
ESIPT-Related Origin of Dual Fluorescence in the Selected Model 1,3,4-Thiadiazole Derivatives

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
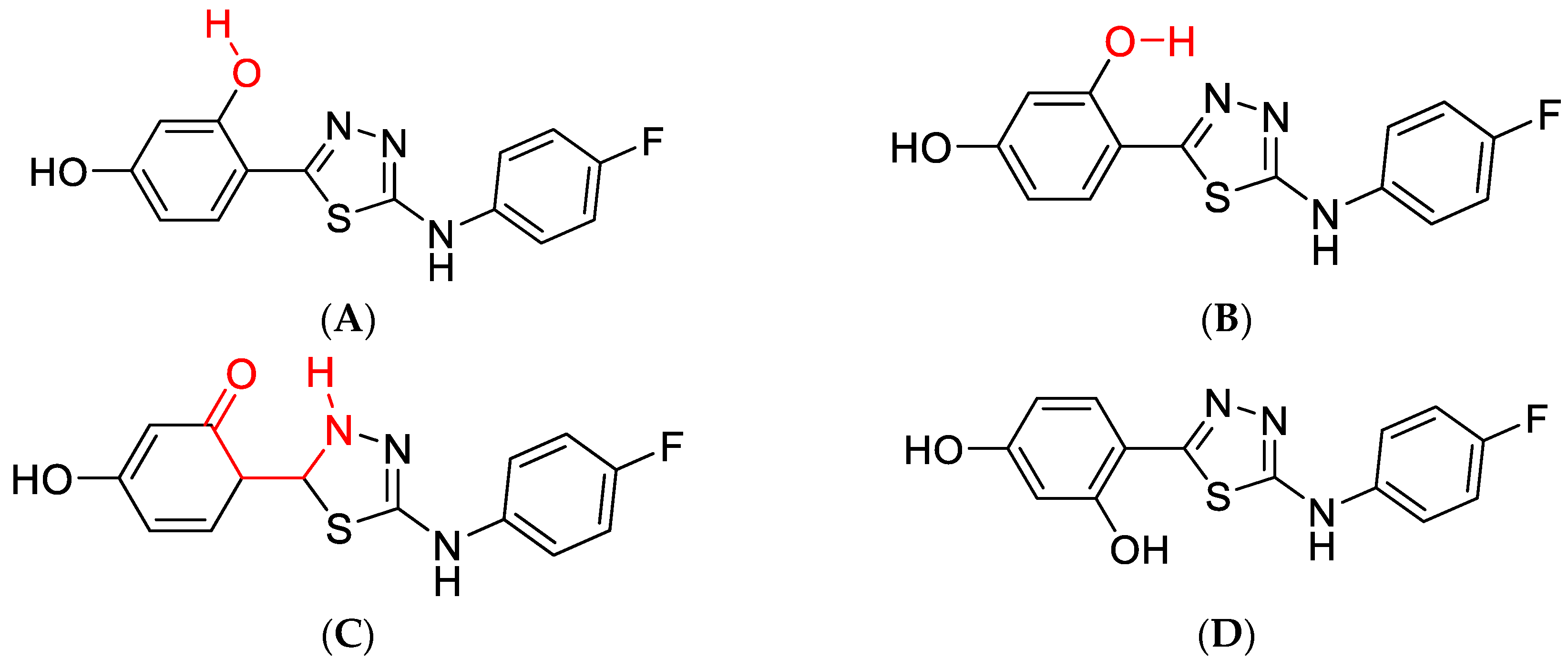
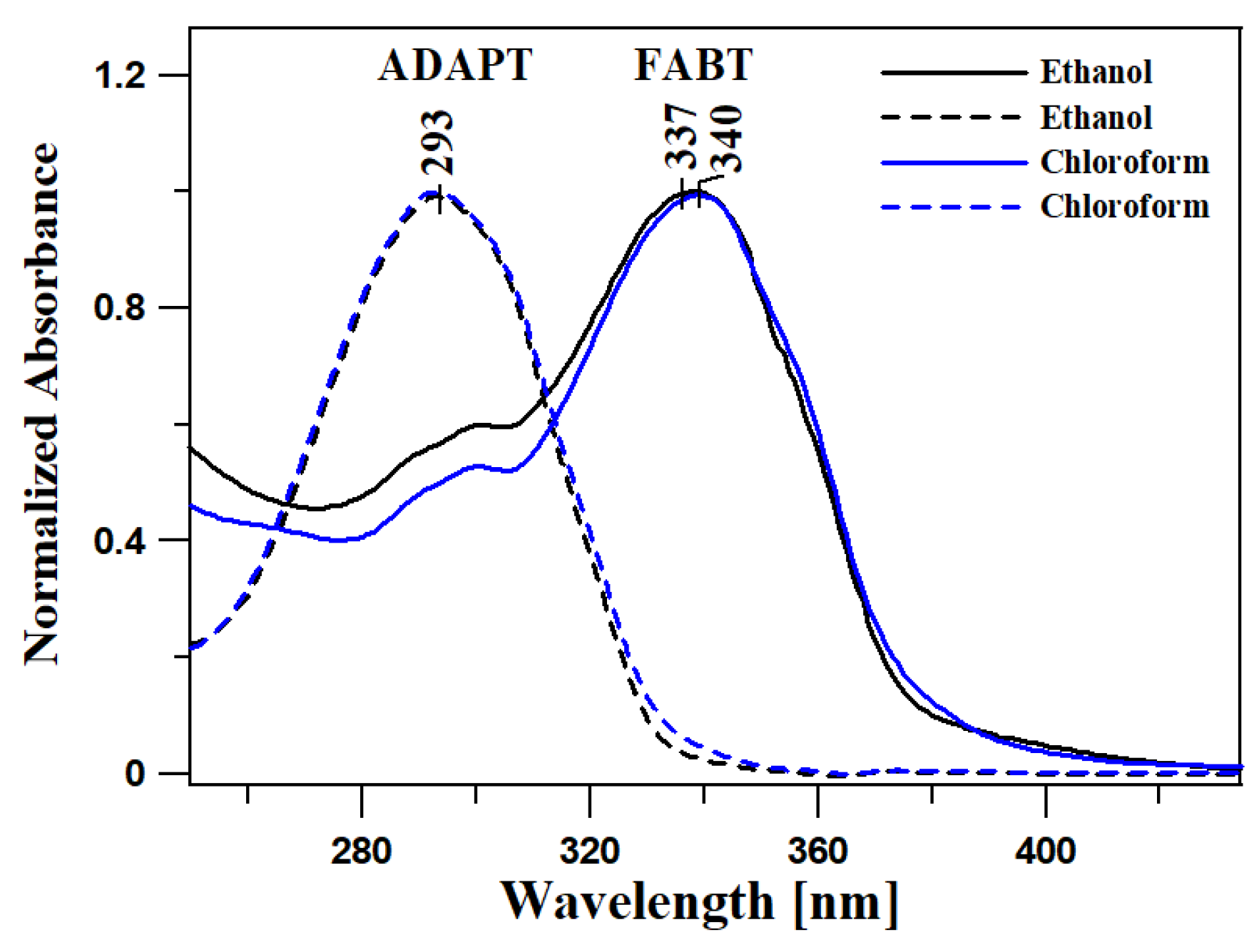
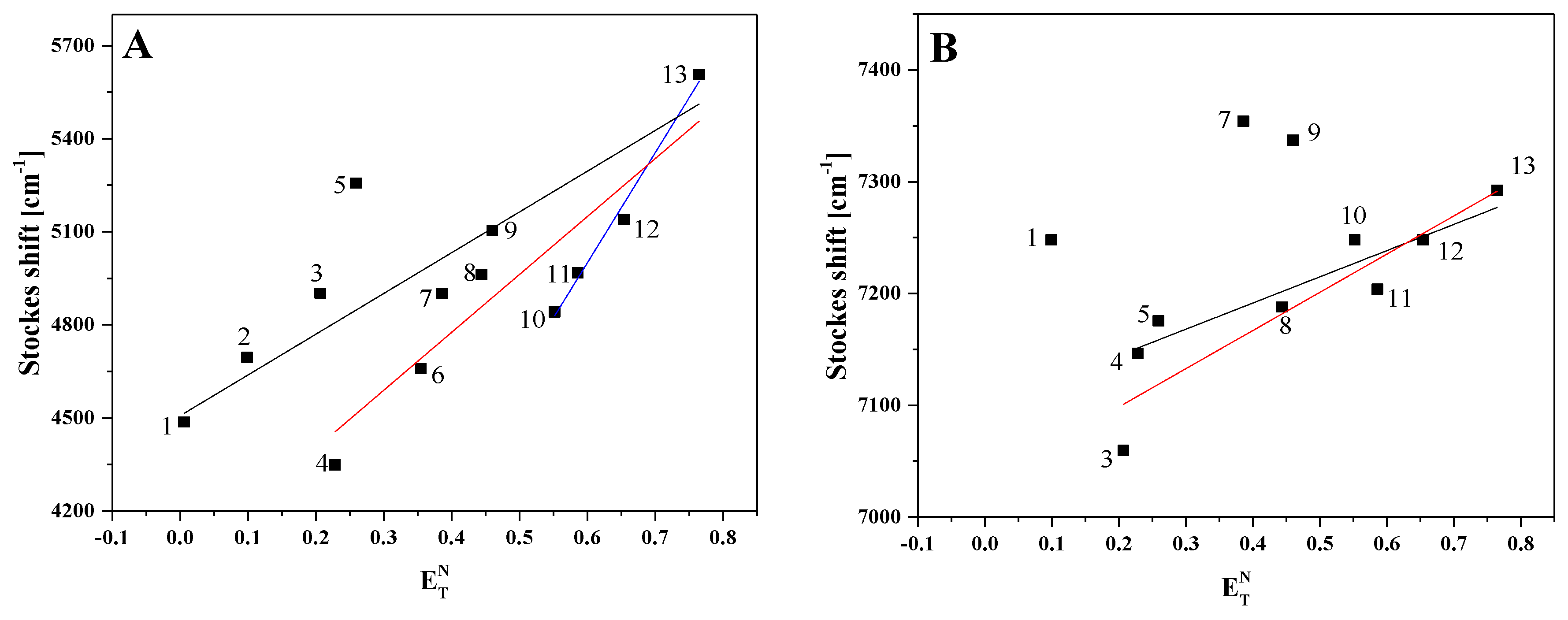
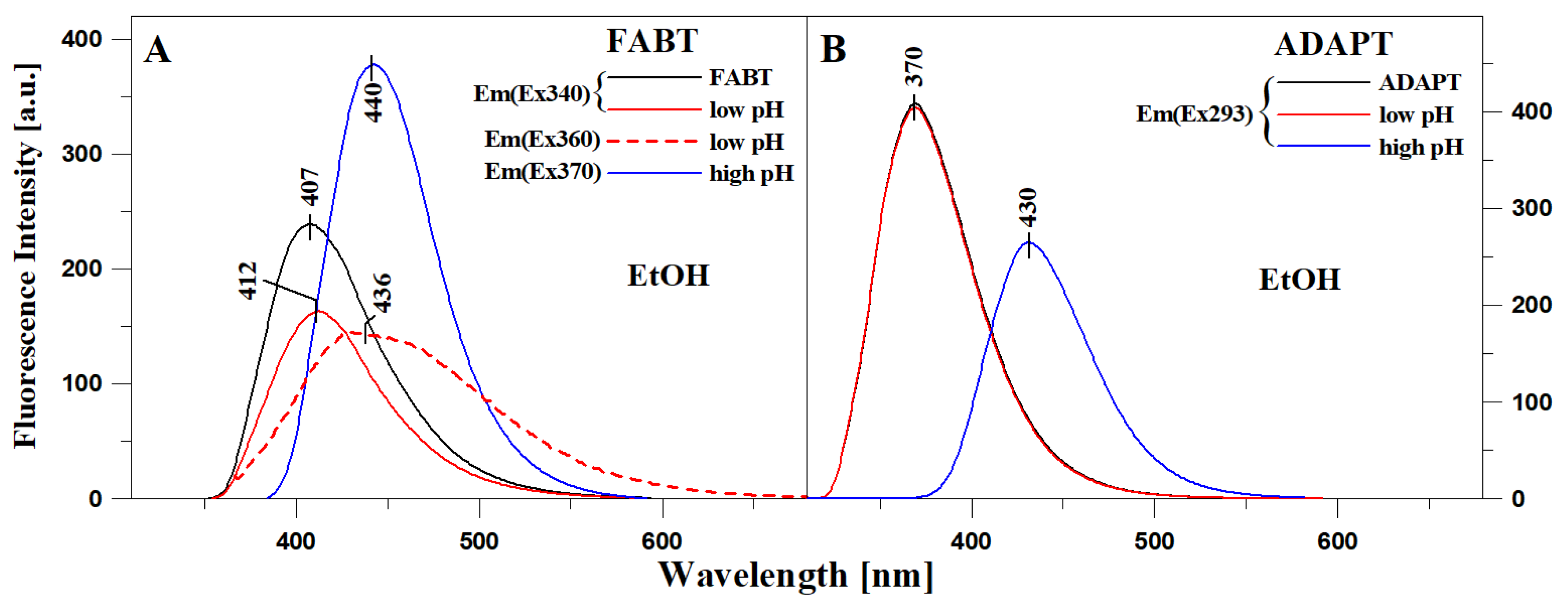
2.1. Analysis of Spectroscopic Effects
2.2. Quantum Mechanical Calculations—[TD]DFT
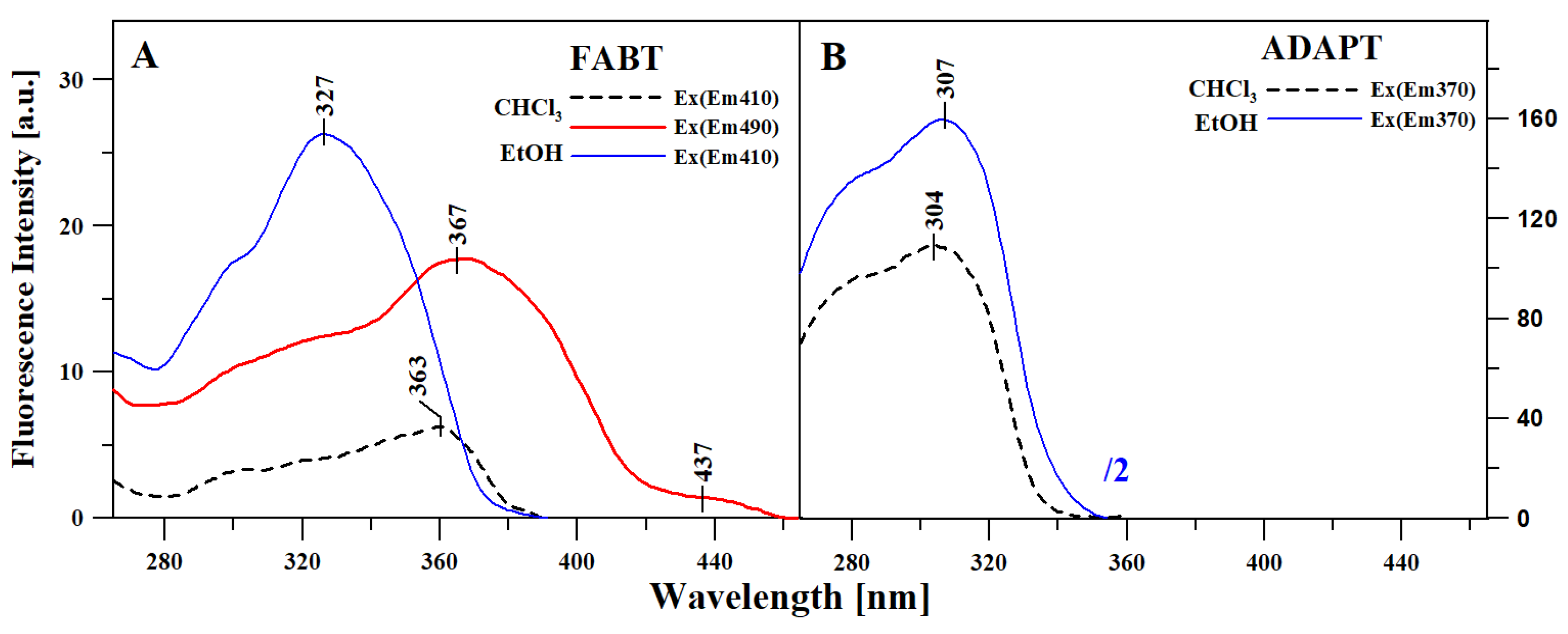
2.3. Excitation Spectra
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Electronic Absorption and Fluorescence Spectra
3.2.2. Time-Correlated Single Photon Counting (TCSPC)
3.2.3. Methodology and Calculation of Dipole Moments
3.2.4. FTIR Measurements
3.2.5. Raman Measurements
3.2.6. Fluorescence Quantum Yield
3.2.7. Computational Details
3.2.8. pH Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Czernel, G.; Matwijczuk, A.; Karcz, D.; Górecki, A.; Niemczynowicz, A.; Szcześ, A.; Gladyszewski, G.; Matwijczuk, A.; Gładyszewska, B.; Niewiadomy, A. Spectroscopic studies of dual fluorescence in 2-(4-Fluorophenylamino)-5-(2,4-dihydroxybenzeno)-1,3,4-thiadiazole: Effect of molecular aggregation in a micellar system. Molecules 2018, 23, 2861. [Google Scholar] [CrossRef] [Green Version]
- Matwijczuk, A.; Górecki, A.; Makowski, M.; Pustuła, K.; Skrzypek, A.; Waś, J.; Niewiadomy, A.; Gagos, M. Spectroscopic and theoretical studies of fluorescence effects in 2-Methylamino-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazole induced by molecular aggregation. J. Fluoresc. 2017, 28, 65–77. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Kluczyk, D.; Górecki, A.; Niewiadomy, A.; Gagos, M. Spectroscopic studies of fluorescence effects in bioactive 4-(5-heptyl-1,3,4-thiadiazol-2-yl) benzene-1,3-diol and 4-(5-Methyl-1,3,4-thiadiazol-2-yl) benzene-1,3-diol molecules induced by ph changes in aqueous solutions. J. Fluoresc. 2017, 27, 1201–1212. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Kamiński, D.; Górecki, A.; Ludwiczuk, A.; Niewiadomy, A.; Maćkowski, S.; Gagos, M. spectroscopic studies of dual fluorescence in 2-((4-fluorophenyl) amino)-5-(2,4-dihydroxybenzeno)-1,3,4-thiadiazole. J. Phys. Chem. A 2015, 119, 10791–10805. [Google Scholar] [CrossRef] [PubMed]
