Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation




Abstract
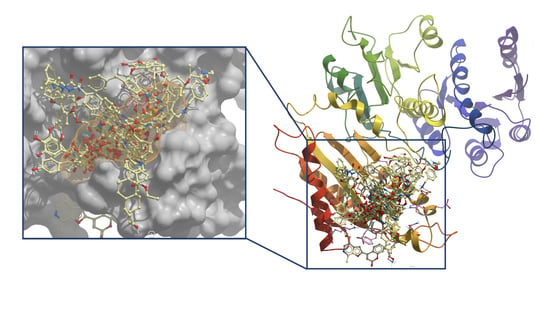
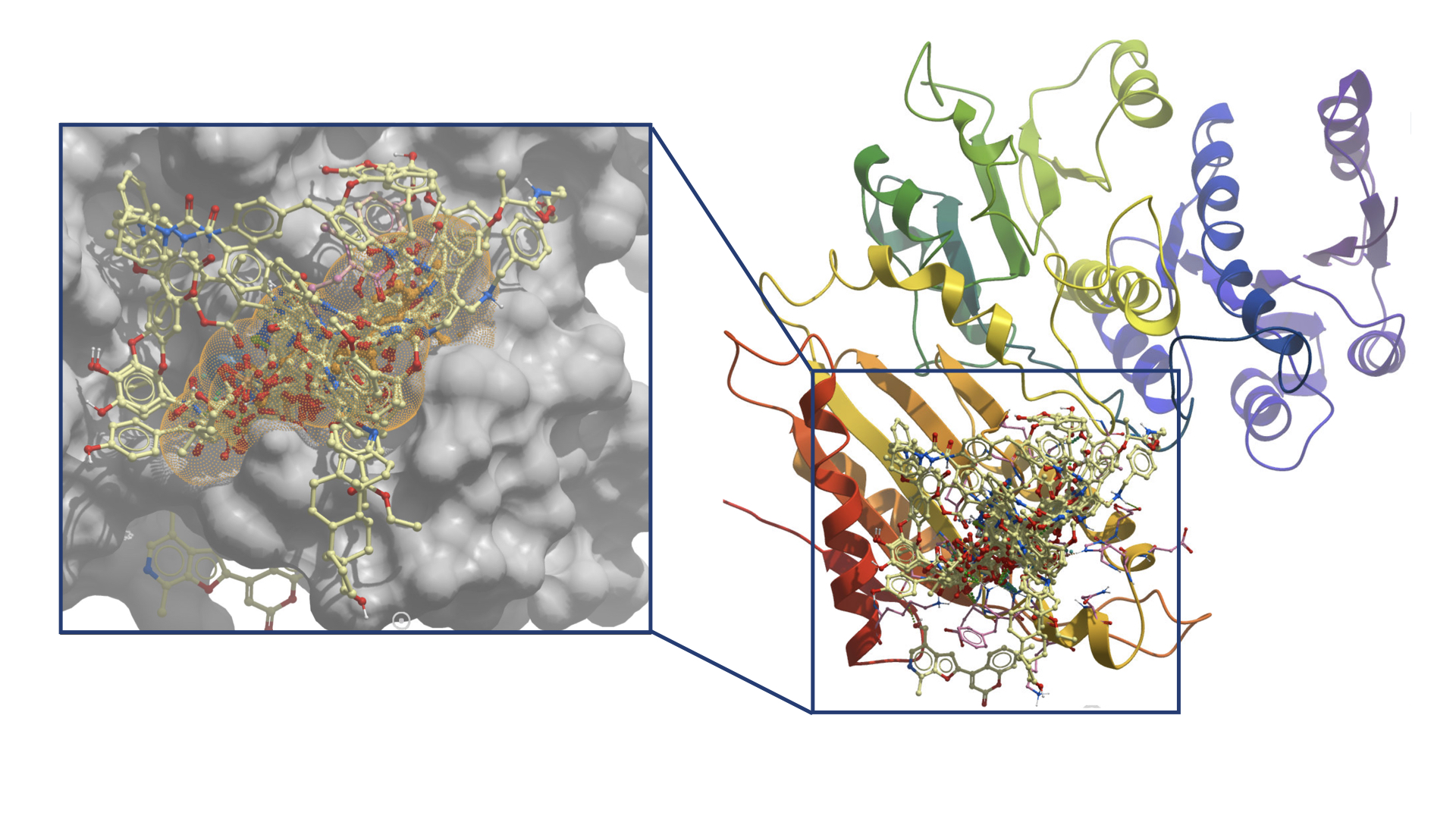
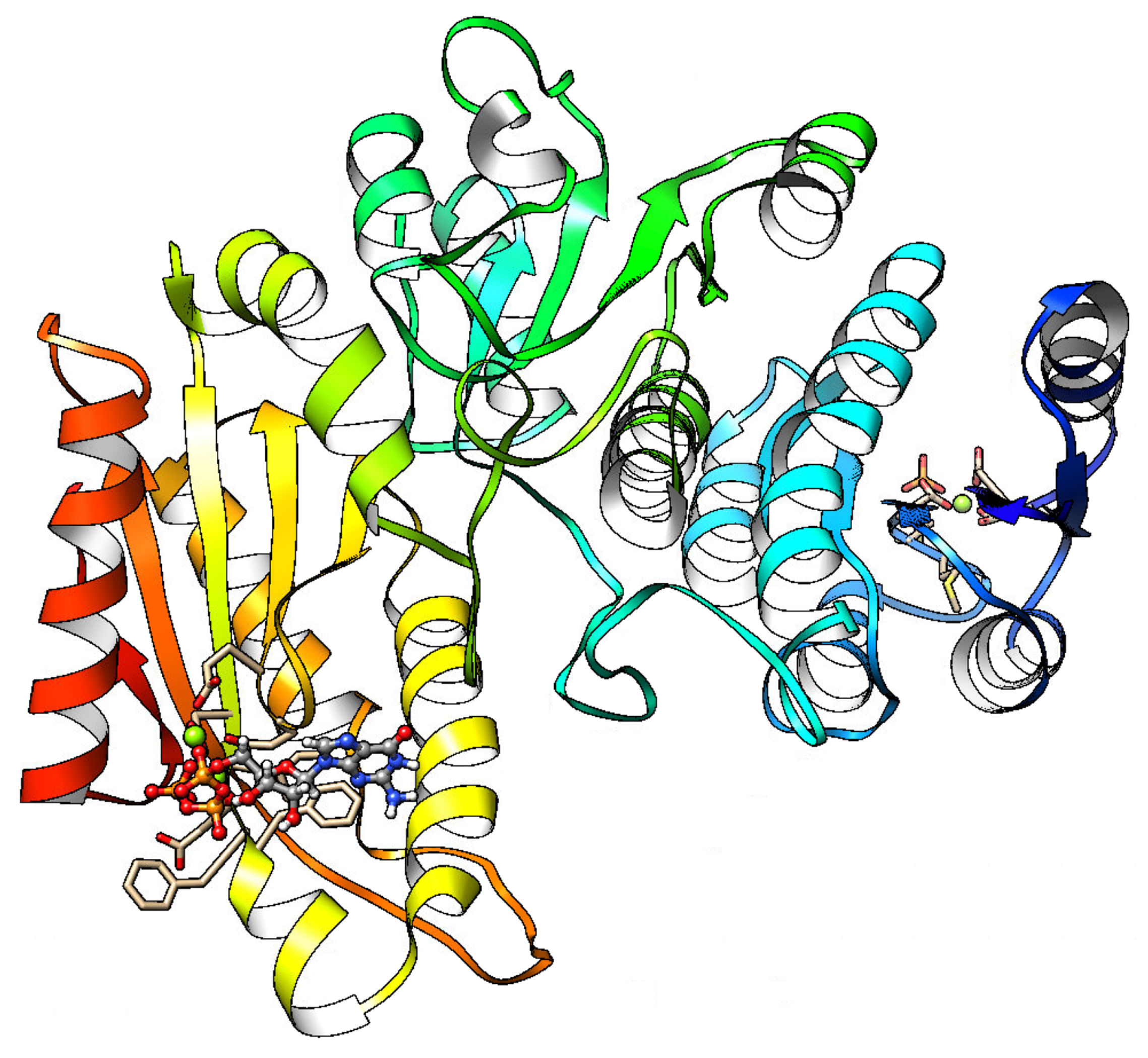
1. Introduction
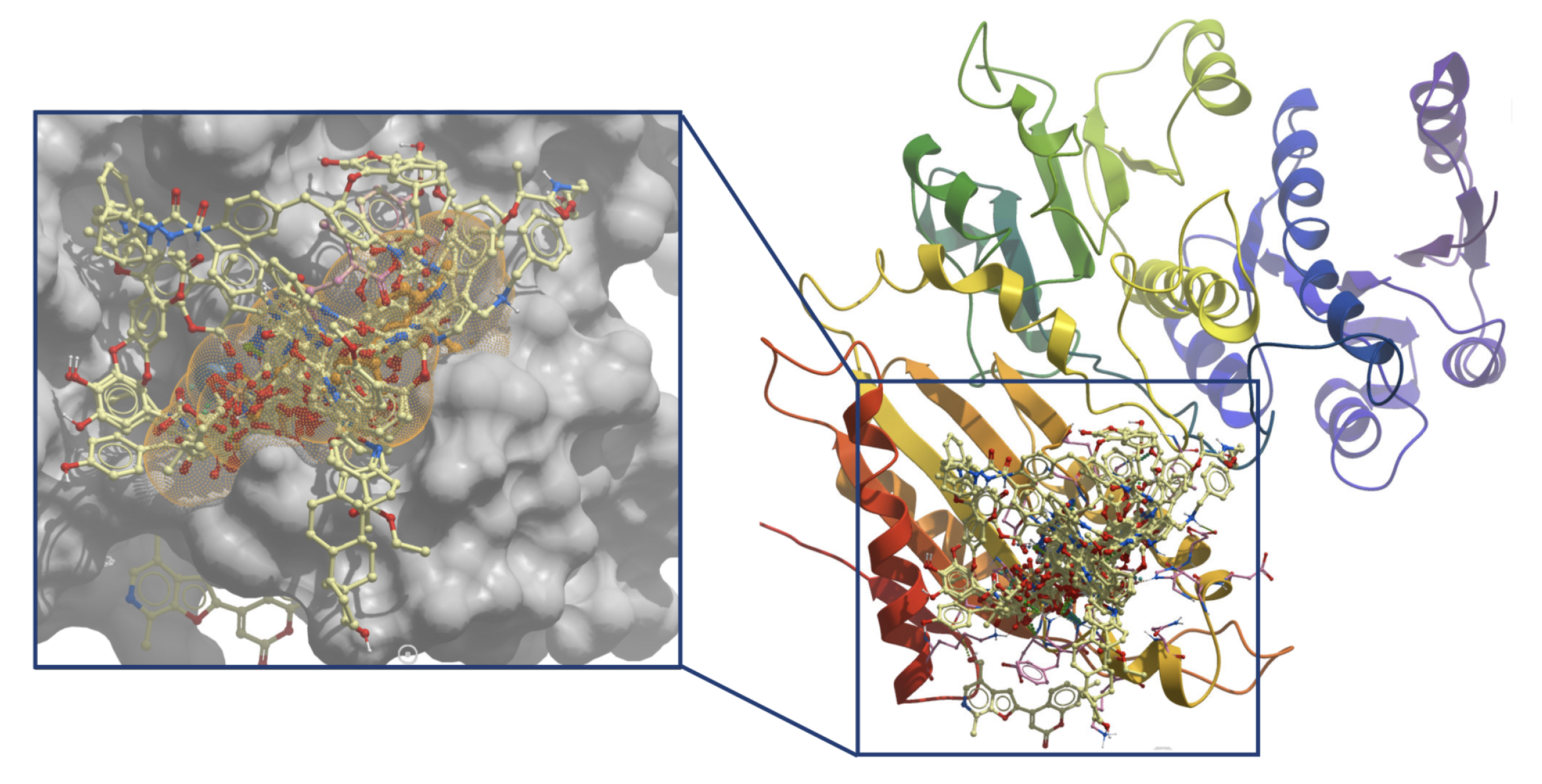
2. Results and Discussion
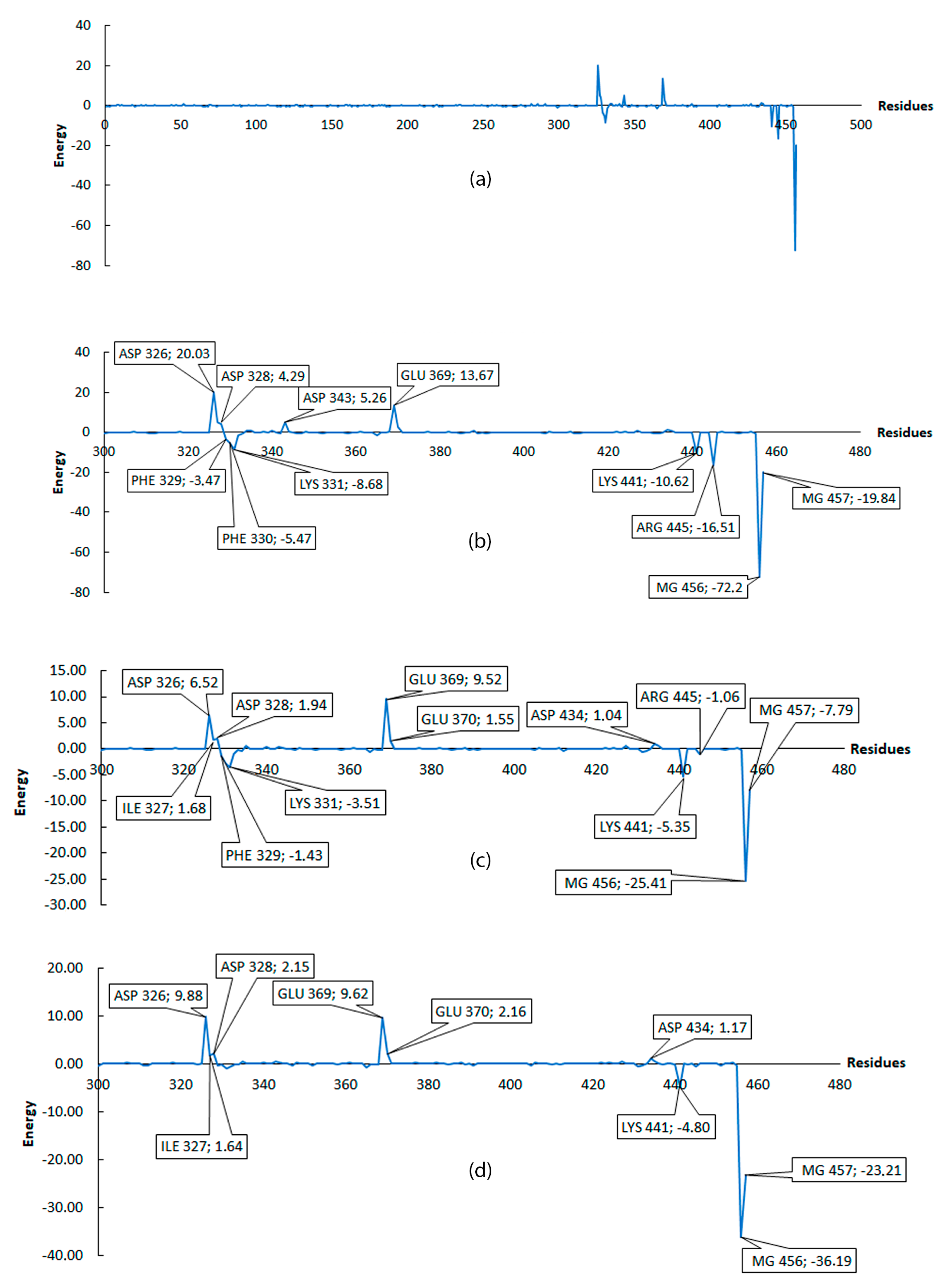
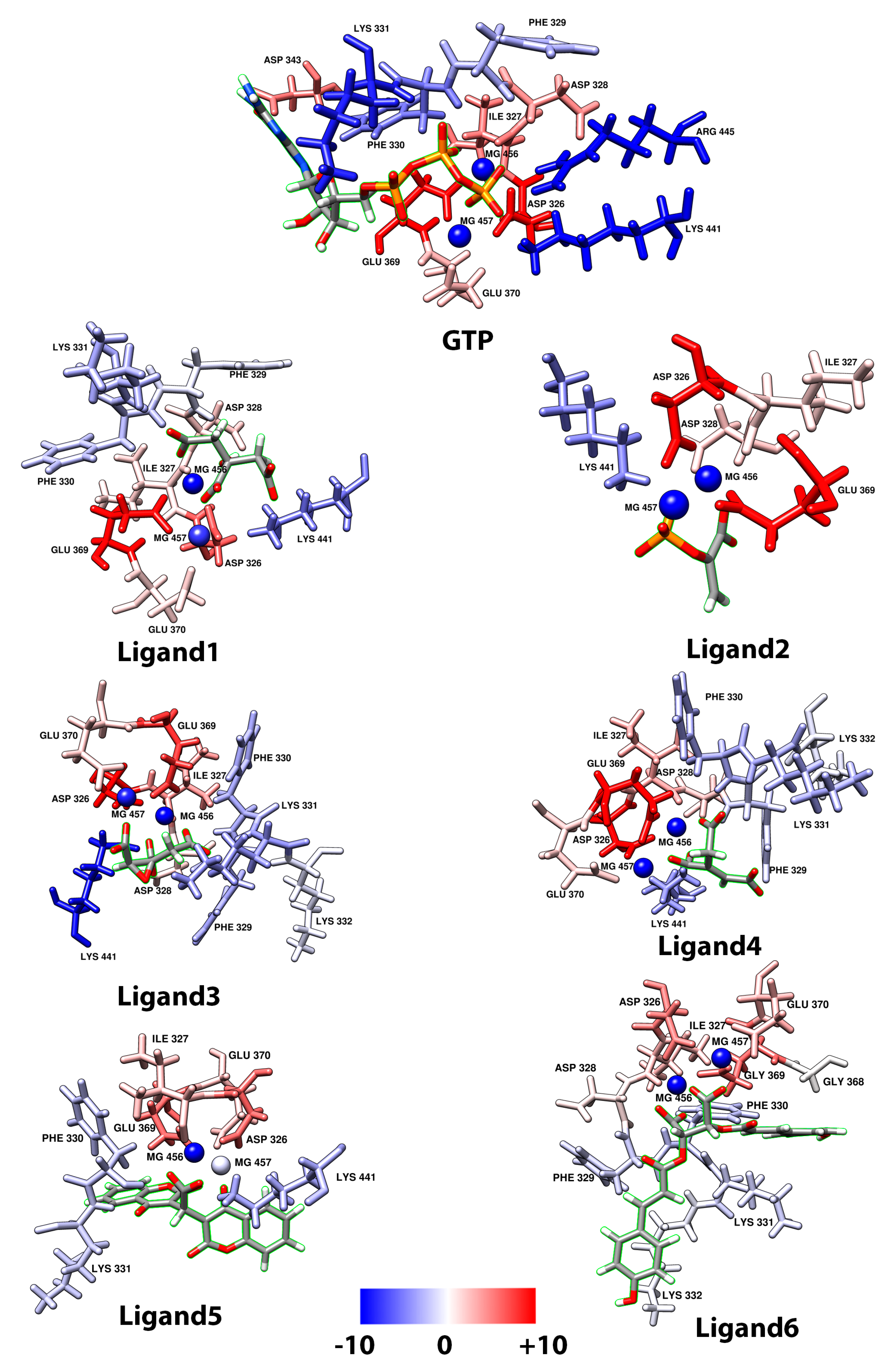
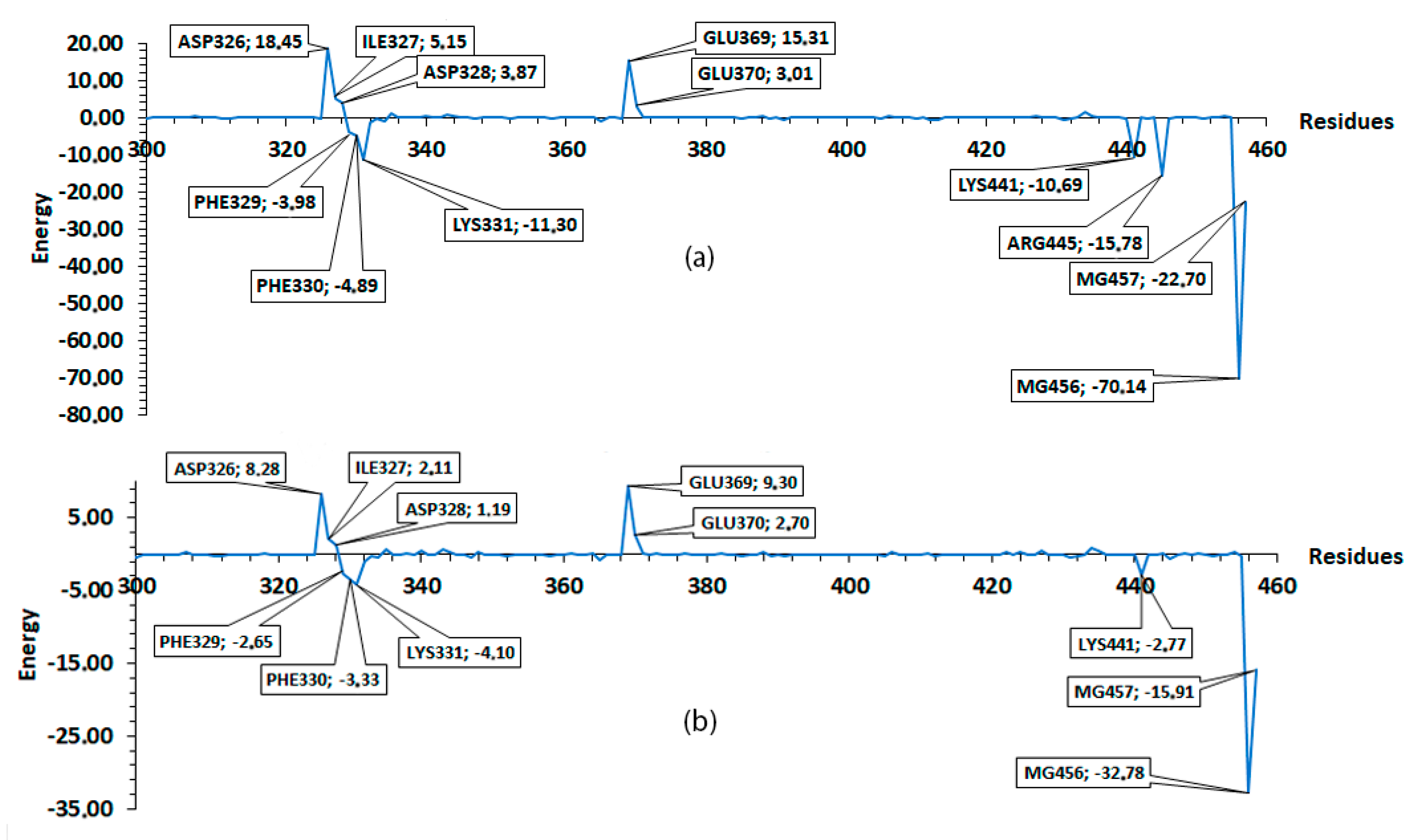
2.1. Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA)
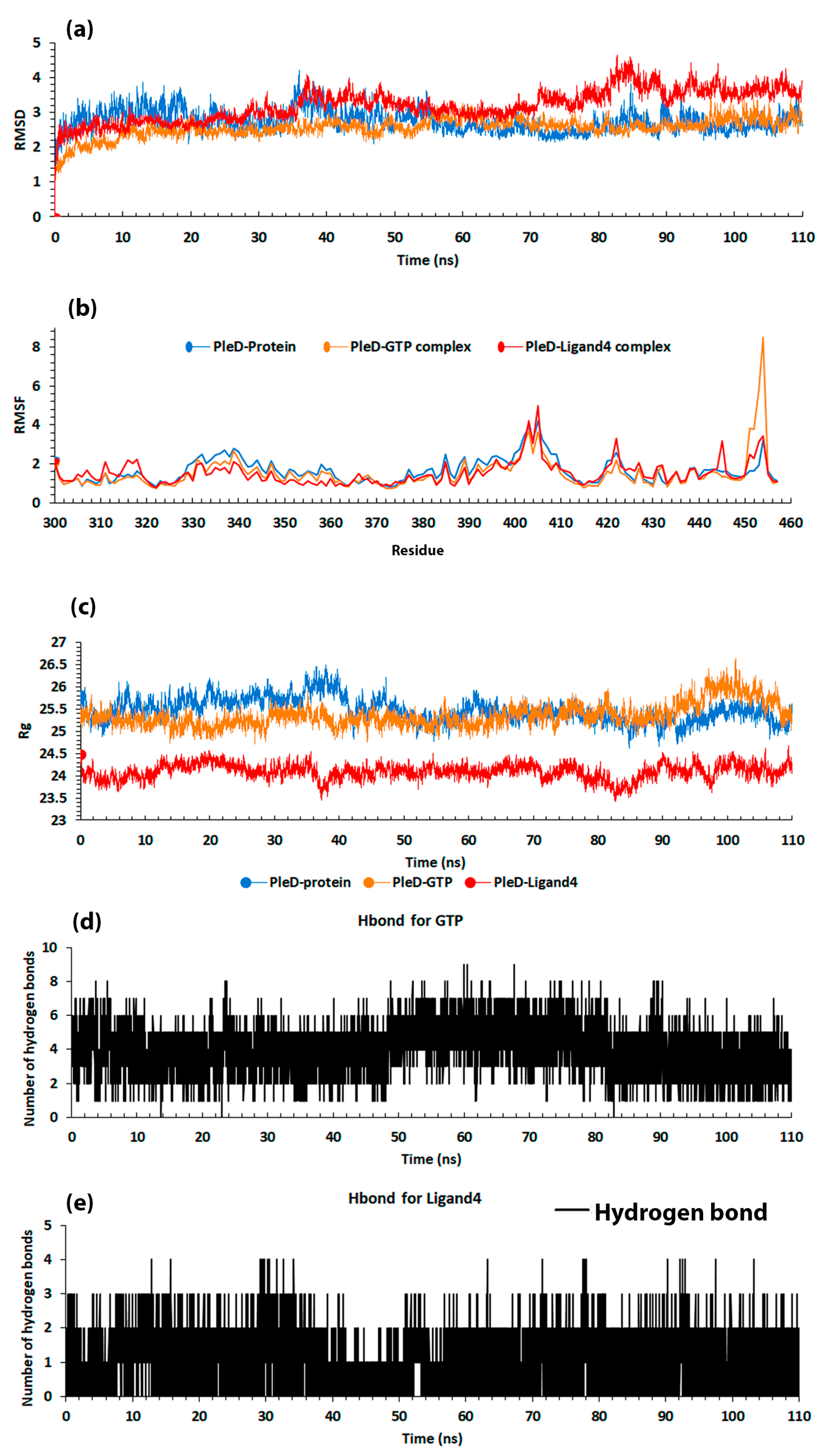
2.1.1. Root Mean Square Deviation (RMSD)
2.1.2. Hydrogen Bonds (H-Bonds)
2.2. Trans-Aconitic Acid (Ligand4)
2.2.1. Root Mean Square Deviation
2.2.2. Root Mean Square Fluctuation
2.2.3. Radius of Gyration
2.2.4. Hydrogen Bond (H-Bond)
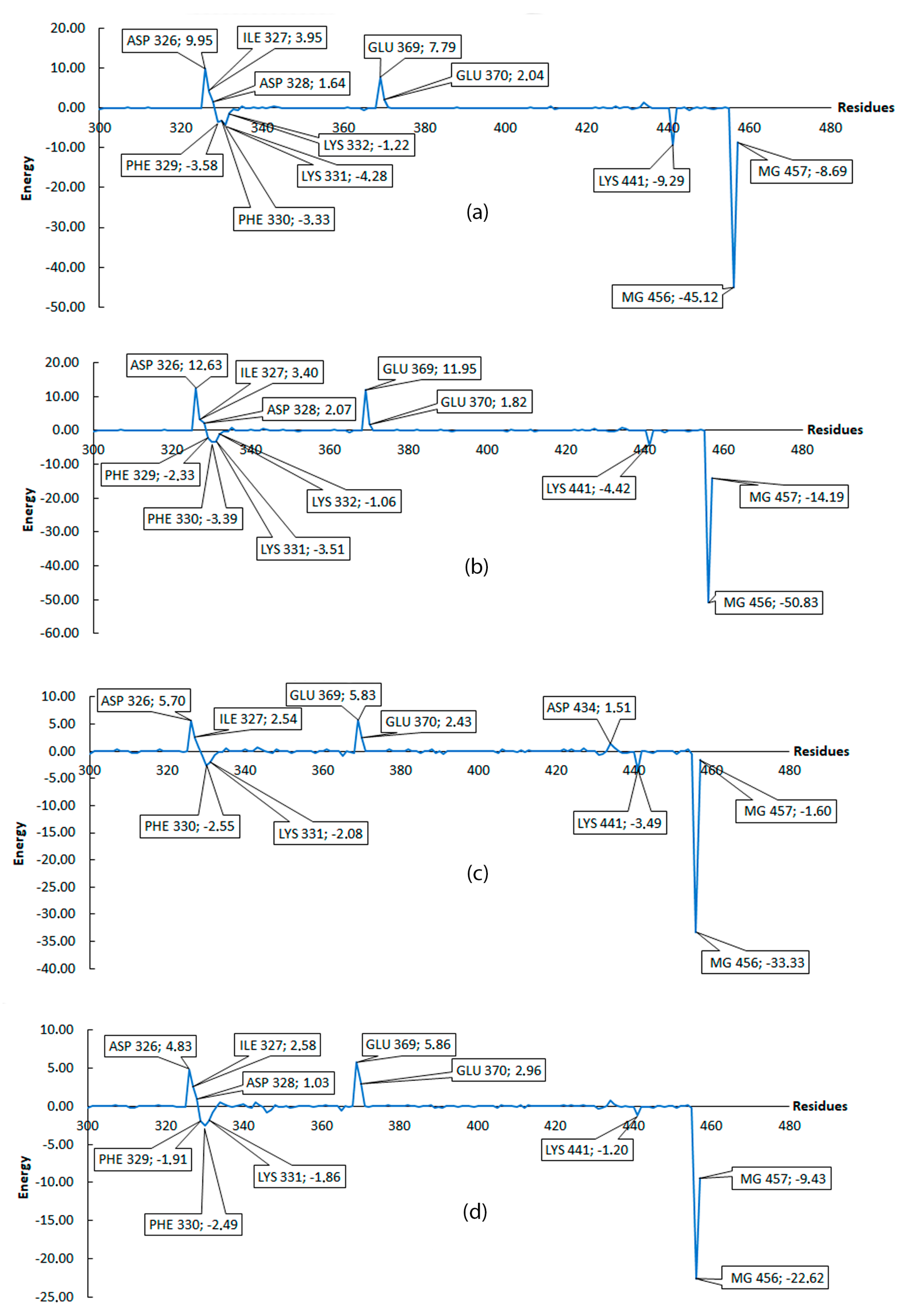
2.2.5. Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA)
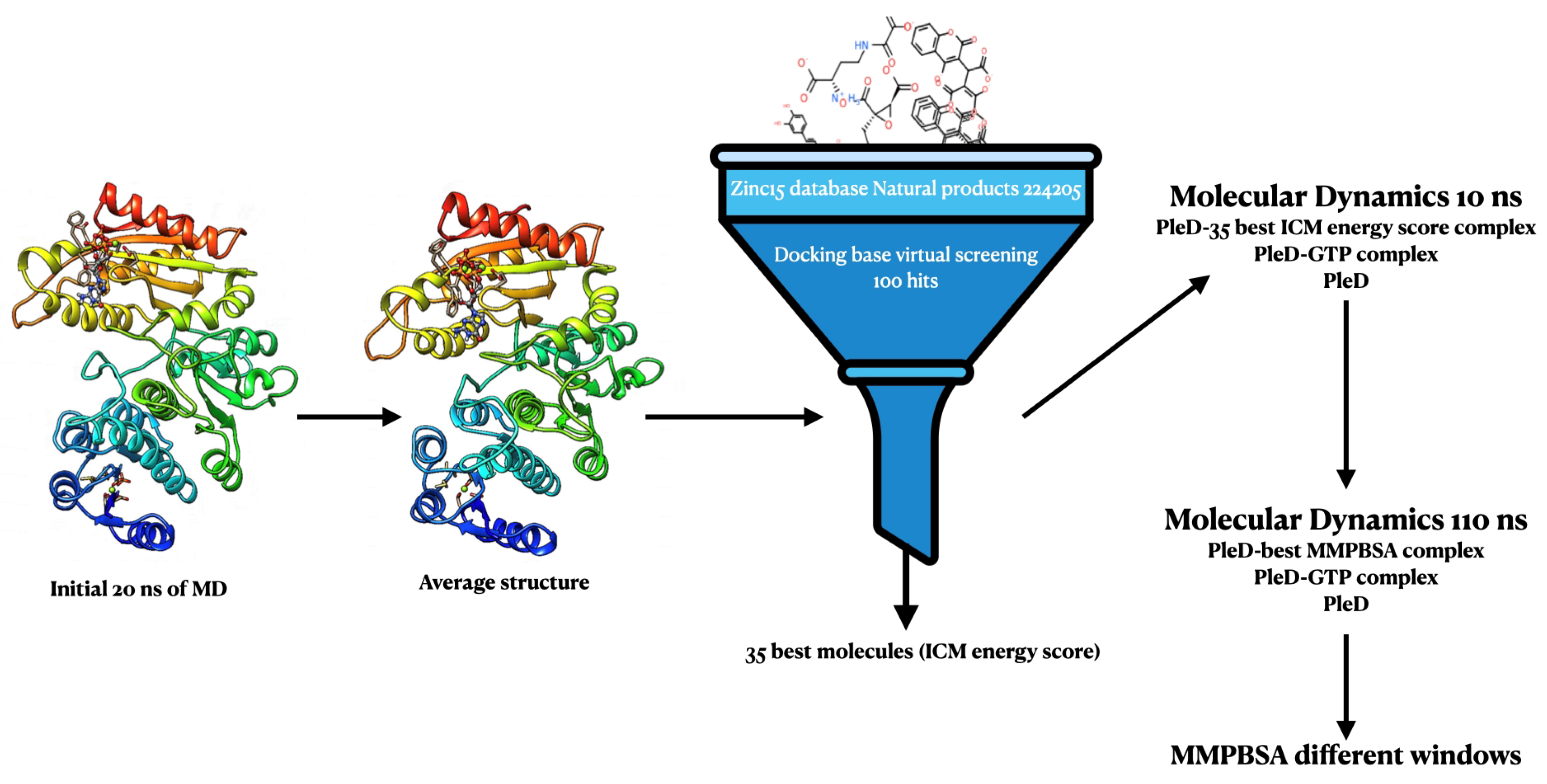
3. Materials and Methods
3.1. Data Collection
3.2. Virtual Screening
3.3. Ligand Binding Free Energy Calculations
3.4. System Preparation
3.5. Molecular Dynamics
3.6. Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) Calculation
3.7. RMSD, RMSF, Radius of Gyration, and H-Bond
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EPS | Extracellular polymeric substances |
| c-di-GMP | Cyclic diguanylate |
| DGC | Diguanylate cyclases |
| PDEs | Phosphodiesterases |
| GTP | Guanosine-5’-triphosphate |
| TTA | trans-aconitic acid |
| VS | virtual screening |
| MD | molecular dynamics |
| MM/PBSA | Molecular Mechanics Poisson-Boltzmann Surface Area |
| MMFF | Merck Molecular Force Field |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| Rg | Radius of gyration |
| H-bond | Hydrogens Bond |
References
