Computational Design of Nitrile Hydratase from Pseudonocardia thermophila JCM3095 for Improved Thermostability


Abstract
:1. Introduction
2. Results and Discussion
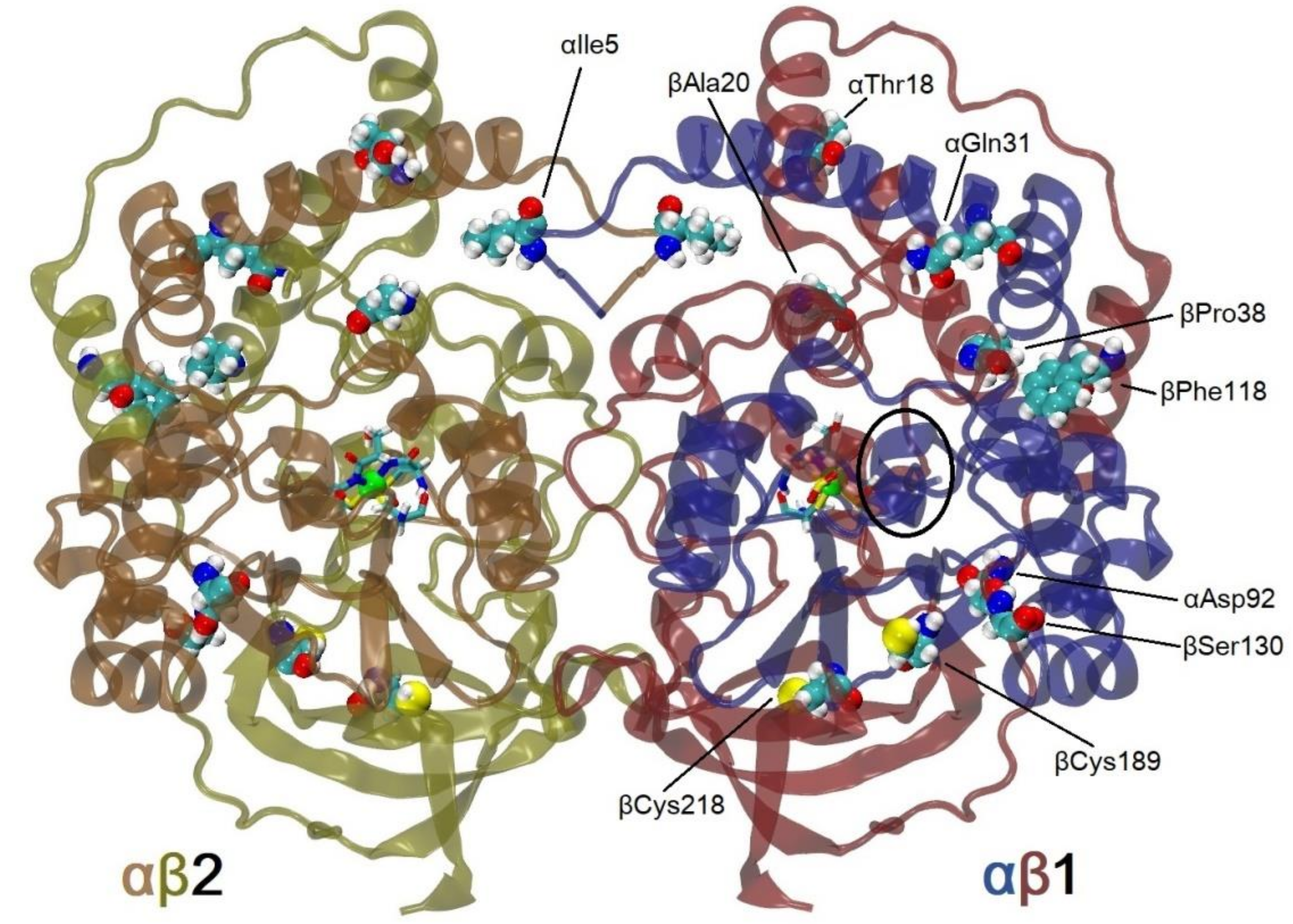
2.1. Rational Design of a Potential Thermostable PtNHase Mutant
2.2. Construction of a Thermostable PtNHase Mutant
2.3. M10 Mutant Shows Higher Thermostability
2.4. In Silico Docking Shows better Affinity of the Substrate with the M10 Mutant
2.5. MD Simulation Justified Higher Thermostability for M10 Mutant
2.6. Influence of a Particular Mutation on the Thermostability of PtNHase
2.6.1. αI5P
2.6.2. αT18Y
2.6.3. αQ31L
2.6.4. αD92H
2.6.5. βA20P
2.6.6. βP38L
2.6.7. βF118W
2.6.8. βS130Y
2.6.9. βC189N
2.6.10. βC218V
2.6.11. M10 PtNHase in the Context of Mechanisms of Increasing Thermostability
3. Materials and Methods
3.1. In Silico Design of NHase
3.2. Protein Expression and Purification
3.3. Enzymatic Assay
3.4. Measurement of the Kinetic Parameters of WT NHase and its M10 Mutant
3.5. Circular Dichroism (CD) and Thermal Denaturation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NHase | Nitrile hydratase |
| PtNHase | NHase from Pseudonocardia thermophila JCM3095 |
| M10 | αI5P/αT18Y/αQ31L/αD92H/βA20P/βP38L/βF118W/βS130Y/βC189N/βC218V mutant of PtNHase |
| WT | Wild-type |
| MD | Molecular dynamics |
| CD | Circular dichroism |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| PC | Principal component |
| CNA | Constrained network analysis |
| Rg | Radius of gyration |
| CEA | Cysteine-sulfenic acid |
| CSD | Cysteine-sulfinic acid |
| H-bond | Hydrogen bond |
| KPB | Kalium phosphate buffer |
References
- Chen, J.; Zheng, R.-C.; Zheng, Y.-G.; Shen, Y.-C. Microbial Transformation of Nitriles to High-Value Acids or Amides. Biotechnol. China I 2009, 113, 33–77. [Google Scholar] [CrossRef]
- Sharma, M.; Akhter, Y.; Chatterjee, S. A review on remediation of cyanide containing industrial wastes using biological systems with special reference to enzymatic degradation. World J. Microbiol. Biotechnol. 2019, 35, 70. [Google Scholar] [CrossRef]
- Prasad, S.; Bhalla, T.C. Nitrile hydratases (NHases): At the interface of academia and industry. Biotechnol. Adv. 2010, 28, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Cui, W.; Xia, Y.; Pepłowski, Ł.; Kobayashi, M.; Zhou, Z. Modulation of Nitrile Hydratase Regioselectivity towards Dinitriles by Tailoring the Substrate Binding Pocket Residues. ChemCatChem 2017, 10, 449–458. [Google Scholar] [CrossRef]
- Cheng, Z.; Peplowski, L.; Cui, W.; Xia, Y.; Liu, Z.; Zhang, J.; Kobayashi, M.; Zhou, Z. Identification of key residues modulating the stereoselectivity of nitrile hydratase toward rac-mandelonitrile by semi-rational engineering. Biotechnol. Bioeng. 2017, 115, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.-S.; Shi, J.-S.; Lu, Z.-M.; Li, H.; Zhou, Z.-M.; Xu, Z.-H. Nitrile-converting enzymes as a tool to improve biocatalysis in organic synthesis: Recent insights and promises. Crit. Rev. Biotechnol. 2015, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Xia, Y.; Zhou, Z. Recent Advances and Promises in Nitrile Hydratase: From Mechanism to Industrial Applications. Front. Bioeng. Biotechnol. 2020, 8, 352. [Google Scholar] [CrossRef]