- Budziak, I.; Karcz, D.; Makowski, M.; Rachwał, K.; Starzak, K.; Matwijczuk, A.; Myśliwa-Kurdziel, B.; Oniszczuk, A.; Combrzyński, M.; Podleśna, A.; et al. Non-typical fluorescence effects and biological activity in selected 1,3,4-thiadiazole derivatives: Spectroscopic and theoretical studies on substituent, molecular aggregation, and pH effects. Int. J. Mol. Sci. 2019, 20, 5494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzeski, W.; Matysiak, J.; Kandefer-Szerszeń, M. Anticancer, neuroprotective activities and computational studies of 2-amino-1,3,4-thiadiazole based compound. Bioorganic Med. Chem. 2007, 15, 3201–3207. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, M.; Matysiak, J.; Szeliga, M.; Pożarowski, P.; Niewiadomy, A.; Albrecht, J.; Rzeski, W. 2-Amino-1,3,4-thiadiazole derivative (FABT) inhibits the extracellular signal-regulated kinase pathway and induces cell cycle arrest in human non-small lung carcinoma cells. Bioorganic Med. Chem. Lett. 2012, 22, 5466–5469. [Google Scholar] [CrossRef]
- Skrzypek, A.; Matysiak, J.; Niewiadomy, A.; Bajda, M.; Szymański, P. Synthesis and biological evaluation of 1,3,4-thiadiazole analogues as novel AChE and BuChE inhibitors. Eur. J. Med. Chem. 2013, 62, 311–319. [Google Scholar] [CrossRef]
- Gadad, A.K.; Mahajanshetti, C.S.; Nimbalkar, S.; Raichurkar, A. ChemInform abstract: Synthesis and antibacterial activity of some 5-guanylhydrazone/thiocyanato-6-arylimidazo [2,1-b]-1,3,4-thiadiazole-2-sulfonamide derivatives. ChemInform 2000, 31, 853–857. [Google Scholar] [CrossRef]
- Karcz, D.; Starzak, K.; Matwijczuk, A.; Ciszkowicz, E.; Lecka-Szlachta, K.; Ciupak, A.; Gładyszewska, B.; Niewiadomy, A. Otrzymywanie, spektroskopia i biologiczna aktywność kompleksów Cu (II) i Zn (II) z pochodnymi 1, 3, 4-tiadiazolu. Przem. Chem. 2018, 1, 97–103. [Google Scholar] [CrossRef]
- Karcz, D. Otrzymywanie, spektroskopia i właściwości antyoksydacyjne nowych pochodnych 1,3,4-tiadiazolu w postaci hybryd tiadiazolo-kumarynowych. Przem. Chem. 2017, 1, 155–159. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Yang, M.-Y.; Sun, Z.-H.; Tan, C.-X.; Weng, J.; Wu, H.-K.; Liu, X.-H. Synthesis and antifungal activity of 1,3,4-thiadiazole derivatives containing pyridine group. Lett. Drug Des. Discov. 2014, 11, 1107–1111. [Google Scholar] [CrossRef]
- Yan, J.; Si, W.; Hu, H.; Zhao, X.; Chen, M.; Wang, X. Design, synthesis and antimicrobial activities of novel 1,3,5-thiadiazine-2-thione derivatives containing a 1,3,4-thiadiazole group. PeerJ 2019, 7, e7581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budziak, I.; Karcz, D.; Makowski, M.; Myśliwa-Kurdziel, B.; Kasprzak, K.; Matwijczuk, A.; Chruściel, E.; Oniszczuk, A.; Adwent, L.; Matwijczuk, A. Spectroscopic and theoretical investigation into substituent and aggregation-related dual fluorescence effects in the selected 2-amino-1,3,4-thiadiazoles. J. Mol. Liq. 2019, 291, 111261. [Google Scholar] [CrossRef]
