3.1. Chemical Synthesis
MS and/or analytical data were obtained using chromatographically homogeneous samples.
1H NMR (500 MHz), 13C NMR (126 MHz), and 31P (202 MHz) spectra were recorded a Bruker Avance or Avance II (Bruker BioSpin, Wissembourg, France) in the given solvents unless otherwise indicated. Chemical shifts (δ) are reported in ppm and coupling constants are given in Hz. Low resolution mass spectra (LRMS) were recorded with an ion trap mass analyzer (LCQ Advantage, ThermoFisher, Bremen, Germany) under electrospray ionization (ESI) in positive and negative ionization mode detection. High resolution mass sprectra (HRMS) were recorded with an Orbitrap mass analyzer (Exactive, ThermoFisher, Bremen, Germany) under electrospray ionization (ESI) in positive or negative ionization mode detection, atmospheric pressure chemical ionization. All reactions were carried out under an argon atmosphere, and were monitored by thin-layer chromatography with Merck 60F-254 precoated silica (0.2 mm, Merck, Darmstadt, Germany) on glass. Flash chromatography was performed with Merck Kieselgel 60 (200–500 µm, VWR, Leuven, Belgium); the solvent systems were given v/v.
3.1.1. Diethyl {[ethoxy(methyl)phosphoryl]methyl}phosphonate 14
A solution of diethyl methylphosphonate (7.6 g, 49.6 mmol) in anhydrous THF (15 mL) under argon atmosphere was cooled by a dry ice/acetone bath. To this solution was added dropwise a solution of n-butyllithium (1.6 M in hexanes, 32 mL, 51.2 mmol) during 30 min. Then the reaction mixture was allowed to warm to room temperature. After 2 h of stirring, the reaction was quenched by addition of an aqueous solution of hydrochloric acid (3N, 25 mL). The reaction mixture was extracted two times with dichloromethane (2 × 75 mL). The organic layers were combined, dried over anhydrous sodium sulfate, filtered, and concentrated under vacuo to obtain the crude product (5.74 g, 22.2 mmol, 89%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 4.22–3.99 (m, 6H, 3 × OCH2CH3), 2.38 (dd, J = 20.7, 16.9 Hz, 2H, PCH2P), 1.66 (d, J = 15.0 Hz, 3H, CH3P), 1.32 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 1.31 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C NMR (126 MHz, CDCl3) δ 62.6 (d, J = 6.4 Hz, OCH2CH3), 62.5 (d, J = 6.5 Hz, OCH2CH3), 60.8 (d, J = 6.4 Hz, OCH2CH3), 29.0 (dd, J = 134.5, 81.2 Hz, PCH2P), 16.6 (d, J = 6.4 Hz, OCH2CH3), 16.4 (d, J = 6.3 Hz, OCH2CH3), 16.3 (d, J = 6.3 Hz, OCH2CH3), 15.8 (d, J = 100.7 Hz, CH3P); 31P NMR (202 MHz, CDCl3) δ 44.5 (d, J = 3.4 Hz, CH3PCH2P), 19.9 (d, J = 3.4 Hz, CH3PCH2P); ESI-MS: Calcd for C8H20NaO5P2: 281.1 [M + Na]+ Found: 281.1; ESI-HRMS: Calcd for C8H21O5P2: 259.0864 [M + H]+ Found: 259.0865.
3.1.2. 3,4,5,7-Tetra-O-benzyl-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy)phosphoryl}-α-d-manno-2-heptulopyranose 15
A solution of diethyl {[ethoxy(methyl)phosphoryl]methyl}phosphonate 14 (1.725 g, 6.68 mmol) in anhydrous THF (9 mL) under argon atmosphere was cooled by a dry ice/acetone bath. To this solution was added dropwise a solution of n-butyllithium (1.6 M in hexanes, 7 mL, 11.2 mmol) during 20 min. After stirring during two hours at −78 °C, a solution of 2,3,4,5-tetra-O-benzyl-mannono-1,5-lactone 11 (1.2 g, 2.23 mmol) in anhydrous THF (6 mL) was added dropwise to the reaction mixture. After stirring during two hours at −78 °C, the reaction was quenched by addition of a solution of acetic acid in THF (33% v/v in THF, 6 mL). The reaction mixture was diluted with 30 mL of ethyl acetate and 45 mL of water. To this mixture was added 12 g of sodium chloride. The organic layer was collected and the aqueous layer was extracted with 45 mL of ethyl acetate. The organic layers were combined, dried over anhydrous sodium sulfate, filtered, and concentrated. Column chromatography (silica gel 125 mL, EtOAc then EtOAc/EtOH, 98/2, v/v) of the residue afforded the product 15 as a mixture of stereoisomers (1.04 g, 1.30 mmol, 58%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.47–7.10 (m, 20H, H aromatic), 5.01–4.42 (m, 8H, 4 × OCH2Ph), 4.25–3.99 (m, 8H, H-4, 3 × POCH2CH3, H-6), 3.94, 3.91 (2t, J = 9.7 Hz, 1H, H-5), 3.77, 3.76 (2d, J = 2.7 Hz, 1H, H-3), 3.74–3.67 (m, 1H, H-7), 3.67, 3.65 (2dd, J = 11.7, 2.2 Hz, J = 10.8, 1.8 Hz, 1H, H-7′), 2.92, 2.66 (2td, J = 20.5, 15.8 Hz, J = 20.0, 15.8 Hz, 1H, PCH2P), 2.85, 2.74 (2dd, J = 15.1, 11.5 Hz, J = 15.3, 13.6 Hz, 1H, H-1), 2.52–2.30 (m, 1H, PCH2P), 2.23, 1.77 (2dd, J = 15.3, 11.5 Hz, J = 19.8, 15.2 Hz, 1H, H-1′), 1.33–1.17 (m, 9H, 3 × POCH2CH3); 13C NMR (126 MHz, CDCl3) δ 138.75, 138.72, 138.66, 138.64, 138.59, 138.50, 138.46 (4 × Cq aromatic), 128.56, 128.54, 128.42, 128.39, 128.3, 128.13, 128.08, 127.91, 127.88, 127.8, 127.71, 127.69, 127.60 (20 × CH aromatic), 98.2, 97.8 (2d, J = 8.8 Hz, J = 5.5 Hz, C-2), 81.5, 81.4 (2d, J = 2.5 Hz, J = 2.3 Hz, C-4), 78.9, 78.4 (2d, J = 9.7 Hz, J = 9.2 Hz, C-3), 75.2 (OCH2Ph), 75.0, 74.9, 74.8, 73.3, 73.2, 73.03, 72.97 (4 × OCH2Ph), 75.1 (C-5), 72.4, 71.9 (C-6), 69.8, 69.7 (C-7), 63.0, 62.8, 62.7, 62.6, 62.44, 61.39 (6d, J = 6.4 Hz, J = 6.3 Hz, J = 6.5 Hz, J = 6.6 Hz, J = 6.4 Hz, J = 6.7 Hz, 3 × OCH2CH3), 37.3, 35.2 (2d, J = 92 Hz, J = 94 Hz, C-1), 30.2, 29.1 (2dd, J = 135, 87 Hz, J = 134, 84 Hz, PCH2P), 16.58–16.29 (m, 3 × POCH2CH3); 31P NMR (202 MHz, CDCl3) δ 48.3, 46.7 (2d, J = 11.5 Hz, J = 3.4 Hz, CH2PCH2P), 20.0, 19.6 (2d, J = 11.5 Hz, J = 3.4 Hz, 1P, CH2PCH2P); ESI-MS: Calcd for C42H54NaO11P2: 819.8 [M + Na]+ Found: 819.2; ESI-HRMS: Calcd for C42H54NaO11P2: 819.3034 [M + Na]+ Found: 819.3035.