- Chen, J.; Yu, H.; Liu, C.; Liu, J.; Shen, Z. Improving stability of nitrile hydratase by bridging the salt-bridges in specific thermal-sensitive regions. J. Biotechnol. 2013, 164, 354–362. [Google Scholar] [CrossRef]
- Pei, X.; Wang, J.; Wu, Y.; Zhen, X.; Tang, M.; Wang, Q.; Wang, A. Evidence for the Participation of an Extra Alpha-Helix at Beta-Subunit Surface in the Thermal Stability of Co-Type Nitrile Hydratase. Appl. Microbiol. Biotechnol. 2018, 102, 7891–7900. [Google Scholar] [CrossRef]
- Cui, Y.; Cui, W.; Liu, Z.; Zhou, L.; Kobayashi, M.; Zhou, Z. Improvement of stability of nitrile hydratase via protein fragment swapping. Biochem. Biophys. Res. Commun. 2014, 450, 401–408. [Google Scholar] [CrossRef]
- Xia, Y.; Cui, W.; Liu, Z.; Zhou, L.; Cui, Y.; Kobayashi, M.; Zhou, Z. Construction of a subunit-fusion nitrile hydratase and discovery of an innovative metal ion transfer pattern. Sci. Rep. 2016, 6, 19183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.A.; Graham, D.; Rainey, F.A.; Cowan, D. A novel thermostable nitrile hydratase. Extremophiles 1998, 2, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pandey, D.; Dhariwal, S.; Sood, P.; Chand, D. Bioconversion of acrylonitrile using nitrile hydratase activity of Bacillus sp. APB-6. 3 Biotech 2018, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pandey, D.; Devi, N.; Chand, D. Bench scale production of butyramide using free and immobilized cells of Bacillus sp. APB-6. Bioprocess. Biosyst. Eng. 2018, 41, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Cramp, R.A.; Cowan, D. Molecular characterisation of a novel thermophilic nitrile hydratase. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1431, 249–260. [Google Scholar] [CrossRef]
- Musil, M.; Stourac, J.; Bendl, J.; Brezovsky, J.; Prokop, Z.; Zendulka, J.; Martinek, T.; Bednar, D.; Damborsky, J. FireProt: Web server for automated design of thermostable proteins. Nucleic Acids Res. 2017, 45, W393–W399. [Google Scholar] [CrossRef]
- Miyanaga, A.; Fushinobu, S.; Ito, K.; Wakagi, T. Crystal Structure of Cobalt-Containing Nitrile Hydratase. Biochem. Biophys. Res. Commun. 2001, 288, 1169–1174. [Google Scholar] [CrossRef]
- Xia, Y.; Cui, W.; Cheng, Z.; Peplowski, L.; Liu, Z.; Kobayashi, M.; Zhou, Z. Improving the Thermostability and Catalytic Efficiency of the Subunit-Fused Nitrile Hydratase by Semi-Rational Engineering. ChemCatChem 2018, 10, 1370–1375. [Google Scholar] [CrossRef]
- Hopmann, K.H. Full Reaction Mechanism of Nitrile Hydratase: A Cyclic Intermediate and an Unexpected Disulfide Switch. Inorg. Chem. 2014, 53, 2760–2762. [Google Scholar] [CrossRef]