- Starzak, K.; Matwijczuk, A.; Creaven, B.S.; Matwijczuk, A.; Wybraniec, S.; Karcz, D. Fluorescence quenching-based mechanism for determination of hypochlorite by coumarin-derived sensors. Int. J. Mol. Sci. 2019, 20, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemczynowicz, A.; Czernel, G.; Matwijczuk, A.; Makowski, M.; Pustuła, K.; Karcz, D.; Matwijczuk, A.; Górecki, A.; Piotrowicz-Cieślak, A.I. Spectroscopic and theoretical studies of dual fluorescence in 2-hydroxy-n-(2-phenylethyl)benzamide induced by ESIPT process—Solvent effects. J. Lumin. 2019, 208, 125–134. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Janik, E.; Luchowski, R.; Niewiadomy, A.; Gruszecki, W.I.; Gagos, M. Spectroscopic studies of the molecular organization of 4-([1,2,4] triazolo [4,3-a] pyridin-3-yl)-6-methylbenzene-1,3-diol in selected solvents. J. Lumin. 2018, 194, 208–218. [Google Scholar] [CrossRef]
- Matwijczuk, A.; Karcz, D.; Pustuła, K.; Makowski, M.; Górecki, A.; Kluczyk, D.; Karpińska, M.M.; Niewiadomy, A.; Gagos, M. Spectroscopic and theoretical studies of fluorescence effects in bio-active: 4-(5-(methyl-1,3,4-thiadiazol-2-yl))benzene-1,3-diol and 4-(5-(methylamino-1,3,4-thiadiazol-2-yl)) benzene-1,3-diol compounds: Effect of molecular aggregation and amino group position. J. Lumin. 2018, 201, 44–56. [Google Scholar] [CrossRef]
- Hoser, A.A.; Kamiński, D.; Skrzypek, A.; Matwijczuk, A.; Niewiadomy, A.; Gagos, M.; Woźniak, K. Interplay of Inter and intramolecular interactions in crystal structures of 1,3,4-thiadiazole resorcinol derivatives. Cryst. Growth Des. 2018, 18, 3851–3862. [Google Scholar] [CrossRef]
- Kluczyk, D.; Matwijczuk, A.; Górecki, A.; Karpińska, M.M.; Szymanek, M.; Niewiadomy, A.; Gagoś, M. Molecular organization of dipalmitoylphosphatidylcholine bilayers containing bioactive compounds 4-(5-heptyl-1,3,4-thiadiazol-2-yl) benzene-1,3-diol and 4-(5-methyl-1,3,4-thiadiazol-2-yl) benzene-1,3-diols. J. Phys. Chem. B 2016, 120, 12047–12063. [Google Scholar] [CrossRef]
- Karcz, D.; Matwijczuk, A.; Boroń, B.; Creaven, B.; Fiedor, L.; Niewiadomy, A.; Gagoś, M. Isolation and spectroscopic characterization of Zn (II), Cu (II), and Pd (II) complexes of 1, 3, 4-thiadiazole-derived ligand. J. Mol. Struct. 2017, 1128, 44–50. [Google Scholar] [CrossRef]