3.1.3. 3,4,5,7-Tetra-O-benzyl-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy)phosphoryl} -α-d-gluco-2-heptulopyranose 16
A solution of diethyl {[ethoxy(methyl)phosphoryl]methyl}phosphonate 14 (2.87 g, 11.1 mmol) in anhydrous THF (15 mL) under argon atmosphere was cooled by a dry ice/acetone bath. To this solution was added dropwise a solution of n-butyllithium (1.6 M in hexanes, 11.6 mL, 18.6 mmol) during 20 min. After stirring during two hours at −78 °C, a solution of 2,3,4,5-tetra-O-benzyl-glucono-1,5-lactone 12 (2 g, 3.71 mmol) in anhydrous THF (10 mL) was added dropwise to the reaction mixture. After stirring during two hours at −78 °C, the reaction was quenched by addition of a solution of acetic acid in THF (28% v/v in THF, 7 mL). The reaction mixture was diluted with 50 mL of ethyl acetate and 75 mL of water. To this mixture was added 20 g of sodium chloride. The organic layer was collected and the aqueous layer was extracted with 75 mL of ethyl acetate. The organic layers were combined, dried over anhydrous sodium sulfate, filtered, and concentrated. Column chromatography (silica gel 200 mL, EtOAc) of the residue afforded the product 16 as a mixture of stereoisomers (1.736 g, 2.17 mmol, 58%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.41–7.12 (m, 20H, H aromatic), 4.98, 4.96, 4.92, 4.91, 4.89, 4.88, 4.84, 4.83, 4.69, 4.65, 4.57, 4.55, 4.53, 4.50, 4.45, 4.44 (16d, J = 11.7 Hz, J = 11.4 Hz, J = 11.0 Hz, J = 10.9 Hz, J = 10.9 Hz, J = 11.0 Hz, J = 11.0 Hz, J = 11.0 Hz, J = 11.4 Hz, J = 11.7 Hz, J = 9.8 Hz, J = 9.8 Hz, J = 11.8 Hz, J = 11.8 Hz, J = 11.8 Hz, J = 11.8 Hz, 8H, 4 × OCH2Ph), 4.22–3.94 (m, 8H, 3 × OCH2CH3, H-6, H-4), 3.75–3.56 (m, 3H, H-7, H-7′, H-5), 3.34, 3.27 (2d, J = 9.5 Hz, 1H, H-3), 2.92–2.02 (m, 4H, H-1, PCH2P, H-1′), 1.37–1.20 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, CDCl3) δ 138.8, 138.7, 138.4, 138.3, 138.2, 138.1 (4 × Cq aromatic), 128.62, 128.58, 128.56, 128.53, 128.52, 128.51, 128.49, 128.04, 128.03, 128.00, 127.94, 127.92, 127.90, 127.87, 127.86, 127.82, 127.80, 127.76, 127.70 (20 × CH aromatic), 97.7, 97.3 (2d, J = 8.4 Hz, J = 6.8 Hz, C-2), 83.7, 83.6 (2d, J = 11.1 Hz, J = 10.0 Hz, C-3), 83.2 (2d, J = 3.0 Hz, J = 3.6 Hz, C-4), 79.0, 78.6 (C-5), 75.8, 75.7, 75.6, 75.5, 75.0, 73.5, 73.2 (4 × OCH2Ph), 71.2, 70.6 (C-6), 69.4, 69.1 (C-7), 62.8, 62.7, 62.6, 62.3, 62.1, 61.4 (6d, J = 6.4 Hz, J = 6.4 Hz, J = 6.4 Hz, J = 6.5 Hz, J = 6.3 Hz, J = 6.6 Hz, 3 × OCH2CH3), 37.5, 34.8 (2d, J = 91.7 Hz, J = 91.3 Hz, C-1), 30.2, 29.3 (2dd, J = 133.8, 86.3 Hz, J = 134.1, 85.4 Hz, PCH2P), 16.6–16.3 (m, 3 × OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 48.3, 45.8 (2d, J = 11.6 Hz, J = 6.6 Hz, CH2PCH2P), 19.6, 19.5 (2d, J = 11.6 Hz, J = 6.6 Hz, CH2PCH2P); ESI-MS: Calcd for C42H54NaO11P2: 819.8 [M + Na]+ Found: 819.1; ESI-HRMS: Calcd for C42H55O11P2: 797.3214 [M + H]+ Found: 797.3228.
