Mimicking the Nucleosomal Context in Peptide-Based Binders of a H3K36me Reader Increases Binding Affinity While Altering the Binding Mode


Abstract
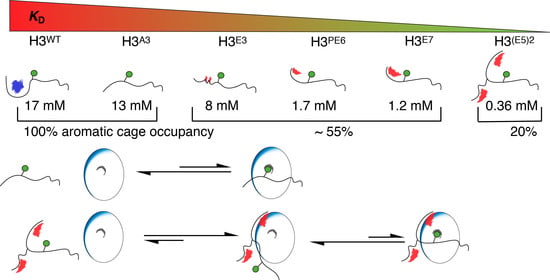
{kind=link}
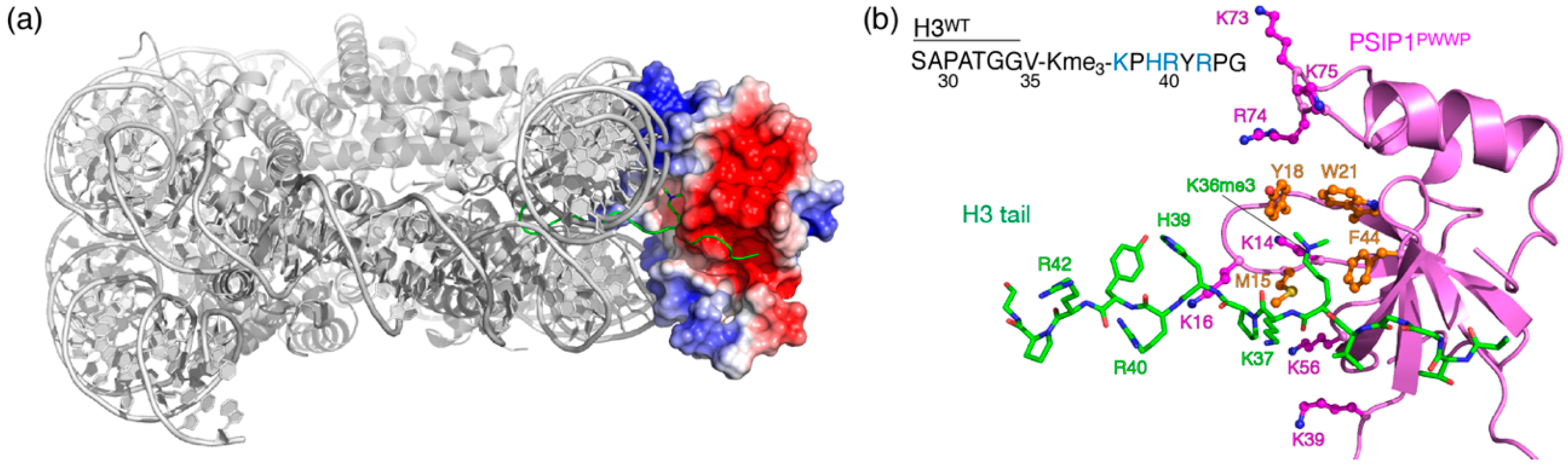{kind=link}
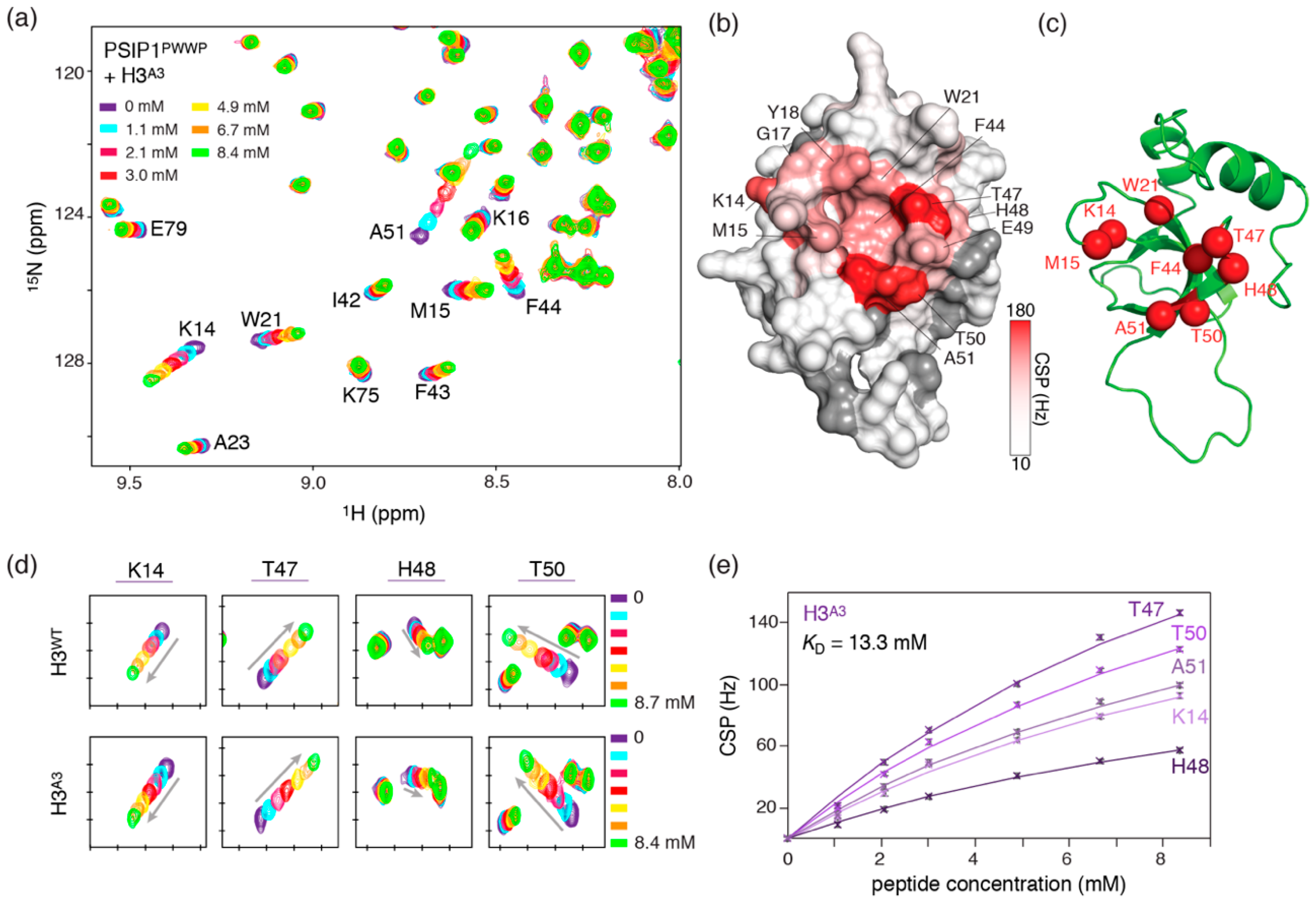{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Electrostatic Repulsion Decreases the Affinity of H3K36me3 Peptides for PSIP1PWWP
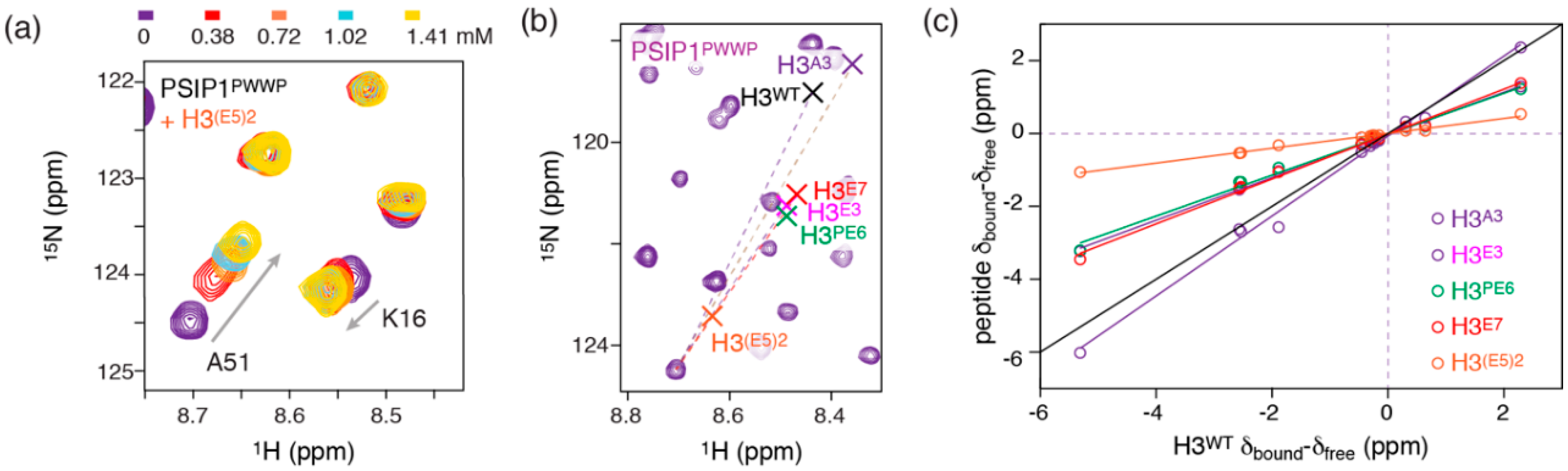
2.2. Introduction of Negative Charges Enhances Peptide Binding to PSIP1PWWP
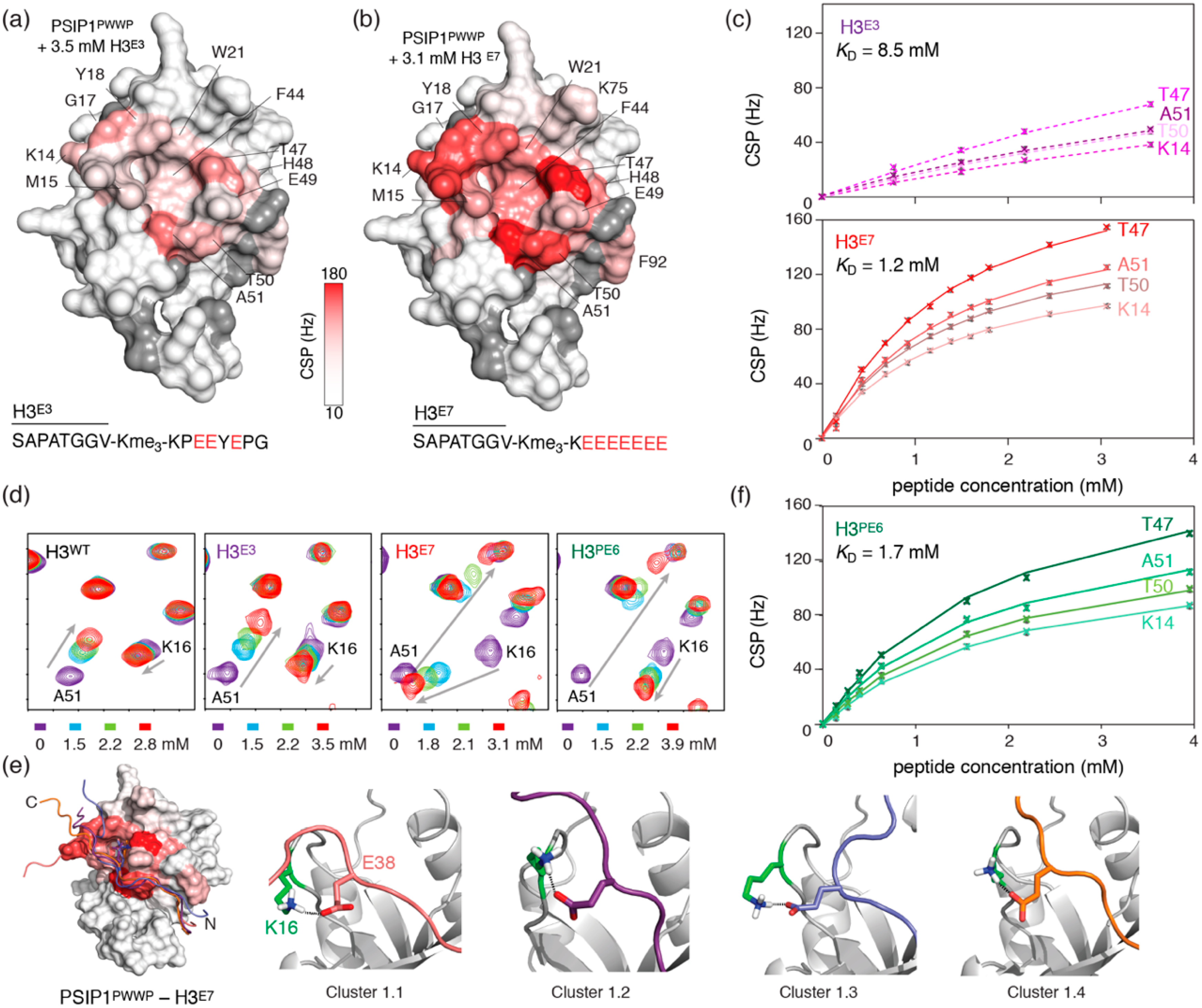
2.3. Dynamic Attractive Electrostatics Drive the Increased Affinity for Glu-Rich H3 Tail Peptides
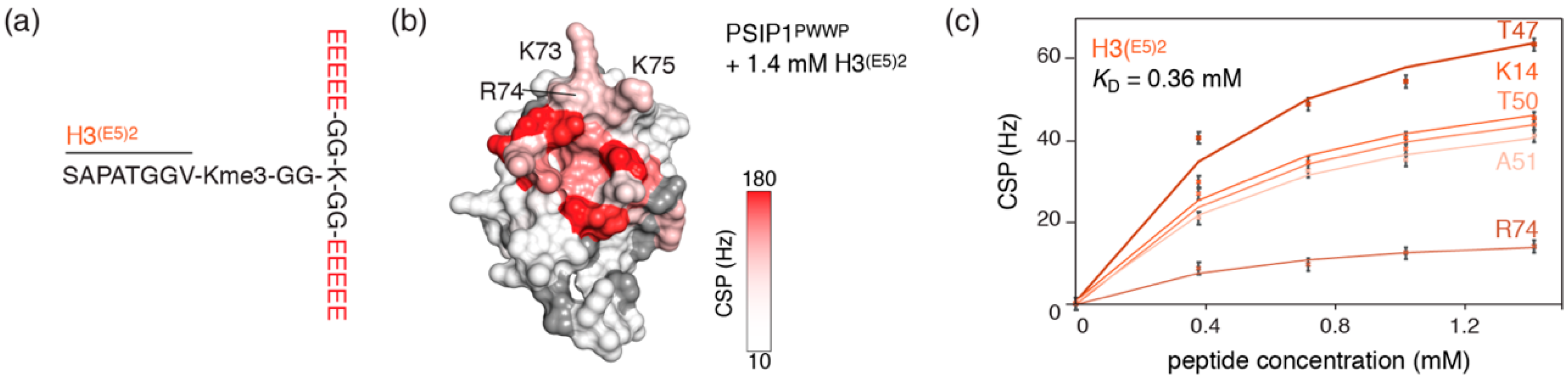
2.4. Nucleosome-Like Charge Distribution in a Branched H3 Peptide Boosts Affinity
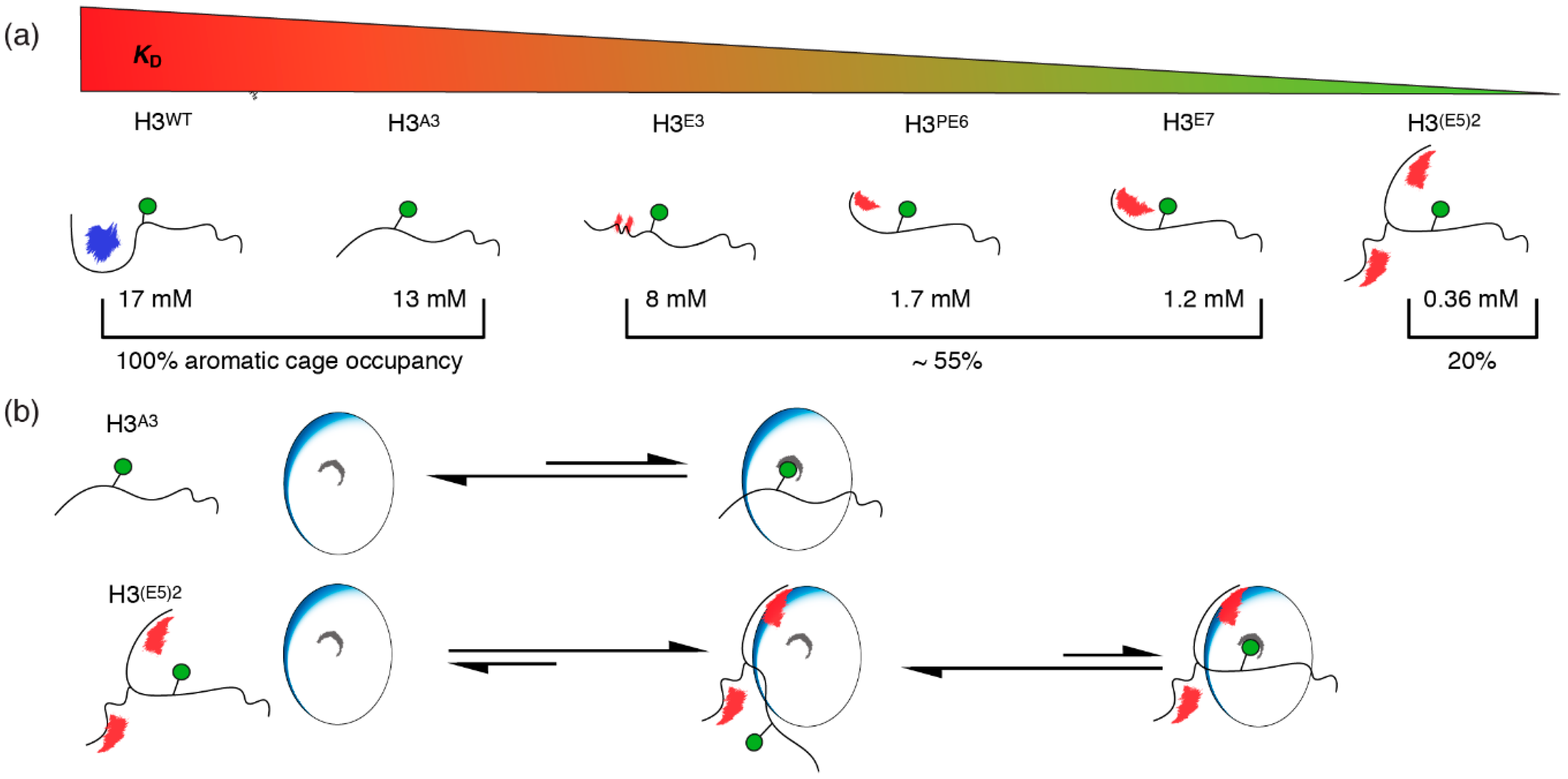
2.5. Electrostatic Interactions Alter Aromatic Cage Binding
3. Discussion
4. Materials and Methods
4.1. Protein Expression
4.2. Peptides
4.3. Synthesis H3(E5)2
4.4. NMR Spectroscopy
4.5. Titration Data Analysis
4.6. Data-Driven Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.; Bountra, C.; Fish, P.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Helin, K.; Dhanak, D. Chromatin proteins and modifications as drug targets. Nature 2013, 502, 480–488. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef]