- Gupta, R.; Mozumdar, S.; Chaudhury, N. Fluorescence spectroscopic studies to characterize the internal environment of tetraethyl-orthosilicate derived sol-gel bulk and thin films with aging. Biosens. Bioelectron. 2005, 20, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, G.; Ramakrishnan, S. Fluorescence spectroscopic studies of solvent and temperature-induced conformational transition in segmented poly [2-methoxy-5-(2 ‘-ethylhexyl) oxy-1, 4-phenylenevinylene] (MEHPPV). J. Phys. Chem. B 2004, 108, 14933–14941. [Google Scholar] [CrossRef]
- Wolfbeis, O.S.; Knierzinger, A.; Schipfer, R. pH-dependent fluorescence spectroscopy XVII: First excited singlet state dissociation constants, phtootautomerism and dual fluorescence of flavonol. J. Photochem. 1983, 21, 67–79. [Google Scholar] [CrossRef]
- Yoon, M.; Cheon, Y.; Kim, N. Absorption and fluorescence spectroscopic studies on dimerization of chloroaluminum (iii) phthalocyanine tetrasulfonate in aqueous alcoholic solutions. Photochem. Photobiol. 1993, 58, 31–36. [Google Scholar] [CrossRef]
- Mishina, S.; Takayanagi, M.; Nakata, M.; Otsuki, J.; Araki, K. Dual fluorescence of 4-dimethylaminopyridine and its derivatives. J. Photochem. Photobiol. A Chem. 2001, 141, 153–158. [Google Scholar] [CrossRef]
- Salassa, L.; Garino, C.; Albertino, A.; Volpi, G.; Nervi, C.; Gobetto, R.; Hardcastle, K.I. Computational and spectroscopic studies of new rhenium (i) complexes containing pyridylimidazo [1,5-a] pyridine ligands: Charge transfer and dual emission by fine-tuning of excited states. Organometallics 2008, 27, 1427–1435. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Von Der Haar, T.; Hebecker, A.; Leinhos, U.; Kühnle, W. Intramolecular charge transfer in aminobenzonitriles: Requirements for dual fluorescence. Pure Appl. Chem. 1993, 65, 1745–1750. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gao, Y.; Liu, H.; Bai, Q.; Li, J.; Liu, L.; Wu, C.; Yang, B.; Wang, K.; Zou, B.; et al. Dual fluorescence polymorphs: Wide-range emission from blue to red regulated by TICT and their dynamic electron state behavior under external pressure. Dye. Pigment. 2017, 145, 294–300. [Google Scholar] [CrossRef]
- Gao, S.; Wei, G.; Zhang, S.; Zheng, B.; Xu, J.; Chen, G.; Li, M.; Song, S.; Fu, W.; Xiao, Z.; et al. Albumin tailoring fluorescence and photothermal conversion effect of near-infrared-II fluorophore with aggregation-induced emission characteristics. Nat. Commun. 2019, 10, 2206. [Google Scholar] [CrossRef]
- Dutta, A.K.; Kamada, K.; Ohta, K. Spectroscopic studies of nile red in organic solvents and polymers. J. Photochem. Photobiol. A Chem. 1996, 93, 57–64. [Google Scholar] [CrossRef]