3.1.4. 3-Acetamido-4,5,7-tri-O-benzyl-1,3-di-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy) phosphoryl}-α-d-gluco-2-heptulopyranose 17
A solution of diethyl {[ethoxy(methyl)phosphoryl]methyl}phosphonate 14 (1.845 g, 7.15 mmol) in anhydrous THF (9 mL) under argon atmosphere was cooled by a dry ice/acetone bath. To this solution was added dropwise a solution of n-butyllithium (1.6 M in hexanes, 7.6 mL, 12.25 mmol) during 20 min. After stirring during two hours at −78 °C, a solution of 2-acetamido-3,4,5-tri-O-benzyl-2-deoxy-glucono-1,5-lactone 13 (1 g, 2.04 mmol) in anhydrous THF (6 mL) was added dropwise to the reaction mixture. After stirring during two hours at −78 °C, the reaction was quenched by addition of a solution of acetic acid in THF (11% v/v in THF, 4.5 mL). The reaction mixture was diluted with 45 mL of ethyl acetate and 45 mL of water. To this mixture was added 12 g of sodium chloride. The organic layer was collected and the aqueous layer was extracted with 45 mL of ethyl acetate. The organic layers were combined, dried over anhydrous sodium sulfate, filtered, and concentrated. Column chromatography (silica gel 150 mL, EtOAc/EtOH, 95/5 to 9/1, v/v) of the residue afforded the product 17 as a mixture of stereoisomers (387 mg, 517 µmol, 25%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.39–7.14 (m, 15H, H aromatic), 6.42, 6.35 (2s, 1H, C-OH), 5.58, 5.44 (2d, J = 10.0 Hz, J = 10.1 Hz, 1H, NHCOCH3), 4.83, 4.82, 4.81, 4.64, 4.56, 4.55, 4.55, 4.49, 4.48, 4.45 (10d, J = 10.9 Hz, J = 11.5 Hz, J = 10.9 Hz, J = 11.5 Hz, J = 10.9 Hz, J = 10.9 Hz, J = 12.0 Hz, J = 12.0 Hz, J = 12.0 Hz, J = 12.0 Hz, 6H, 3 × OCH2Ph), 4.21–3.98 (m, 8H, 3 × OCH2CH3, H-6, H-3), 3.77–3.59 (m, 4H, H-7, H-7′, H-4, H-5), 2.92–2.32 (m, 3H, PCH2P, H-1), 2.21, 2.12 (t, dd, J = 14.7 Hz, J = 19.7, 15.3 Hz, 1H, H-1′), 1.88, 1.86 (2s, 3H, NHCOCH3), 1.38–1.23 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, CDCl3) δ 170.3, 170.1 (NHCOCH3), 138.7, 138.6, 138.3, 138.21, 138.17, 138.1 (3 × Cq aromatic), 128.62, 128.57, 128.56, 128.55, 128.52, 128.2, 128.13, 128.11, 127.93, 127.89, 127.81, 127.77 (15 × CH aromatic), 98.1, 97.4 (2d, J = 9.2 Hz, J = 6.5 Hz, C-2), 80.9, 80.8 (2d, J = 3.7 Hz, J = 3.0 Hz, C-4), 78.7, 75.2, 75.0, 73.4, 73.3 (3 × OCH2Ph), 71.3, 71.0 (C-6), 69.3, 69.1 (C-7), 63.0, 62.8, 62.6, 62.4, 61.7 (5d, J = 6.3 Hz, J = 6.5 Hz, J = 6.5 Hz, J = 6.3 Hz, J = 6.3 Hz, OCH2CH3), 57.4, 57.2 (2d, J = 11.2 Hz, J = 11.7 Hz, C-3), 37.0, 35.6 (2d, J = 90.3 Hz, J = 94.0 Hz, C-1), 30.1, 29.2 (2dd, J = 134.7, 88.3 Hz, J = 133.7, 83.4 Hz, PCH2P), 23.63, 23.61 (NHCOCH3), 16.6–16.3 (m, 3 × OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 48.2, 46.5 (2d, J = 12.5 Hz, J = 1.8 Hz, CH2PCH2P), 19.4, 19.1 (2d, J = 12.5 Hz, J = 1.8 Hz, CH2PCH2P); ESI-MS: Calcd for C37H51NNaO11P2: 770.3 [M + Na]+ Found: 770.1; ESI-HRMS: Calcd for C37H52NO11P2: 748.3010 [M + H]+ Found: 748.3036.
3.1.5. 2,6-Anhydro-3,4,5,7-tetra-O-benzyl-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy) phosphoryl}-d-manno-hept-1-enitol 18
To a solution of heptulopyranose 15 (0.9 g, 1.13 mmol) in anhydrous dichlormethane (35 mL) under argon atmopshere and cooled by an ice bath, was added dropwise pyridine (3.5 mL, 43.3 mmol) then trifluoroacetic anhydride (0.66 mL, 4.75 mmol). After stirring three hours at 0 °C, the reaction is quenched by the addition of an aqueous saturated solution of sodium bicarbonate (70 mL). The organic layer was collected and the aqueous layer was extracted two times with 50 mL of dichloromethane. The organic layers were combined, washed with 70 mL of an aqueous solution of hydrochloric acid 1N, then 70 mL of brine, dried over anhydrous sodium sulfate, filtered, and concentrated. Column chromatography (silica gel 100 mL, EtOAc/EtOH, 98/2 to 9/1, v/v) of the residue afforded three fractions of diastereoisomers 18 isolated as colorless oils (94 mg, 120 µmol, 10%; 77 mg, 99 µmol, 9%; 537 mg, 689 µmol, 61%). The physicochemical characteristics of the major fraction are reported. 1H NMR (500 MHz, CDCl3) δ 7.43–7.16 (m, 20H, H aromatic), 5.20 (2d, J = 11.9 Hz, J = 9.7 Hz, 1H, H-1), 4.79–4.45 (m, 8H, 4 × OCH2Ph), 4.21–4.06 (m, 1H, H-3), 4.16–3.91 (m, 9H, 3 × POCH2CH3, H-2, H-5, H-6), 3.85–3.66 (m, 3H, H-4, H-7, H-7′), 2.85–2.52 (m, 2H, PCH2P), 1.30–1.23 (m, 9H, 3 × POCH2CH3); 13C NMR (126 MHz, CDCl3) δ 164.6, 164.0 (s, d, J = 1.3 Hz, C-2), 138.2, 138.1, 138.0, 137.95, 137.91, 137.8, 137.42, 137.36 (4 × Cq aromatic), 128.64, 128.60, 128.55, 128.54, 128.50, 128.2, 128.1, 128.03, 128.00, 127.94, 127.92, 127.82, 127.78 (20 × CH aromatic), 100.5, 97.7 (2d, J = 136.4 Hz, J = 137.5 Hz, C-1), 79.7, 78.9 (C-6), 78.8, 77.9 (C-4), 75.2, 74.6 (2d, J = 12.0 Hz, J = 12.2 Hz, C-3), 74.9, 74.4 (C-5), 74.2, 73.52, 73.48, 73.4, 72.5, 72.1, 71.6, 71.0 (4 × OCH2Ph), 69.5 (C-7), 62.5, 62.44, 62.39, 62.3, 61.00, 60.98 (6d, J = 6.3 Hz, J = 6.3 Hz, J = 6.5 Hz, J = 6.2 Hz, J = 6.1 Hz, J = 5.8 Hz, 3 xOCH2CH3), 29.2, 28.9 (2dd, J = 134, 92.5 Hz, J = 134, 94 Hz, PCH2P), 16.62, 16.56, 16.48, 16.47, 16.45 (5d, J = 7.9 Hz, J = 7.1 Hz, J = 6.3 Hz, J = 5.9 Hz, J = 6.4 Hz, OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 31.1, 30.4 (2d, J = 3.6 Hz, J = 7.8 Hz, CH2PCH2P), 20.5, 20.4 (2d, J = 3.6 Hz, J = 7.8 Hz, CH2PCH2P); ESI-MS: Calcd for C42H52NaO10P2: 801.8 [M + Na]+ Found: 801.1; ESI-HRMS: Calcd for C42H53O10P2: 779.3108 [M + H]+ Found: 779.3140.