- Greschik, H.; Schüle, R.; Günther, T. Selective targeting of epigenetic reader domains. Expert Opin. Drug Discov. 2017, 12, 449–463. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.-M. Chemical modulators for epigenome reader domains as emerging epigenetic therapies for cancer and inflammation. Curr. Opin. Chem. Biol. 2017, 39, 116–125. [Google Scholar] [CrossRef]
- Pérez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef]
- James, L.I.; Barsyte-Lovejoy, D.; Zhong, N.; Krichevsky, L.; Korboukh, V.K.; Herold, J.M.; MacNevin, C.J.; Norris, J.L.; Sagum, C.A.; Tempel, W.; et al. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat. Chem. Biol. 2013, 9, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Greschik, H.; Johansson, C.; Seifert, L.; Bacher, J.; Park, K.; Babault, N.; Martini, M.; Fagan, V.; Li, F.; et al. Discovery of a Potent and Selective Fragment-like Inhibitor of Methyllysine Reader Protein Spindlin 1 (SPIN1). J. Med. Chem. 2019, 62, 8996–9007. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Selvaraju, S.; Curtin, M.L.; Jakob, C.G.; Zhu, H.; Comess, K.M.; Shaw, B. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017, 13, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef]
- James, L.I.; Frye, S.V. Chemical probes for methyl lysine reader domains. Curr. Opin. Chem. Biol. 2016, 33, 135–141. [Google Scholar] [CrossRef]
- Hauser, A.T.; Robaa, D.; Jung, M. Epigenetic small molecule modulators of histone and DNA methylation. Curr. Opin. Chem. Biol. 2018, 45, 73–85. [Google Scholar] [CrossRef]