- Yang, Y.; Zhai, H.; Liu, Y.; Jia, X.; He, Y.; Ma, Q.; Jiang, K.; Liu, Y. Excited state intramolecular proton transfer induced fluorescent change and decay pathway of salicylideneaniline. J. Lumin. 2019, 216, 116736. [Google Scholar] [CrossRef]
- He, L.; Dong, B.; Liu, Y.; Lin, W. Fluorescent chemosensors manipulated by dual/triple interplaying sensing mechanisms. Chem. Soc. Rev. 2016, 45, 6449–6461. [Google Scholar] [CrossRef]
- Niemczynowicz, A.; Budziak, I.; Kulesza, S.; Górecki, A.; Makowski, M.; Karcz, D.; Starzak, K.; Gładyszewska, B.; Podleśny, J.; Piotrowicz-Cieślak, A.I.; et al. Spectroscopic and theoretical studies of fluorescence effects induced by the ESIPT process in a new derivative 2-Hydroxy-N-(2-phenylethyl) benzamide—Study on the effects of pH and medium polarity changes. PLoS ONE 2020, 15, e0229149. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Pahari, B.; Sengupta, P.K. Ground and excited state proton transfer and antioxidant activity of 7-hydroxyflavone in model membranes: Absorption and fluorescence spectroscopic studies. Biophys. Chem. 2009, 139, 29–36. [Google Scholar] [CrossRef]
- Wu, C.; Xu, H.; Li, Y.; Xie, R.; Li, P.; Pang, X.; Zhou, Z.; Gu, B.; Li, H.; Zhang, Y. An ESIPT-based fluorescent probe for the detection of phosgene in the solution and gas phases. Talanta 2019, 200, 78–83. [Google Scholar] [CrossRef]
- Karsili, T.N.V.; Marchetti, B.; Ashfold, M.N.R. Mechanistic insights into excited state intramolecular proton transfer in isolated and metal chelated supramolecular chemosensors. Dalton Trans. 2016, 45, 18921–18930. [Google Scholar] [CrossRef] [Green Version]
- Benelhadj, K.; Muzuzu, W.; Massue, J.; Retailleau, P.; Charaf-Eddin, A.; Laurent, A.D.; Jacquemin, D.; Ulrich, G.; Ziessel, R. White emitters by tuning the excited-state intramolecular proton-transfer fluorescence emission in 2-(2’-hydroxybenzofuran) benzoxazole dyes. Chem. A Eur. J. 2014, 20, 12843–12857. [Google Scholar] [CrossRef]
- Heyer, E.; Benelhadj, K.; Budzák, S.; Jacquemin, D.; Massue, J.; Ulrich, G. On the fine-tuning of the excited-state intramolecular proton transfer (esipt) process in 2-(2′-hydroxybenzofuran) benzazole (hbbx) dyes. Chem. A Eur. J. 2017, 23, 7324–7336. [Google Scholar] [CrossRef]
- Li, B.; Zhou, L.; Cheng, H.; Huang, Q.; Lan, J.; Zhou, L.; You, J. Dual-emissive 2-(2’-hydroxyphenyl) oxazoles for high performance organic electroluminescent devices: Discovery of a new equilibrium of excited state intramolecular proton transfer with a reverse intersystem crossing process. Chem. Sci. 2018, 9, 1213–1220. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Fukazawa, A.; Nagura, K.; Saito, S.; Yokogawa, D.; Irle, S.; Yamaguchi, S.; Kitoh-Nishioka, H. A strap strategy for construction of an excited-state intramolecular proton transfer (esipt) system with dual fluorescence. Angew. Chem. 2014, 126, 8370–8374. [Google Scholar] [CrossRef]