3.1.6. 2,6-Anhydro-3,4,5,7-tetra-O-benzyl-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy) phosphoryl}-d-gluco-hept-1-enitol 19
To a solution of heptulopyranose 16 (1.1 g, 1.38 mmol) in anhydrous dichloromethane (41 mL) under argon atmosphere and cooled by an ice bath, was added dropwise pyridine (4.1 mL, 50.7 mmol) then trifluoroacetic anhydride (0.77 mL, 5.52 mmol). After stirring three hours at 0 °C, the reaction is quenched by the addition of an aqueous saturated solution of sodium bicarbonate (80 mL). The organic layer was collected and the aqueous layer was extracted two times with 60 mL of dichloromethane. The organic layers were combined, washed with 80 mL of an aqueous solution of hydrochloric acid 1N, then 80 mL of brine, dried over anhydrous sodium sulfate, filtered, and concentrated. Column chromatography (silica gel 100 mL, EtOAc/EtOH, 98/2 to 9/1, v/v) of the residue afforded the product 19 as a mixture of stereoisomers (0.75 g, 0.96 mmol, 70%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.52–6.98 (m, 20H, H aromatic), 5.29, 5.18 (2d, J = 11.2 Hz, J = 10.1 Hz, 1H, H-1), 4.79–4.49 (m, 8H, 4 × OCH2Ph), 4.27–3.97 (m, 7H, 3 × OCH2CH3, H-6), 3.97–3.93 (m, 1H, H-3), 3.84–3.76 (m, 3H, H-4, H-5, H-7), 3.73, 3.71 (2dd, J = 12.3, 4.1 Hz, J = 12.2, 4.0 Hz, 1H, H-7′), 2.93–2.47 (m, 2H, PCH2P), 1.31–1.25 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, CDCl3) δ 164.36, 164.2 (d, s, J = 1.1 Hz, C-2), 138.0, 137.92, 137.86, 137.8, 137.7, 137.3, 137.2 (4 × Cq aromatic), 128.7, 128.62, 128.56, 128.55, 128.53, 128.50, 128.47, 128.2, 128.13, 128.09, 128.08, 128.05, 128.02, 128.00, 127.98, 127.96, 127.91, 127.90, 127.83, 127.81 (20C, CH aromatic), 99.3, 99.0 (2d, J = 136.8 Hz, J = 136.9 Hz, C-1), 83.6, 82.9 (C-4), 78.7, 77.9 (2d, J = 11.9 Hz, J = 12.1 Hz, C-3), 78.2, 77.6 (C-6), 77.4, 77.10 (C-5), 74.3, 73.90, 73.85, 73.56, 73.51, 73.49, 72.8, 72.2 (4 × OCH2Ph), 68.5, 68.4 (C-7), 62.5, 62.42, 62.35, 62.33, 61.00, 60.98 (6d, J = 6.2 Hz, J = 6.2 Hz, J = 6.3 Hz, J = 6.2 Hz, J = 6.1 Hz, J = 6.1 Hz, 3 × OCH2CH3), 29.1 (dd, J = 134.4, 93.1 Hz, CHPCH2P), 16.7–16.4 (m, 3 × OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 30.8, 30.6 (2d, J = 7.7 Hz, J = 6.5 Hz, CH2PCH2P), 20.5, 20.5 (2d, J = 7.7 Hz, J = 6.5 Hz, CH2PCH2P); ESI-MS: Calcd for C42H53O10P2: 779.8 [M + H]+ Found: 779.0, Calcd for C42H54KO10P2: 819.9 [M + 2H + K]+ Found: 819.2; ESI-HRMS: Calcd for C42H53O10P2: 779.3108 [M + H]+ Found: 779.3127.
3.1.7. 3-Acetamido-2,6-Anhydro-4,5,7-tri-O-benzyl-1,3-di-deoxy-1-{[(diethoxyphosphoryl) methyl](ethoxy)phosphoryl}-d-gluco-hept-1-enitol 20
To a solution of heptulopyranose 17 (340 mg, 455 µmol) in anhydrous dichlormethane (14 mL) under argon atmosphere and cooled by an ice bath, was added dropwise pyridine (1.4 mL, 17.3 mmol) then trifluoroacetic anhydride (250 µL, 1.82 mmol). After stirring two hours at 0 °C, the reaction is quenched by the addition of an aqueous saturated solution of sodium bicarbonate (26 mL). The organic layer was collected and the aqueous layer was extracted two times with 26 mL of dichloromethane. The organic layers were combined, washed with 26 mL of an aqueous solution of hydrochloric acid 1N, then 26 mL of brine, dried over anhydrous sodium sulfate, filtered, and concentrated. Column chromatography (silica gel 30 mL, EtOAc/EtOH, 9/1 to 8/2, v/v) of the residue afforded the product 20 as a mixture of stereoisomers and rotamers (210 mg, 288 µmol, 63%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.52–6.92 (m, 30H, H aromatic), 5.14, 5.09, 5.03, 4.94 (4d, J = 11.3 Hz, J = 11.4 Hz, J = 9.3 Hz, J = 11.7 Hz, 1H, H-1), 4.84–4.34 (m, 6H, 3 × OCH2Ph), 4.29–3.55 (m, 12H, H-6, 3 × OCH2CH3, H-3, H-4, H-5, H-7, H-7′), 3.27–2.23 (m, 2H, PCH2P), 2.00, 1.97, 1.94, 1.92, 1.91, 1.90, 1.87, (7 s, 3H, NHCOCH3), 1.36–1.15 (m, 9H, 3 × OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 31.3, 30.8 (2broad s, CH2PCH2P), 20.7, 20.6, 20.2, 20.1, 19.8, 19.6 (6d, J = 4.7 Hz, J = 5.7 Hz, J = 2.1 Hz, J = 2.9 Hz, J = 5.8 Hz, J = 3.6 Hz, CH2PCH2P); ESI-MS: Calcd for C37H49NNaO10P2: 752.7327 [M + Na]+ Found: 752.2; ESI-HRMS: Calcd for C37H49NNaO10P2: 752.2724 [M + Na]+ Found: 752.2730.