- Flemming, H.C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Yin, W.; Wang, Y.; Liu, L.; He, J. Biofilms: The Microbial “Protective Clothing” in Extreme Environments. Int. J. Mol. Sci. 2019, 20, 3423. [Google Scholar] [CrossRef]
- Nadell, C.D.; Drescher, K.; Foster, K.R. Spatial structure, cooperation and competition in biofilms. Nat. Rev. Microbiol. 2016, 14, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo, J.L. Biofilm-related disease. Expert Rev. Anti-Infect. Ther. 2018, 16, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.; Rumbaugh, K.P. Approaches to Dispersing Medical Biofilms. Microorganisms 2017, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Fernicola, S.; Paiardini, A.; Giardina, G.; Rampioni, G.; Leoni, L.; Cutruzzolà, F.; Rinaldo, S. In Silico Discovery and In Vitro Validation of Catechol-Containing Sulfonohydrazide Compounds as Potent Inhibitors of the Diguanylate Cyclase PleD. J. Bacteriol. 2016, 198, 147–156. [Google Scholar] [CrossRef]
- Cai, Y.M.; Hutchin, A.; Craddock, J.; Walsh, M.A.; Webb, J.S.; Tews, I. Differential impact on motility and biofilm dispersal of closely related phosphodiesterases in Pseudomonas aeruginosa. Sci. Rep. 2020, 10, 6232. [Google Scholar] [CrossRef]
- Seshasayee, A.S.; Fraser, G.M.; Luscombe, N.M. Comparative genomics of cyclic-di-GMP signalling in bacteria: Post-translational regulation and catalytic activity. Nucleic Acids Res. 2010, 38, 5970–5981. [Google Scholar] [CrossRef]
- Galperin, M.Y. A census of membrane-bound and intracellular signal transduction proteins in bacteria: Bacterial IQ, extroverts and introverts. BMC Microbiol. 