- Suzuki, N.; Suda, K.; Yokogawa, D.; Kitoh-Nishioka, H.; Irle, S.; Ando, A.; Abegão, L.; Kamada, K.; Fukazawa, A.; Yamaguschi, S. Near infrared two-photon-excited and emissive dyes based on a strapped excited-state intramolecular proton-transfer (ESIPT) scaffold. Chem. Sci. 2018, 9, 2666–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; He, Z.; Kausar, F.; Chen, G.; Zhang, Y.; Yuan, W.Z. Aggregation-induced dual emission and unusual luminescence beyond excimer emission of poly (ethylene terephthalate). Macromolecules 2018, 51, 9035–9042. [Google Scholar] [CrossRef]
- Brancato, G.; Signore, G.; Neyroz, P.; Polli, D.; Cerullo, G.; Abbandonato, G.; Nucara, L.; Barone, V.; Beltram, F.; Bizzarri, R. Dual fluorescence through kasha’s rule breaking: An unconventional photomechanism for intracellular probe design. J. Phys. Chem. B 2015, 119, 6144–6154. [Google Scholar] [CrossRef]
- Shafikov, M.Z.; Brandl, F.; Dick, B.; Czerwieniec, R. Can coumarins break Kasha’s rule? J. Phys. Chem. Lett. 2019, 10, 6468–6471. [Google Scholar] [CrossRef]
- Liu, J.; Koslowski, A.; Thiel, W. Analytic gradient and derivative couplings for the spin-flip extended configuration interaction singles method: Theory, implementation, and application to proton transfer. J. Chem. Phys. 2018, 148, 244108. [Google Scholar] [CrossRef] [Green Version]
- Jacquemin, D.; Khelladi, M.; De Nicola, A.; Ulrich, G. Turning ESIPT-based triazine fluorophores into dual emitters: From theory to experiment. Dye. Pigment. 2019, 163, 475–482. [Google Scholar] [CrossRef]
- Sobolewski, A.L.L.; Domcke, W. Ab initio study of excited-state intramolecular proton dislocation in salicylic acid. Chem. Phys. 1998, 232, 257–265. [Google Scholar] [CrossRef]
- Böhnke, H.; Bahrenburg, J.; Ma, X.; Röttger, K.; Näther, C.; Rode, M.F.; Sobolewski, A.L.; Temps, F. Ultrafast dynamics of the ESIPT photoswitch N-(3-pyridinyl)-2-pyridinecarboxamide. Phys. Chem. Chem. Phys. 2018, 20, 2646–2655. [Google Scholar] [CrossRef]
- Budzák, Š.; Jacquemin, D. Mechanism of fluorescence switching in one ESIPT-based Al3+ probe. J. Phys. Chem. B 2016, 120, 6730–6738. [Google Scholar] [CrossRef]