3.1.8. 2,6-Anhydro-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy)phosphoryl}-d-glycero-d-galacto-heptitol 21
To a solution of a mixture of the previous d-manno-hept-1-enitol 18 (476 mg, 611 µmol) in ethanol (28 mL) under argon atmosphere was added a catalytic amount of palladium 10% on charcoal (140 mg, 132 µmol). Then, the reaction mixture was purged with hydrogen gas and stirred overnight under hydrogen atmosphere. The resulting solution was degassed with argon atmosphere and it was filtered through a celite© (ThermoFisher, Kandel, Germany) patch which was rinsed with 10 mL of ethanol. The concentration under vacuum furnished the product 21 as a mixture of stereoisomers with a R configuration for the C2 stereogenic centre (253 mg, 602 µmol, quant.) as a white solid. 1H NMR (500 MHz, MeOD) δ 4.24–4.10 (m, 6H, 3 × POCH2CH3), 3.94–3.86 (m, 1H, H-1), 3.87, 3.86 (2dd, J = 11.7, 2.3 Hz, J = 11.3, 2.4 Hz, 1H, H-7), 3.72, 3.71 (2d, J = 3.0 Hz, 1H, H-3), 3.66, 3.65 (2dd, J = 11.3, 6.7 Hz, J = 11.7, 6.3 Hz, 1H, H-7′), 3.53 (2t, J = 9.4 Hz, 1H, H-5), 3.49, 3.48 (2dd, J = 9.4, 3.0 Hz, 1H, H-4), 3.26, 3.23 (2ddd, J = 9.4, 6.3, 2.3 Hz, J = 9.4, 6.7, 2.4 Hz, 1H, H-6), 2.97–2.75 (m, 2H, PCH2P), 2.61–2.51 (m, 1H, H-1), 2.20–2.02 (m, 1H, H-1′), 1.35, 1.34 (2t, J = 7.0 Hz, 9H, 3 × POCH2CH3); 13C NMR (126 MHz, MeOD) δ 82.33, 82.28 (C-6), 76.3 (C-4), 74.7, 74.5 (2d, J = 3.7 Hz, J = 4.6 Hz, C-2), 73.3, 73.2 (d, J = 12.2 Hz, J = 11.4 Hz, C-3), 68.6, 68.5 (C-5), 64.2, 64.08, 64.02, 63.98, 62.9, 62.8 (6d, J = 6.5 Hz, POCH2CH3), 63.06, 63.00 (C-7), 33.5, 33.4 (2d, J = 99 Hz, C-1), 28.6, 28.4 (2dd, J = 135, 83 Hz, PCH2P), 16.83, 16.79, 16.7, 16.6 (4d, J = 6.3 Hz, 3 × POCH2CH3); 31P NMR (202 MHz, MeOD) δ 48.8, 47.7 (2d, J = 11.4 Hz, J = 7.5 Hz, CH2PCH2P), 21.2, 21.0 (2d, J = 7.5 Hz, J = 11.4 Hz, CH2PCH2P); ESI-MS: Calcd for C28H59NaO20P4: 862.7 [2M – H + Na]+ Found: 862.7; ESI-HRMS: Calcd for C14H30NaO10P2: 443.1206 [M + Na]+ Found: 443.1223.
3.1.9. 2,6-Anhydro-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy)phosphoryl}-d-glycero-d-gulo-heptitol 22
To a solution of a mixture of the d-gulo-hept-1-enitol 19 (720 mg, 925 µmol) in ethanol (42 mL) under argon atmosphere was added a catalytic amount of palladium 10% on charcoal (210 mg, 190 µmol). Then, the reaction mixture was purged with hydrogen gas and stirred overnight under hydrogen atmosphere. The resulting solution was degassed with argon atmosphere and it was filtered through a celite© patch. The concentration under vacuum furnished the product 22 as a mixture of stereoisomers with a R configuration for the C2 stereogenic centre (377 mg, 897 µmol, 97%) as a white solid. 1H NMR (500 MHz, MeOD) δ 4.24–4.08 (m, 6H, 3 × OCH2CH3), 3.87, 3.86 (2dd, J = 11.9, 2.2 Hz, J = 11.8, 1.4 Hz, 1H, H-7), 3.64–3.50 (m, 2H, H-7′, H-2), 3.38–3.22 (m, 3H, H-4, H-6, H-5), 3.11, 3.08 (2t, J = 9.0 Hz, J = 9.1 Hz, 1H, H-3), 2.99–2.74 (m, 2H, PCH2P), 2.57–2.40 (m, 1H, H-1), 2.26–2.16 (m, 1H, H-1′), 1.41–1.28 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, MeOD) δ 82.04, 81.96 (C-6), 79.44, 79.42 (d, J = 3,8 Hz, J = 4.0 Hz, C-4), 76.2, 76.1 (d, J = 5.3 Hz, J = 6.0 Hz, C-2), 75.9, 75.7 (d, J = 13.7 Hz, J = 12.7 Hz, C-3), 71.9, 71.8 (C-5), 64.1, 64.03, 63.99, 63.65, 62.81, 62.75 (6d, J = 6.4 Hz, J = 6.3 Hz, J = 6.3 Hz, J = 6.3 Hz, J = 6.5 Hz, J = 6.6 Hz, OCH2CH3), 63.0, 62.9 (C-7), 33.3, 32.9 (2d, J = 99.1 Hz, J = 99.2 Hz, C-1), 29.1, 28.5 (2dd, J = 135.0, 24.4 Hz, J = 135.0, 24.9 Hz, PCH2P), 16.9–16.53 (m, 3 × OCH2CH3); 31P NMR (202 MHz, MeOD) δ 47.2, 46.3 (2d, J = 11.4 Hz, J = 7.9 Hz, CH2PCH2P), 19.8, 19.6 (d, J = 7.9 Hz, J = 11.4 Hz, CH2PCH2P); ESI-MS: Calcd for C28H59NaO20P4: 862.7 [2M – H + Na]+ Found: 862.7; ESI-HRMS: Calcd for C14H31O10P2: 421.1387 [M + H]+ Found: 421.1394.
3.1.10. 3-Acetamido-2,6-anhydro-1,3-di-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy) phosphoryl}-d-glycero-d-gulo-heptitol 23
To a solution of a mixture of the d-gluco-hept-1-enitol 20 (210 mg, 288 µmol) in ethanol (14 mL) under argon atmosphere was added a catalytic amount of palladium 10% on charcoal (70 mg, 66 µmol). Then, the reaction mixture was purged with hydrogen gas and stirred overnight under hydrogen atmosphere. The resulting solution was degassed with argon atmosphere and it was filtered through a celite© patch. The concentration under vacuum furnished the product 23 as a mixture of stereoisomers with a R configuration for the C2 stereogenic centre (132 mg, 288 µmol, quant.) as a white solid. 1H NMR (500 MHz, MeOD) δ 4.23–4.10 (m, 6H, 3 × OCH2CH3), 3.91–3.85 (m, 1H, H-7), 3.70–3.56 (m, 3H, H-7′, H-3, H-2), 3.45–3.39 (m, 1H, H-4), 3.34–3.25 (m, 1H, H-5), 3.26 (ddd, J = 9.7, 5.8, 2.2 Hz, 1H, H-6), 3.05–2.71 (m, 2H, PCH2P), 2.30–2.18 (m, 1H, H-1), 2.15–2.06 (m, 1H, H-1′), 2.00 (s, 3H, NHCOCH3), 1.37–1.32 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, MeOD) δ 173.93, 173.89 (NHCOCH3), 82.0, 81.9 (C-6), 76.8 (2d, J = 2.7 Hz, J = 2.4 Hz, C-4), 75.4, 75.0 (2d, J = 5.0 Hz, J = 6.3 Hz, C-2), 72.23, 72.15 (C-5), 64.1, 64.04, 63.98, 63.93, 62.85, 62.84 (6d, J = 6.3 Hz, J = 6.9 Hz, J = 6.3 Hz, J = 6.1 Hz, J = 7.7 Hz, J = 7.7 Hz, 3 × OCH2CH3), 62.92, 62.88 (C-6), 57.7, 57.6 (d, J = 13.6 Hz, J = 14.2 Hz, C-3), 33.8, 33.4 (2d, J = 99.4 Hz, J = 100.2 Hz, C-1), 28.7, 28.5 (2dd, J = 134.8, 82.5 Hz, J = 134.8, 83.2 Hz, PCH2P), 23.0, 22.9 (NHCOCH3), 16.81, 16.76, 16.64, 16.63 (4d, J = 6.2 Hz, J = 6.3 Hz, J = 6.3 Hz, J = 6.1 Hz, 3 × OCH2CH3); 31P NMR (202 MHz, MeOD) δ 48.2, 47.1 (2d, J = 12.0 Hz, J = 7.1 Hz, CH2PCH2P), 21.3, 21.0 (2d, J = 7.1 Hz, J = 12.0 Hz, CH2PCH2P); ESI-MS: Calcd for C32H65N2NaO20P4: 944.8 [2M − H + Na]+ Found: 944.7, Calcd for C16H33NNaO10P2: 484.4 [M + Na]+ Found: 484.1; ESI-HRMS: Calcd for C16H34O10NP2: 462.1662 [M + H]+ Found: 462.1667.