2005, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Römling, U.; Galperin, M.Y.; Gomelsky, M. Cyclic di-GMP: The First 25 Years of a Universal Bacterial Second Messenger. Microbiol. Mol. Biol. Rev. 2013, 77, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Feirer, N.; Kim, D.; Xu, J.; Fernandez, N.; Waters, C.M.; Fuqua, C. The Agrobacterium tumefaciens CheY-like protein ClaR regulates biofilm formation. Microbiology 2017, 163, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Alviz-Gazitua, P.; Fuentes-Alburquenque, S.; Rojas, L.A.; Turner, R.J.; Guiliani, N.; Seeger, M. The Response of Cupriavidus metallidurans CH34 to Cadmium Involves Inhibition of the Initiation of Biofilm Formation, Decrease in Intracellular c-di-GMP Levels, and a Novel Metal Regulated Phosphodiesterase. Front. Microbiol. 2019, 10, 1499. [Google Scholar] [CrossRef]
- Jenal, U.; Malone, J. Mechanisms of cyclic-di-GMP signaling in bacteria. Annu. Rev. Genet. 2006, 40, 385–407. [Google Scholar] [CrossRef]
- Paul, R.; Weiser, S.; Amiot, N.C.; Chan, C.; Schirmer, T.; Giese, B.; Jenal, U. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 2004, 18, 715–727. [Google Scholar] [CrossRef]
- Skerker, J.M.; Laub, M.T. Cell-cycle progression and the generation of asymmetry in Caulobacter crescentus. Nat. Rev. Microbiol. 2004, 2, 325–337. [Google Scholar] [CrossRef]
- Entcheva-Dimitrov, P.; Spormann, A.M. Dynamics and Control of Biofilms of the Oligotrophic Bacterium Caulobacter crescentus. J. Bacteriol. 2004, 186, 8254–8266. [Google Scholar] [CrossRef]
- Valentini, M.; Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef]
- Lage, O.M.; Ramos, M.C.; Calisto, R.; Almeida, E.; Vasconcelos, V.; Vicente, F. Current screening methodologies in drug discovery for selected human diseases. Mar. Drugs 2018, 16, 279. [Google Scholar] [CrossRef]