- Jankowska, J.; Barbatti, M.; Sadlej, J.; Sobolewski, A.L. Tailoring the Schiff base photoswitching—A non-adiabatic molecular dynamics study of substituent effect on excited state proton transfer. Phys. Chem. Chem. Phys. 2017, 19, 5318–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matwijczuk, A.; Kluczyk, D.; Górecki, A.; Niewiadomy, A.; Gagos, M. solvent effects on molecular aggregation in 4-(5-heptyl-1,3,4-thiadiazol-2-yl) benzene-1,3-diol and 4-(5-methyl-1,3,4-thiadiazol-2-yl) benzene-1,3-diol. J. Phys. Chem. B 2016, 120, 7958–7969. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J.; Opolski, A. Synthesis and antiproliferative activity of N-substituted 2-amino-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazoles. Bioorganic Med. Chem. 2006, 14, 4483–4489. [Google Scholar] [CrossRef]
- Pasternack, R.; Collings, P. Resonance light scattering: A new technique for studying chromophore aggregation. Science 1995, 269, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Kawski, A. On the estimation of excited-state dipole moments from solvatochromic shifts of absorption and fluorescence spectra. Z. Für Nat. A 2002, 57, 255–262. [Google Scholar] [CrossRef]
- Suppan, P. Excited-state dipole moments from absorption/fluorescence solvatochromic ratios. Chem. Phys. Lett. 1983, 94, 272–275. [Google Scholar] [CrossRef]
- Thiaré, D.D.; Khonté, A.; Diop, A.; Cissé, L.; Coly, A.; Tine, A.; Delattre, F. Determination of ground and excited state dipole moments of amino-benzimidazole by solvatochromic shift methods and theoretical calculations. J. Mol. Liq. 2015, 211, 640–646. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Aslan, C.; Berber, H.; Sıdır, I. The electronic structure, solvatochromism, and electric dipole moments of new Schiff base derivatives using absorbance and fluorescence spectra. Struct. Chem. 2018, 30, 835–851. [Google Scholar] [CrossRef]
- Sıdır, I. Solvatochromism and intramolecular hydrogen-bonding assisted dipole moment of phenyl 1-hydroxy-2-naphthoate in the ground and excited states. J. Mol. Liq. 2016, 221, 972–985. [Google Scholar] [CrossRef]
- Martins, S.; Candeias, A.; Caldeira, A.; Pereira, A.M.D.R.L. 7-(diethylamino)-4-methyl-3-vinylcoumarin as a new important intermediate to the synthesis of photosensitizers for DSSCs and fluorescent labels for biomolecules. Dye. Pigment. 2020, 174, 108026. [Google Scholar] [CrossRef]