3.1.11. 3,4,5,7-Tetra-O-acetyl-2,6-anhydro-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy) phosphoryl}-d-glycero-d-galacto-heptitol 24
To a solution of a mixture of the previous d-glycero-d-galacto-heptitol 21 (235 mg, 559 µmol) in pyridine (9 mL) cooled by an ice bath, was added dropwise acetic anhydride (4.5 mL, 48 mmol). The reaction mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the reaction is stopped by evaporation of the reactants. Column chromatography (silica gel 60 mL, CH2Cl2/EtOH, 95/5 to 9/1, v/v) of the residue afforded 24 as a mixture of stereoisomers with a R configuration for the C2 stereogenic centre (265 mg, 450 µmol, 80%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.32, 5.31 (2broad d, J = 3.4 Hz, J = 3.6 Hz, 1H, H-3), 5.17, 5.15 (2t, J = 10.0 Hz, 1H, H-5), 5.07, 5.06 (2dd, J = 10.0, 3.6 Hz, J = 10.0, 3.4 Hz, 1H, H-4), 4.25–4.07 (m, 9H, H-2, 3 × POCH2CH3, H-7, H-7′), 3.75, 3.71 (2ddd, J = 10.0, 7.0, 2.3 Hz, J = 10.0, 6.3, 2.4 Hz, 1H, H-6), 2.70–2.35 (m, 3H, 1.5 × PCH2P, H-1), 2.17, 2.08, 2.04, 1.96 (4s, 12H, 4 × CH3CO), 2.06–1.77 (m, 1H, H-1′), 1.38–1.28 (m, 9H, 3 × POCH2CH3); 13C NMR (126 MHz, CDCl3) δ 170.7, 170.49, 170.47, 170.1, 170.0, 169.92, 169.85 (4 × CH3CO), 76.6, 76.4 (C-6), 72.4, 72.16 (2d, J = 2.7 Hz, J = 3.4 Hz, C-2), 72.17, 72.10 (2d, J = 1.7 Hz, J = 2.0 Hz, C-4), 70.62, 70.55 (2d, J = 12.9 Hz, J = 12.4 Hz, C-3), 66.1, 66.0 (C-5), 63.2, 63.1 (C-7), 62.8, 62.62, 62.60, 62.36, 61.58, 61.56 (6d, J = 6.5 Hz, 3 × POCH2CH3), 32.2, 31.1 (2d, J = 98.5 Hz, J = 99.7 Hz, C-1), 29.3, 28.9 (2dd, J = 134.5, 83.1 Hz, J = 134.6, 81.9 Hz, PCH2P), 20.86, 20.84, 20.83, 20.69, 20.68 (4 × CH3CO), 16.8–16.4 (m, 3 × POCH2CH3); 31P NMR (202 MHz, CDCl3) δ 44.1, 42.2 (2d, J = 9.8 Hz, J = 5.9 Hz, CH2PCH2P), 19.9, 19.7 (d, J = 9.8 Hz, J = 5.9 Hz, CH2PCH2P); ESI-MS: Calcd for C44H75NaO28P4: 1198.9 [2M – H + Na]+ Found: 1198.4; ESI-HRMS: Calcd for C22H39O14P2: 589.1810 [M + H]+ Found: 589.1818.
3.1.12. 3,4,5,7-Tetra-O-acetyl-2,6-anhydro-1-deoxy-1-{[(diethoxyphosphoryl)methyl](ethoxy) phosphoryl}-d-glycero-d-gulo-heptitol 25
To a solution of a mixture of the previous d-glycero-d-gulo-heptitol 22 (372 mg, 885 µmol) in pyridine (15 mL) cooled by an ice bath, was added dropwise acetic anhydride (7.5 mL, 79 mmol). The reaction mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the reaction is stopped by evaporation of the reactants. Column chromatography (silica gel 80 mL, CH2Cl2/EtOH, 95/5 to 9/1, v/v) of the residue afforded 25 as a mixture of stereoisomers with a R configuration for the C2 stereogenic centre (503 mg, 854 µmol, 96%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.14, 5.13 (2t, J = 9.6 Hz, 1H, H-4), 4.98, 4.96 (2t, J = 9.6 Hz, 1H, H-5), 4.87, 4.86 (2t, J = 9.6 Hz, 1H, H-3), 4.20–4.00 (m, 8H, 3 × OCH2CH3, H-7, H-7′), 3.99–3.81 (m, 1H, H-2), 3.74, 3.69 (2ddd, J = 10.0, 6.3, 2.2 Hz, 1H, J = 10.0, 5.6, 2.2 Hz, H-6), 2. 70–2.39 (m, 2H, PCH2P), 2.32–1.86 (m, 2H, H-1, H-1′), 2.03, 2.01, 2.00, 1.98, 1.95, 1.94 (6s, 12H, 4 × CH3CO), 1.34–1.25 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, CDCl3) δ 170.5, 170.3, 170.2, 169.7, 169.6, 169.5 (4 × CH3CO), 76.0, 75.8 (C-6), 74.0, 73.9 (2d, J = 2.2 Hz, J = 1.7 Hz, C-4), 73.4, 73.2 (2d, J = 4.0 Hz, J = 6.5 Hz, C-2), 72.2, 72.1 (d, J = 14.6 Hz, J = 13.8 Hz, C-3), 68.6, 68.5 (C-5), 62.7, 62.5, 62.4, 62.3, 61.4 (5d, J = 6.3 Hz, J = 6.1 Hz, J = 4.5 Hz, J = 6.4 Hz, Hz, J = 6.5 Hz, 3 × OCH2CH3), 62.5, 62.4 (C-7), 32.4, 31.5 (2d, J = 98.0 Hz, J = 100.0 Hz, C-1), 29.5, 28.9 (2dd, J = 134.5, 83.8 Hz, J = 134.9, 81.7 Hz, PCH2P), 20.7, 20.6 (4 × CH3CO), 16.6–16.3 (m, 3 × OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 43.9, 42.1 (2d, J = 9.0 Hz, J = 3.1 Hz, CH2PCH2P), 19.9, 19.7 (2d, J = 9.0 Hz, J = 3.1 Hz, CH2PCH2P); ESI-MS: Calcd for C44H75NaO28P4: 1198.9 [2M – H + Na]+ Found: 1198.4; ESI-HRMS: Calcd for C22H39O14P2: 589.1810 [M + H]+ Found: 589.1820.