- Hughes, R.M.; Wiggins, K.R.; Khorasanizadeh, S.; Waters, M.L. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. USA 2007, 104, 11184–11188. [Google Scholar] [CrossRef]
- Kamps, J.; Huang, J.; Poater, J.; Xu, C.; Pieters, B.; Dong, A.; Min, J.; Sherman, W.; Beuming, T.; Bickelhaupt, F.; et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat. Commun. 2015, 6, 8911. [Google Scholar] [CrossRef]
- Taverna, S.; Li, H.; Ruthenburg, A.; Allis, C.D.; Paterl, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, J.; Dickson, B.; Cheng, N.; Liu, Y.; Norris, J.; Cholensky, S.; Tempel, W.; Qin, S.; Huber, K.; Sasum, C.; et al. A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol. 2016, 12, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Barnash, K.D.; The, J.; Norris-Drouin, J.L.; Cholensky, S.H.; Worley, B.M.; Li, F.; Stuckey, J.I.; Brown, P.J.; Vedadi, M.; Arrowsmith, C.H.; et al. Discovery of Peptidomimetic Ligands of EED as Allosteric Inhibitors of PRC2. ACS Comb. Sci. 2017, 19, 161–172. [Google Scholar] [CrossRef]
- Lamb, K.N.; Bsteh, D.; Dishman, S.N.; Moussa, H.F.; Fan, H.; Stuckey, J.I.; Norris, J.L.; Cholensky, S.H.; Li, N.; Wang, J.; et al. Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7. Cell Chem. Biol. 2019, 26, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Weaver, T.M.; Morrison, E.A.; Musselman, C.A. Reading More than Histones: The Prevalence of Nucleic Acid Binding among Reader Domains. Molecules 2018, 23, 2614. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Smith, S.G.; Yap, K.; Li, S.; Li, J.; Mezei, M.; Rodriguez, Y.; Vincek, A.; Aguilo, F.; Walsh, M.J.; et al. Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Med. Chem. Lett. 2016, 7, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Ge, H.; Si, Y.Z.; Roeder, R.G. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 1998, 17, 6723–6729. [Google Scholar] [CrossRef]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef]
- LeRoy, G.; Oksuz, O.; Descostes, N.; Aoi, Y.; Ganai, R.; Kara, H.; Yu, J.-R.; Lee, C.-H.; Stafford, J.; Shilatifard, A.; et al. LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription. Sci. Adv. 2019, 5, eaay3068. [Google Scholar] [CrossRef]
- Llano, M.; Vanegas, M.; Hutchins, N.; Thompson, D.; Delgado, S.; Poeschla, E.M. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 2006, 360, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Bushman, F.D. Retroviral DNA integration: HIV and the role of LEDGF/p75. Trends Genet. 2006, 22, 388–395. [Google Scholar] [CrossRef]
- Ganapathy, V.; Casiano, C.A. Autoimmunity to the nuclear autoantigen DFS70 (LEDGF): What exactly are the autoantibodies trying to tell us? Arthritis Rheum. 2004, 50, 684–688. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.S.; Myklebust, L.M.; Kjarland, E.; Gjertsen, B.T.; Pendino, F.; Bruserud, Ø.; Døskeland, S.O.; Lillehaug, J.R. LEDGF/p75 has increased expression in blasts from chemotherapy-resistant human acute myelogenic leukemia patients and protects leukemia cells from apoptosis in vitro. Mol. Cancer 2007, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Van Nuland, R.; Van Schaik, F.M.; Simonis, M.; Van Heesch, S.; Cuppen, E.; Boelens, R.; Timmers, H.M.; Van Ingen, H. Nucleosomal DNA binding drives the recognition of H3K36-methylated nucleosomes by the PSIP1-PWWP domain. Epigenet. Chromatin 2013, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Eidahl, J.O.; Crowe, B.L.; North, J.A.; McKee, C.J.; Shkriabai, N.; Feng, L.; Plumb, M.; Graham, R.L.; Gorelick, R.J.; Hess, S.; et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013, 41, 3924–3936. [Google Scholar] [CrossRef]
- Wang, H.; Farnung, L.; Dienemann, C.; Cramer, P. Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 2020, 27, 8–13. [Google Scholar] [CrossRef]
- Musselman, C.A.; Gibson, M.D.; Hartwick, E.W.; North, J.A.; Gatchalian, J.; Poirier, M.G.; Kutateladze, T.G. Binding of PHF1 Tudor to H3K36me3 enhances nucleosome accessibility. Nat. Commun. 2013, 4, 2969. [Google Scholar] [CrossRef]
- Wu, H.; Zeng, H.; Lam, R.; Tempel, W.; Amaya, M.F.; Xu, C.; Dombrovski, L.; Qiu, W.; Wang, Y.; Min, J. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 2011, 6, e18919. [Google Scholar] [CrossRef]