- Pepłowski, Ł.; Kubiak, K.; Nowak, W. Insights into catalytic activity of industrial enzyme Co-nitrile hydratase. Docking studies of nitriles and amides. J. Mol. Model. 2007, 13, 725–730. [Google Scholar] [CrossRef]
- Liu, J.; Yu, H.; Shen, Z. Insights into thermal stability of thermophilic nitrile hydratases by molecular dynamics simulation. J. Mol. Graph. Model. 2008, 27, 529–535. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Murthy, M. Protein thermal stability: Insights from atomic displacement parameters (B values). Protein Eng. 2000, 13, 9–13. [Google Scholar] [CrossRef]
- Du, J.; Dong, J.; Du, S.; Zhang, K.; Yu, J.; Hu, S.; Yin, H. Understanding Thermostability Factors of Barley Limit Dextrinase by Molecular Dynamics Simulations. Front. Mol. Biosci. 2020, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, S.; Xu, Y.; Yu, X. Enhancing the thermostability of Rhizopus chinensis lipase by rational design and MD simulations. Int. J. Biol. Macromol. 2020, 160, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, A.; Molakarimi, M.; Taghdir, M.; Sajedi, R.H.; Hasannia, S. Exploring single-domain antibody thermostability by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2018, 37, 3686–3696. [Google Scholar] [CrossRef] [PubMed]
- Takarada, H.; Kawano, Y.; Hashimoto, K.; Nakayama, H.; Ueda, S.; Yohda, M.; Kamiya, N.; Dohmae, N.; Maeda, M.; Odaka, M. Mutational study on alphaGln90 of Fe-type nitrile hydratase from Rhodococcus sp. N771. Biosci. Biotechnol. Biochem. 2006, 70, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Krüger, D.M.; Rathi, P.C.; Pfleger, C.; Gohlke, H. CNA web server: Rigidity theory-based thermal unfolding simulations of proteins for linking structure, (thermo-)stability, and function. Nucleic Acids Res. 2013, 41, W340–W348. [Google Scholar] [CrossRef]
- Pfleger, C.; Rathi, P.C.; Klein, D.L.; Radestock, S.; Gohlke, H. Constraint Network Analysis (CNA): A Python Software Package for Efficiently Linking Biomacromolecular Structure, Flexibility, (Thermo-)Stability, and Function. J. Chem. Inf. Model. 2013, 53, 1007–1015. [Google Scholar] [CrossRef]
- Nagashima, S.; Nakasako, M.; Dohmae, N.; Tsujimura, M.; Wei, N.; Odaka, M.; Yohda, M.; Kamiya, N.; Endo, I. Novel non-heme iron center of nitrile hydratase with a claw setting of oxygen atoms. Nat. Genet. 1998, 5, 347–351. [Google Scholar] [CrossRef]
- Piersma, S.R.; Nojiri, M.; Tsujimura, M.; Noguchi, T.; Odaka, M.; Yohda, M.; Inoue, Y.; Endo, I. Arginine 56 Mutation in the Beta Subunit of Nitrile Hydratase: Importance of Hydrogen Bonding to the Non-Heme Iron Center. J. Inorg. Biochem. 2000, 80, 283–288. [Google Scholar] [CrossRef]
- Martinez, S.; Wu, R.; Krzywda, K.; Opalka, V.; Chan, H.; Liu, D.; Holz, R.C. Analyzing the catalytic role of active site residues in the Fe-type nitrile hydratase from Comamonas testosteroni Ni1. JBIC J. Biol. Inorg. Chem. 2015, 20, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepłowski, Ł.; Kubiak, K.; Nowak, W. Mechanical aspects of nitrile hydratase enzymatic activity. Steered molecular dynamics simulations of Pseudonocardia thermophila JCM 3095. Chem. Phys. Lett. 2008, 467, 144–149. [Google Scholar] [CrossRef]