- Rossiter, S.E.; Fletcher, M.H.; Wuest, W.M. Natural Products as Platforms to Overcome Antibiotic Resistance. Chem. Rev. 2017, 117, 12415–12474. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Fayad, A.A.; Müller, R. Natural products from myxobacteria: Novel metabolites and bioactivities. Nat. Prod. Rep. 2017, 34, 135–160. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Nofiani, R.; Weisberg, A.J.; Tsunoda, T.; Panjaitan, R.G.P.; Brilliantoro, R.; Chang, J.H.; Philmus, B.; Mahmud, T. Antibacterial Potential of Secondary Metabolites from Indonesian Marine Bacterial Symbionts. Int. J. Microbiol. 2020, 2020, 8898631. [Google Scholar] [CrossRef] [PubMed]
- Emiru, Y.K.; Siraj, E.A.; Teklehaimanot, T.T.; Amare, G.G. Antibacterial Potential of Aloe weloensis (Aloeacea) Leaf Latex against Gram-Positive and Gram-Negative Bacteria Strains. Int. J. Microbiol. 2019, 2019, 5328238. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Burton, G.J.; Hecht, G.B.; Newton, A. Roles of the histidine protein kinase pleC in Caulobacter crescentus motility and chemotaxis. J. Bacteriol. 1997, 179, 5849–5853. [Google Scholar] [CrossRef]
- Aldridge, P.; Paul, R.; Goymer, P.; Rainey, P.; Jenal, U. Role of the GGDEF regulator PleD in polar development of Caulobacter crescentus. Mol. Microbiol. 2003, 47, 1695–1708. [Google Scholar] [CrossRef]
- Aldridge, P.; Jenal, U. Cell cycle-dependent degradation of a flagellar motor component requires a novel-type response regulator. Mol. Microbiol. 1999, 32, 379–391. [Google Scholar] [CrossRef]
- Jenal, U. Cyclic di-guanosine-monophosphate comes of age: A novel secondary messenger involved in modulating cell surface structures in bacteria? Curr. Opin. Microbiol. 2004, 7, 185–191. [Google Scholar] [CrossRef]