- Vezzoli, A.; Bonadies, N.; Allen, M.D.; Freund, S.M.; Santiveri, C.M.; Kvinlaug, B.T.; Huntly, B.J.; Göttgens, B.; Bycroft, M. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 2010, 17, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cui, G.; Botuyan, M.V.; Mer, G. Structural Basis for the Recognition of Methylated Histone H3K36 by the Eaf3 Subunit of Histone Deacetylase Complex Rpd3S. Structure 2008, 16, 1740–1750. [Google Scholar] [CrossRef]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; Van Dijk, M.; De Vries, S.; Bonvin, A. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Worrall, J.A.; Reinle, W.; Bernhardt, R.; Ubbink, M. Transient Protein Interactions Studied by NMR Spectroscopy: The Case of Cytochrome c and Adrenodoxin. Biochemistry 2003, 42, 7068–7076. [Google Scholar] [CrossRef]
- Schilder, J.; Ubbink, M. Formation of transient protein complexes. Curr. Opin. Struct. Biol. 2013, 23, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Ubbink, M. The courtship of proteins: Understanding the encounter complex. FEBS Lett. 2009, 583, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. USA 1981, 78, 4046–4050. [Google Scholar] [CrossRef]
- Connelly, K.E.; Weaver, T.M.; Alpsoy, A.; Gu, B.X.; Musselman, C.A.; Dykhuizen, E.C. Engagement of DNA and H3K27me3 by the CBX8 chromodomain drives chromatin association. Nucleic Acids Res. 2019, 47, 2289–2305. [Google Scholar] [CrossRef]