- Panja, A.S.; Bandopadhyay, B.; Maiti, S. Protein Thermostability is Owing to Their Preferences to Non-Polar Smaller Volume Amino Acids, Variations in Residual Physico-Chemical Properties and More Salt-Bridges. PLoS ONE 2015, 10, e0131495. [Google Scholar] [CrossRef]
- Haney, P.J.; Badger, J.H.; Buldak, G.L.; Reich, C.I.; Woese, C.R.; Olsen, G.J. Thermal adaptation analyzed by comparison of protein sequences from mesophilic and extremely thermophilic Methanococcus species. Proc. Natl. Acad. Sci. USA 1999, 96, 3578–3583. [Google Scholar] [CrossRef] [Green Version]
- Saelensminde, G.; Halskau, Ø., Jr.; Jonassen, I. Amino Acid Contacts in Proteins Adapted to Different Temperatures: Hydrophobic Interactions and Surface Charges Play a Key Role. Extremophiles 2009, 13, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Tsai, C.-J.; Nussinov, R. Factors enhancing protein thermostability. Protein Eng. Des. Sel. 2000, 13, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Bednar, D.; Beerens, K.; Sebestova, E.; Bendl, J.; Khare, S.; Chaloupkova, R.; Prokop, Z.; Brezovsky, J.; Baker, D.; Damborsky, J. FireProt: Energy- and Evolution-Based Computational Design of Thermostable Multiple-Point Mutants. PLoS Comput. Biol. 2015, 11, e1004556. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- MacKerell, J.A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Skjærven, L.; Yao, X.-Q.; Scarabelli, G.; Grant, B.J. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinform. 2014, 15, 399. [Google Scholar] [CrossRef] [Green Version]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | kcat (s−1) | Km (mM) | kcat/Km (s−1·mM−1) | Specific Activity (U·mg−1) |
|---|---|---|---|---|
| WT | 107.9 ± 4.2 | 0.20 ± 0.03 | 539.5 | 81.1 ± 2.8 |
| M10 | 165.1 ± 6.9 | 0.12 ± 0.02 | 1375.8 | 168.8 ± 5.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Z.; Lan, Y.; Guo, J.; Ma, D.; Jiang, S.; Lai, Q.; Zhou, Z.; Peplowski, L. Computational Design of Nitrile Hydratase from Pseudonocardia thermophila JCM3095 for Improved Thermostability. Molecules 2020, 25, 4806. https://doi.org/10.3390/molecules25204806
Cheng Z, Lan Y, Guo J, Ma D, Jiang S, Lai Q, Zhou Z, Peplowski L. Computational Design of Nitrile Hydratase from Pseudonocardia thermophila JCM3095 for Improved Thermostability. Molecules. 2020; 25(20):4806. https://doi.org/10.3390/molecules25204806
Chicago/Turabian StyleCheng, Zhongyi, Yao Lan, Junling Guo, Dong Ma, Shijin Jiang, Qianpeng Lai, Zhemin Zhou, and Lukasz Peplowski. 2020. "Computational Design of Nitrile Hydratase from Pseudonocardia thermophila JCM3095 for Improved Thermostability" Molecules 25, no. 20: 4806. https://doi.org/10.3390/molecules25204806