3.1.13. 3-Acetamido-4,5,7-tri-O-acetyl-2,6-anhydro-1,3-di-deoxy-1-{[(diethoxyphosphoryl) methyl](ethoxy)phosphoryl}-d-glycero-d-gulo-heptitol 26
To a solution of a mixture of the previous d-glycero-d-gulo-heptitol 23 (132 mg, 288 µmol) in pyridine (5 mL) cooled by an ice bath, was added dropwise acetic anhydride (2.5 mL, 26.4 mmol). The reaction mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the reaction is stopped by evaporation of the reactants. Column chromatography (silica gel 30 mL, CH2Cl2/EtOH, 95/5 to 9/1, v/v) of the residue afforded 26 as a mixture of stereoisomers with a R configuration for the C2 stereogenic centre (124 mg, 211 µmol, 73%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 6.27, 6.20 (2d, J = 9.3 Hz, J = 9.5 Hz, 1H, NHCOCH3), 5.09–4.98 (m, 2H, H-5, H-4), 4.25–3.99 (m, 9H, 3 × OCH2CH3, H-7, H-7′, H-3), 3.92, 3.86 (2ddd, J = 11.9, 9.5, 2.2 Hz, J = 12.1, 9.3, 2.2 Hz, 1H, H-2), 3.75–3.67 (m, 1H, H-6), 2.66–2.01 (m, 4H, H-1, H-1′, PCH2P), 2.07, 2.06, 2.02, 2.01, 2.00 (5s, 9H, 3 × OCOCH3), 1.94 (s, 3H, NHCOCH3), 1.38–1.30 (m, 9H, 3 × OCH2CH3); 13C NMR (126 MHz, CDCl3) δ 171.4, 171.3, 170.75, 170.74, 170.67, 170.6, 169.5 (3 × OCOCH3, NHCOCH3), 76.1, 75.9 (C-6), 74.7, 74.0 (2d, J = 3.6 Hz, J = 5.8 Hz, C-2), 74.33, 74.29 (2d, J = 2.4 Hz, J = 3.4 Hz, C-4), 68.7, 68.6 (C-5), 63.0, 62.64, 62.56, 62.54, 61.5, 61.4 (6d, J = 6.5 Hz, 3 × OCH2CH3), 62.8, 62.52 (C-7), 54.31, 54.25 (2d, J = 13.5 Hz, J = 14.8 Hz, C-3), 32.8, 32.0 (d, J = 98.1 Hz, J = 99.4 Hz, C-1), 29.3, 28.7 (2dd, J = 134.5, 83.6 Hz, J = 135.1, 81.6 Hz, CH2PCH2P), 23.3 (NHCOCH3), 20.84, 20.81, 20.79, 20.74, 20.73 (3 × OCOCH3), 16.7–16.4 (3 × OCH2CH3); 31P NMR (202 MHz, CDCl3) δ 44.8, 43.0 (2d, J = 8.0 Hz, J = 1.4 Hz, CH2PCH2P), 19.8, 19.6 (2d, J = 8.0 Hz, J = 1.4 Hz, CH2PCH2P); ESI-MS: Calcd for C22H39NNaO13P2: 610.5 [M + Na]+ Found: 610.1, Calcd for C44H77N2NaO26P4: 1197.0 [2M − H + Na]+ Found: 1196.5; ESI-HRMS: Calcd for C22H40NO13P2: 588.1969 [M + H]+ Found: 588.1991.
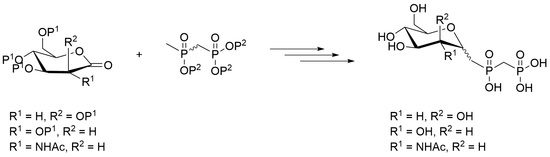
3.1.14. Sodium {[oxido(2,6-anhydro-1-deoxy-d-glycero-d-galacto-heptityl)phosphoryl]methyl} phosphonate 27
To a solution of a mixture of the previous d-glycero-d-galacto-heptitol 24 (53.5 mg, 90 µmol) in anhydrous dichloromethane (2 mL) cooled by an ice bath, was added dropwise bromotrimethylsilane (142 µL, 1.08 mmol). The reaction mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the reaction is stopped by evaporation of the reactants. Then the residue was dissolved in water (4 mL). Sodium hydoxide (28.8 mg, 720 µmol) was added to the reaction mixture. After 16 h of stirring at room temperature, the reaction was stopped with addition of cation exchange resin (DOWEX© 50WX8 resin, H+ form, ACROS, Geel, Belgium). The resin was removed by filtration through a millipore filter (45 µm, AIT, Houilles, France). The aqueous solution was freeze dried. The resulting solid (48 mg) was solubilized in water (0.48 mL). To this solution was added absolute ethanol (4.8 mL). After 16 h at room temperature, the resulting solid was recovered and rinsed with absolute ethanol. It was solubilized in water and freeze dried to afford the phosphonate 27 as a white solid (34.6 mg, 86 µmol, 95%). 1H NMR (500 MHz, CDCl3) δ 4.03 (td, J = 9.3, 3.9 Hz, 1H, H-2), 3.97 (dd, J = 12.3, 2.0 Hz, 1H, H-7), 3.94 (d, J = 3.2 Hz, 1H, H-3), 3.78 (dd, J = 12.3, 6.3 Hz, 1H, H-7′), 3.76 (dd, J = 9.6, 3.2 Hz, 1H, H-4), 3.63 (t, J = 9.6 Hz, 1H, H-5), 3.45 (ddd, J = 9.6, 6.3, 2.0 Hz, 1H, H-6), 2.57–2.34 (m, 3H, PCH2P, H-1), 2.14 (td, J = 16.3, 3.9 Hz, 1H, H-1′); 13C NMR (126 MHz, CDCl3) δ 80.0 (C-6), 74.2 (C-4), 73.6 (d, J = 2.8 Hz, C-2), 71.8 (d, J = 10.5 Hz, C-3), 67.0 (C-5), 61.3 (C-7), 32.2 (d, J = 96.5 Hz, C-1), 30.9 (dd, J = 122.1, 80.7 Hz, PCH2PO); 31P NMR (202 MHz, CDCl3) δ 36.05 (d, J = 4.5 Hz, CH2PCH2P), 11.75–11.65 (m, CH2PCH2P); 31P NMR non decoupled (202 MHz, CDCl3) δ 36.50–35.73 (m, CH2PCH2P), 11.70 (td, J = 18.5, 4.5 Hz, CH2PCH2P); ESI-MS (mobile phase gradient ACN/H2O + 0.1% formic acid): Calcd for C8H19O10P2: 337.0 [M + H]+ Found: 337.0; ESI-HRMS: Calcd for C8H19O10P2: 337.0453 [M + H]+ Found: 337.0443.
