P.J.T.; van Delft, F.L.; Blaauw, R.H. A short and scalable route to orthogonally O-protected 2-deoxystreaptamine. J. Org. Chem. 2007, 72, 3577–3580. [Google Scholar] [CrossRef]
- Alper, P.B.; Hung, S.-C.; Wong, C.-H. Metal catalyzed diazo transfer. Tetrahedron Lett. 1996, 37, 6029–6032. [Google Scholar] [CrossRef]
- Geny, S.; Moreno, P.M.D.; Krzywkowski, T.K.; Gissberg, O.; Andersen, N.K.; Isse, A.J.; El-Madani, A.M.; Lou, C.; Pabon, Y.V.; et al. Next-generation bis-locked nucleic acids with stacking linker and 2′-glycylamino-LNA show enhanced DNA invasion into supercoiled duplexes. Nucleic Acids Res. 2016, 44, 2007–2019. [Google Scholar] [CrossRef]
- Tähtinen, V.; Granqvist, L.; Virta, P. Synthesis of C-5, C-2′ and C-4′-neomycin-conjugated triplex forming oligonucleotides and their affinity to DNA-duplexes. Bioorg. Med. Chem. 2015, 23, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Granqvist, L.; Virta, P. 4′-C-[(4-trifluoromethyl-1H-1,2,3-triazol-1-yl)methyl]thymidine as a sensitive 19F NMR sensor for the detection of oligonucleotide secondary structures. J. Org. Chem. 2014, 79, 3529–3536. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Sugiura, M.; Nishimoto, S. Monitoring of duplex and triplex formation by 19F NMR using oligodeoxynucleotides possessing 5-fluorodeoxyuridine unit as 19F signal transmitter. Bioorg. Med. Chem. 2010, 18, 6690–6694. [Google Scholar] [CrossRef] [PubMed]
- Granqvist, L.; Kraszewski, A.; Tähtinen, V.; Virta, P. Synthesis of aminoglycoside-2′-O-methyl oligoribonucleotide fusions. Molecules 2017, 22, 760. [Google Scholar] [CrossRef] [PubMed]
- Bhuma, N.; Tähtinen, V.; Virta, P. Synthesis and Applicability of Base-Discriminating DNA-Triplex-Forming 19F NMR Probes. Eur. J. Org. Chem. 2018, 605–613. [Google Scholar] [CrossRef]
- Granqvist, L.; Virta, P. 2′-O-[(4-CF3-triazol-1-yl)methyl] uridine—A sensitive 19F NMR sensor for the detection of RNA secondary structures. J. Org. Chem. 2015, 80, 7961–7970. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 23–25 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| RNA Triplex Model | ||
| AG | Tm3(°C) pH 6.0 | Tm2(°C) pH 6.0 |
| none | 41.9 | 60.1 |
| 1 (5 eq) | 51.2 (+9.3) | 68.0 (+7.9) |
| 23 (5 eq) | 53.2 (+11.3) | 66.7 (+6.6) |
| 24 (5 eq) | 52.5 (+10.6) | 67.7 (+7.6) |
| 25 (5 eq) | 51.4 (+9.5) | 68.0 (+7.9) |
| AG | Tm3(°C) pH 7.0 | Tm2(°C) pH 7.0 |
| none | 22.6 | 60.1 |
| 1 (5 eq) | 29.4 (+6.9) | 62.0 (+1.9) |
| 23 (5 eq) | 30.4 (+7.8) | 61.2 (+1.1) |
| 24 (5 eq) | 30.4 (+7.8) | 61.6 (+1.5) |
| 25 (5 eq) | 29.4 (+6.9) | 61.9 (+1.8) |
| DNA Triplex Model | ||
| AG | Tm3(°C) pH 6.0 | Tm2(°C) pH 6.0 |
| none | 25.5 | 54.6 |
| 1 (5 eq) | 40.1 (+14.6) | 56.3 (+1.7) |
| 23 (5 eq) | 38.1 (+12.6) | 55.5 (+0.9) |
| 24 (5 eq) | 36.6 (+11.1) | 56.3 (+1.7) |
| 25 (5 eq) | 38.6 (+13.1) | 55.3 (+0.7) |
| 1 (10 eq) | 44.1 (+18.6) | 56.3 (+1.7) |
| 23 (10 eq) | 41.3 (+15.8) | 55.9 (+1.3) |
| 24 (10 eq) | 39.4 (+13.9) | 56.9 (+2.3) |
| 25 (10 eq) | 42.9 (+17.4) | 56.5 (+1.9) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granqvist, L.; Tähtinen, V.; Virta, P. Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability. Molecules 2019, 24, 580. https://doi.org/10.3390/molecules24030580
Granqvist L, Tähtinen V, Virta P. Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability. Molecules. 2019; 24(3):580. https://doi.org/10.3390/molecules24030580
Chicago/Turabian StyleGranqvist, Lotta, Ville Tähtinen, and Pasi Virta. 2019. "Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability" Molecules 24, no. 3: 580. https://doi.org/10.3390/molecules24030580
APA StyleGranqvist, L., Tähtinen, V., & Virta, P. (2019). Synthesis of Glycosidic (β-1′′→6, 3′ and 4′) Site Isomers of Neomycin B and Their Effect on RNA and DNA Triplex Stability. Molecules, 24(3), 580. https://doi.org/10.3390/molecules24030580