- Tamayo, R.; Pratt, J.T.; Camilli, A. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu. Rev. Microbiol. 2007, 61, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Yosa Reyes, J.; Nagy, T.; Meuwly, M. Competitive reaction pathways in vibrationally induced photodissociation of H2SO4. Phys. Chem. Chem. Phys. 2014, 16, 18533–18544. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, P.; Chan, C.; Paul, R.; Beck, A.; Heerklotz, H.; Jenal, U.; Schirmer, T. Structure of BeF3−-Modified Response Regulator PleD: Implications for Diguanylate Cyclase Activation, Catalysis, and Feedback Inhibition. Structure 2007, 15, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, U.T.; Nageswara Rao, G.V.S.; Mohan, K.M.; Ramanaviciene, A.; Ramanavicius, A. Antibacterial and antifungal activity of silver nanospheres synthesized by tri-sodium citrate assisted chemical approach. Vacuum 2017, 146, 259–265. [Google Scholar] [CrossRef]
- Choudhury, R.; Majumdar, M.; Biswas, P.; Khan, S.; Misra, T.K. Kinetic study of functionalization of citrate stabilized silver nanoparticles with catechol and its anti-biofilm activity. Nano-Struct. Nano-Objects 2019, 19, 100326. [Google Scholar] [CrossRef]
- Du, C.; Cao, S.; Shi, X.; Nie, X.; Zheng, J.; Deng, Y.; Ruan, L.; Peng, D.; Sun, M. Genetic and Biochemical Characterization of a Gene Operon for trans-Aconitic Acid, a Novel Nematicide from Bacillus thuringiensis. J. Biol. Chem. 2017, 292, 3517–3530. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Holst, M.J.; McCammon, J.A. The adaptive multilevel finite element solution of the Poisson-Boltzmann equation on massively parallel computers. IBM J. Res. Dev. 2001, 45, 427–438. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Konecny, R.B.; McCammon, N.A.; Andrew, J. iAPBS: A programming interface to the adaptive Poisson-Boltzmann solver. Comput. Sci. Discov. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Nittinger, E.; Inhester, T.; Bietz, S.; Meyder, A.; Schomburg, K.T.; Lange, G.; Klein, R.; Rarey, M. Large-Scale Analysis of Hydrogen Bond Interaction Patterns in Protein–Ligand Interfaces. J. Med. Chem. 2017, 60, 4245–4257. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Yuhara, K.; Yonehara, H.; Hattori, T.; Kobayashi, K.; Kirimura, K. Enzymatic characterization and gene identification of aconitate isomerase, an enzyme involved in assimilation of trans-aconitic acid, from Pseudomonas sp. WU-0701. FEBS J. 2015, 282, 4257–4267. [Google Scholar] [CrossRef]
- Bortolo, T.d.S.C.; Marchiosi, R.; Viganó, J.; Siqueira-Soares, R.d.C.; Ferro, A.P.; Barreto, G.E.; Bido, G.d.S.; Abrahão, J.; dos Santos, W.D.; Ferrarese-Filho, O. Trans-aconitic acid inhibits the growth and photosynthesis of Glycine max. Plant Physiol. Biochem. 2018, 132, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, M.; Petereit, F.; Nahrstedt, A. Trans-Aconitic acid, glucosylflavones and hydroxycinnamoyltartaric acids from the leaves of Echinodorus grandiflorus ssp. aureus, a Brazilian medicinal plant. Rev. Bras. Farmacogn. 2007, 17, 149–154. [Google Scholar] [CrossRef]
- Kanitkar, A.; Aita, G.; Madsen, L. The recovery of polymerization grade aconitic acid from sugarcane molasses. J. Chem. Technol. Biotechnol. 2013, 88, 2188–2192. [Google Scholar] [CrossRef]