- Barnash, K.D.; Lamb, K.N.; James, L.I.; Frye, S.V. Peptide Technologies in the Development of Chemical Tools for Chromatin-Associated Machinery. Drug Dev. Res. 2017, 78, 300–312. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reason. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Trellet, M.; Melquiond, A.; Bonvin, A. A Unified Conformational Selection and Induced Fit Approach to Protein-Peptide Docking. PLoS ONE 2013, 8, e58769. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horn, V.; Jongkees, S.A.K.; van Ingen, H. Mimicking the Nucleosomal Context in Peptide-Based Binders of a H3K36me Reader Increases Binding Affinity While Altering the Binding Mode. Molecules 2020, 25, 4951. https://doi.org/10.3390/molecules25214951
Horn V, Jongkees SAK, van Ingen H. Mimicking the Nucleosomal Context in Peptide-Based Binders of a H3K36me Reader Increases Binding Affinity While Altering the Binding Mode. Molecules. 2020; 25(21):4951. https://doi.org/10.3390/molecules25214951
Chicago/Turabian StyleHorn, Velten, Seino A. K. Jongkees, and Hugo van Ingen. 2020. "Mimicking the Nucleosomal Context in Peptide-Based Binders of a H3K36me Reader Increases Binding Affinity While Altering the Binding Mode" Molecules 25, no. 21: 4951. https://doi.org/10.3390/molecules25214951
APA StyleHorn, V., Jongkees, S. A. K., & van Ingen, H. (2020). Mimicking the Nucleosomal Context in Peptide-Based Binders of a H3K36me Reader Increases Binding Affinity While Altering the Binding Mode. Molecules, 25(21), 4951. https://doi.org/10.3390/molecules25214951