3.1.15. Sodium {[oxido(2,6-anhydro-1-deoxy-d-glycero-d-gulo-heptityl)phosphoryl]methyl} phosphonate 28
To a solution of a mixture of the previous d-glycero-d-gulo-heptitol 25 (63.5 mg, 116 µmol) in anhydrous dichloromethane (2 mL) cooled by an ice bath, was added dropwise bromotrimethylsilane (183 µL, 1.39 mmol). The reaction mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the reaction is stopped by evaporation of the reactants. Then the residue was dissolved in water (4 mL). Sodium hydoxide (37 mg, 925 µmol) was added to the reaction mixture. After 16 h of stirring at room temperature, the reaction was stopped with addition of cation exchange resin (DOWEX© 50WX8 resin, H+ form). The resin was removed by filtration through a millipore filter (45 µm). The aqueous solution was freeze dried. The resulting solid (62 mg) was solubilized in water (0.62 mL). To this solution was added absolute ethanol (6.2 mL). After 16 h at room temperature, the resulting solid was recovered and rinsed with absolute ethanol. It was solubilized in water and freeze dried to afford the phosphonate 28 as a white solid (43 mg, 107 µmol, 92%). 1H NMR (500 MHz, D2O) δ 3.93 (d, J = 12.3 Hz, 1H, H-7), 3.74 (dd, J = 12.3, 4.3 Hz, 1H, H-7′), 3.68 (qd, J = 9.4, 2.5 Hz, 1H, H-2), 3.52 (t, J = 9.4 Hz, 1H, H-4), 3.48–3.40 (m, 2H, H-5, H-6), 3.26 (t, J = 9.4 Hz, 1H, H-3), 2.33–2.01 (m, 4H, H-1, H-1′, PCH2P); 13C NMR (126 MHz, D2O) δ 79.5 (C-6), 77.2 (C-4), 75.6 (d, J = 4.3 Hz, C-2), 74.5 (d, J = 11.2 Hz, C-3), 69.8 (C-5), 60.9 (C-7), 33.71 (d, J = 96.0 Hz, C-1), 32.38 (dd, J = 119.5, 79.8 Hz, PCH2PO); 31P NMR (202 MHz, D2O) δ 34.72 (d, J = 6.1 Hz, CH2PCH2P), 15.25 (d, J = 6.1 Hz, CH2PCH2P); 31P NMR non decoupled (202 MHz, D2O) δ 35.97–32.61 (m, CH2PCH2P), 15.22 (td, J = 19.3, 6.1 Hz, CH2PCH2P); ESI-MS (mobile phase gradient ACN/H2O + 0.1% formic acid): Calcd for C8H19O10P2: 337.0 [M + H]+ Found: 337.0; ESI-HRMS: Calcd for C8H19O10P2: 337.0453 [M + H]+ Found: 337.0448.
3.1.16. Sodium {[oxido(3-acetamido-2,6-anhydro-1,3-di-deoxy-d-glycero-d-gulo-heptityl) phosphoryl]methyl}phosphonate 29
To a solution of a mixture of the previous d-glycero-d-gulo-heptitol 26 (51 mg, 87 µmol) in anhydrous dichloromethane (2 mL) cooled by an ice bath, was added dropwise bromotrimethylsilane (137 µL, 1.04 mmol). The reaction mixture was allowed to warm to room temperature. After stirring overnight at room temperature, the reaction is stopped by evaporation of the reactants. Then the residue was dissolved in water (4 mL). Sodium hydoxide (27.8 mg, 695 µmol) was added to the reaction mixture. After 16 h of stirring at room temperature, the reaction was stopped with addition of cation exchange resin (DOWEX© 50WX8 resin, H+ form). The resin was removed by filtration through a millipore filter (45 µm). The aqueous solution was freeze dried. The resulting solid (51 mg) was solubilized in water (0.51 mL). To this solution was added absolute ethanol (5.1 mL). After 16 h at room temperature, the resulting solid was recovered and rinsed with absolute ethanol. It was solubilized in water and freeze dried to afford the phosphonate 29 as an orange solid (32.9 mg, 74 µmol, 85%). 1H NMR (500 MHz, D2O) δ 3.95 (d, J = 12.4 Hz, 1H, H-7), 3.78 (dd, J = 12.4, 5.1 Hz, 1H, H-7′), 3.77–3.71 (m, 1H, H-2), 3.67 (t, J = 9.3 Hz, 1H, H-3), 3.56 (t, J = 9.3 Hz, 1H, H-4), 3.50 (t, J = 9.3 Hz, 1H, H-5), 3.45 (dd, J = 9.3, 5.1 Hz, 1H, H-6), 2.27 (dt, J = 19.2, 15.7 Hz, 1H, 0.5 × PCH2P), 2.19–2.05 (m, 1H, 0.5 × PCH2P), 2.10 (s, 3H, NHCOCH3), 2.05–1.89 (m, 2H, H-1, H-1′); 13C NMR (126 MHz, D2O) δ 174.7 (NHCOCH3), 79.4 (C-6), 75.4 (C-4), 74.5 (d, J = 5.1 Hz, C-2), 69.9 (C-5), 60.8 (C-7), 56.5 (d, J = 13.2 Hz, C-3), 33.4 (d, J = 96.4 Hz, C-1), 32.0 (dd, J = 119.8, 79.3 Hz, PCH2PO), 22.3 (NHCOCH3); 31P NMR (202 MHz, D2O) δ 34.08 (d, J = 7.5 Hz, CH2PCH2P), 15.76 (d, J = 7.5 Hz, CH2PCH2P); 31P NMR non decoupled (202 MHz, D2O) δ 34.3–33.8 (m, CH2PCH2P), 15.74 (td, J = 19.4, 7.5 Hz, CH2PCH2P); ESI-MS (mobile phase gradient ACN/H2O + 0.1% formic acid): Calcd for C10H22NO10P2: 378.1 [M + H]+ Found: 378.1; ESI-HRMS: Calcd for C10H22NO10P2: 378.0719 [M + H]+ Found: 378.0709.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