- Cigáň, M.; Donovalová, J.; Szöcs, V.; Gašpar, J.; Jakusová, K.; Gáplovský, A. 7-(Dimethylamino) coumarin-3-carbaldehyde and its phenylsemicarbazone: TICT excited state modulation, fluorescent h-aggregates, and preferential solvation. J. Phys. Chem. A 2013, 117, 4870–4883. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Jackson, W.R.; Choi, C.Y.; Bergmark, W.R. Solvent effects on emission yield and lifetime for coumarin laser dyes. Requirements for a rotatory decay mechanism. J. Phys. Chem. 1985, 89, 294–300. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision A. 01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. II. The effect of the Perdew–Wang generalized-gradient correlation correction. J. Chem. Phys. 1992, 97, 9173–9177. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. Theochem. 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | a0[Å] | m1 | Δµ [D] | r2 | n |
|---|---|---|---|---|---|
| FABT | 4.281 | 1866.541 | 2.098 | 0.79 | 9 |
| 1313.541 | 1.760 | 0.92 | 7 | ||
| 3548.132 | 2.893 | 0.98 | 4 | ||
| ADAPT | 4.829 | 234.435 | 0.891 | 0.86 | 7 |
| 341.867 | 1.157 | 0.87 | 7 |
| Buffer | Time [ns] |
|---|---|
| EtOH neutral | 3.346 ± 0.004 |
| EtOH basic | 2.822 ± 0.004 |
| EtOH acidic | 1.26 ± 0.01 |
| Chloroform | 3.44 ± 0.02 |
| Buffer | Constituent | Lifetime [ns] | Fraction [%] |
|---|---|---|---|
| EtOH neutral | τ1 | 1.59 ± 0.054 | (13.9 ± 1.2) |
| τ2 | 0.794 ± 0.003 | (86.1 ± 1.2) | |
| EtOH basic | τ1 | 1.79 ± 0.01 | (31.1 ± 0.4) |
| τ2 | 0.22 ± 0.01 | (68.9 ± 0.4) | |
| EtOH acidic | τ1 | 5.0 ± 0.4 | (1.8 ± 0.1) |
| τ2 | 0.73 ± 0.1 | (98.2 ± 0.1) | |
| Chloroform | τ1 | 27.3 ± 0.2 | (34.7 ± 0.1) |
| τ2 | 1.67 ± 0.02 | (25.4 ± 0.4) | |
| τ3 | 0.22 ± 0.01 | (39.9 ± 0.3) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czernel, G.; Budziak, I.; Oniszczuk, A.; Karcz, D.; Pustuła, K.; Górecki, A.; Matwijczuk, A.; Gładyszewska, B.; Gagoś, M.; Niewiadomy, A.; et al. ESIPT-Related Origin of Dual Fluorescence in the Selected Model 1,3,4-Thiadiazole Derivatives. Molecules 2020, 25, 4168. https://doi.org/10.3390/molecules25184168
Czernel G, Budziak I, Oniszczuk A, Karcz D, Pustuła K, Górecki A, Matwijczuk A, Gładyszewska B, Gagoś M, Niewiadomy A, et al. ESIPT-Related Origin of Dual Fluorescence in the Selected Model 1,3,4-Thiadiazole Derivatives. Molecules. 2020; 25(18):4168. https://doi.org/10.3390/molecules25184168
Chicago/Turabian StyleCzernel, Grzegorz, Iwona Budziak, Anna Oniszczuk, Dariusz Karcz, Katarzyna Pustuła, Andrzej Górecki, Alicja Matwijczuk, Bożena Gładyszewska, Mariusz Gagoś, Andrzej Niewiadomy, and et al. 2020. "ESIPT-Related Origin of Dual Fluorescence in the Selected Model 1,3,4-Thiadiazole Derivatives" Molecules 25, no. 18: 4168. https://doi.org/10.3390/molecules25184168