- De Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P., Jr. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, 8, 93. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Hevener, K.E.; Pesavento, R.; Ren, J.; Lee, H.; Ratia, K.; Johnson, M.E. Chapter Twelve—Hit-to-Lead: Hit Validation and Assessment. In Modern Approaches in Drug Discovery; Methods in Enzymology; Lesburg, C.A., Ed.; Academic Press: New York, NY, USA, 2018; Volume 610, pp. 265–309. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased Probability Monte Carlo Conformational Searches and Electrostatic Calculations for Peptides and Proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Rapid boundary element solvation electrostatics calculations in folding simulations: Successful folding of a 23-residue peptide. Pept. Sci. 2001, 60, 124–133. [Google Scholar] [CrossRef]
- An, J.; Totrov, M.; Abagyan, R. Pocketome via Comprehensive Identification and Classification of Ligand Binding Envelopes. Mol. Cell. Proteom. 2005, 4, 752–761. [Google Scholar] [CrossRef]
- Fernandez-Recio, J.; Totrov, M.; Skorodumov, C.; Abagyan, R. Optimal docking area: A new method for predicting protein–protein interaction sites. PROTEINS Struct. Funct. Bioinform. 2005, 58, 134–143. [Google Scholar] [CrossRef]
- Fernandez-Recio, J.; Totrov, M.; Abagyan, R. ICM-DISCO docking by global energy optimization with fully flexible side-chains. PROTEINS Struct. Funct. Bioinform. 2003, 52, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Méndez, R.; Leplae, R.; Lensink, M.F.; Wodak, S.J. Assessment of CAPRI predictions in rounds 3—5 shows progress in docking procedures. PROTEINS Struct. Funct. Bioinform. 2005, 60, 150–169. [Google Scholar] [CrossRef]
- Méndez, R.; Leplae, R.; De Maria, L.; Wodak, S.J. Assessment of blind predictions of protein—protein interactions: Current status of docking methods. PROTEINS Struct. Funct. Bioinform. 2003, 52, 51–67. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Su, P.C.; Tsai, C.C.; Mehboob, S.; Hevener, K.E.; Johnson, M.E. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (FabI). J. Comput. Chem. 2015, 36, 1859–1873. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff 14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff 99SB. J. Chem. Theory Comput. 2015, 18, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Izadi, S. Water models for biomolecular simulations. WIREs Comput. Mol. Sci. 2018, 8, e1347. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Ben-Shalom, I.Y.; Pfeiffer-Marek, S.; Baringhaus, K.H.; Gohlke, H. Efficient Approximation of Ligand Rotational and Translational Entropy Changes upon Binding for Use in MM-PBSA Calculations. J. Chem. Inf. Model. 2017, 57, 170–189. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate Ligand-Binding Energies. J. Chem. Theory Comput. 2011, 7, 3768–3778. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
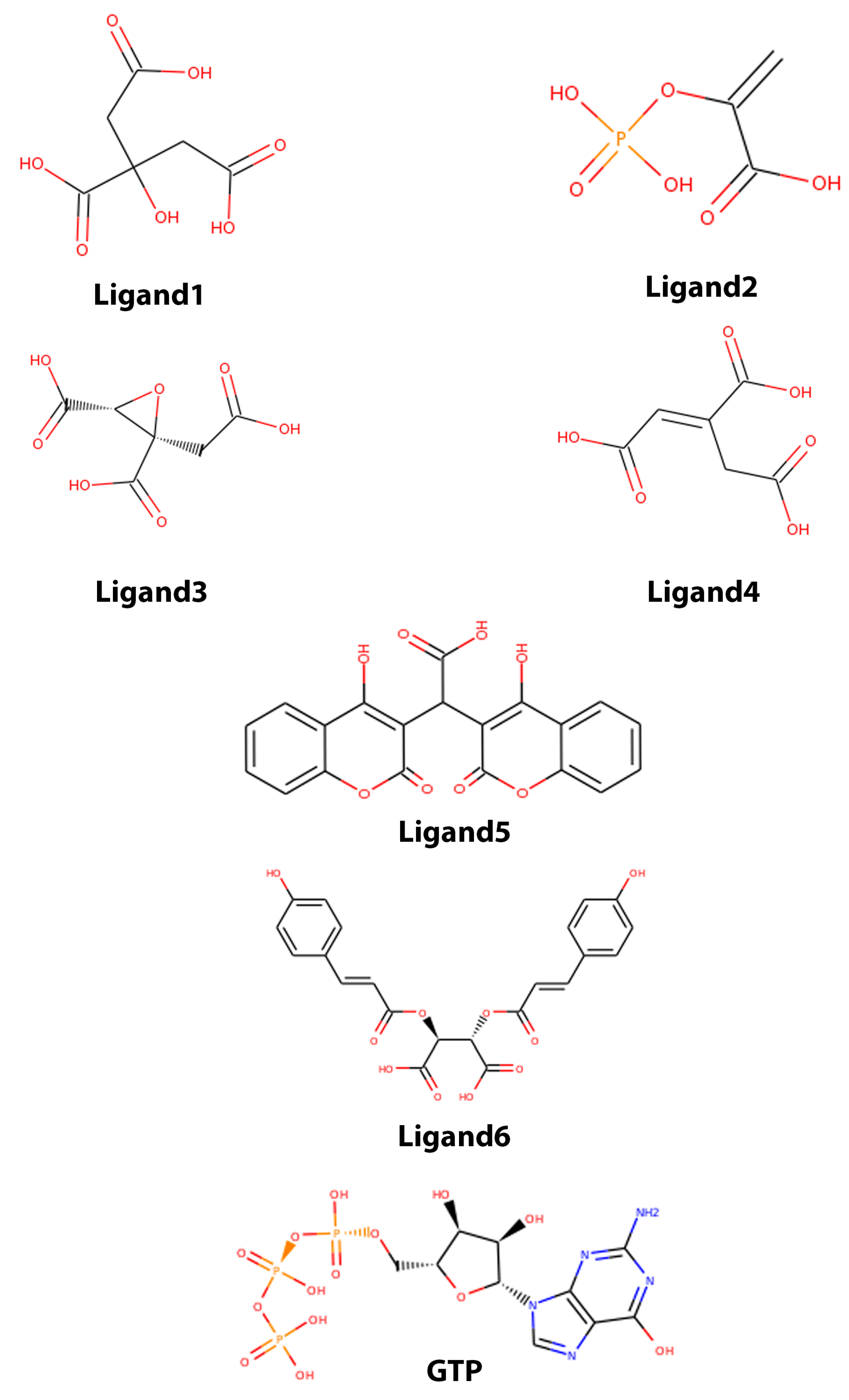
| Zinc15 ID | Name in this Work | Common Name |
|---|---|---|
| ZINC00895081 | Ligand1 | Citrate |
| ZINC03870145 | Ligand2 | Phosphoenolpyruvic acid |
| ZINC04028701 | Ligand3 | 3-carboxy-2-(carboxymethyl)oxirane-2-carboxylate |
| ZINC04501392 | Ligand4 | trans-Aconitic acid |
| ZINC19336068 | Ligand5 | bis(4-hydroxy-2-oxo-2H-chromen-3-yl)acetic acid |
| ZINC27558828 | Ligand6 | 2,3-Bis[(2E)-3-(4-hydroxyphenyl)-2-propenoyl]oxysuccinic acid |
| Ligand | VDWAALS | EEL | EPB | ENPOLAR | ΔG |
|---|---|---|---|---|---|
| Ligand1 | 2.92 ± 4.30 | −867.68 ± 21.67 | 766.94 ± 18.20 | −1.64 ± 0.06 | −99.46 ± 5.67 |
| Ligand2 | 19.03 ± 4.87 | −736.68 ± 24.60 | 615.55 ± 21.83 | −1.22 ± 0.06 | −103.32 ± 4.81 |
| Ligand3 | 8.72 ± 5.14 | −776.77 ± 38.28 | 650.15 ± 35.30 | −1.67 ± 0.06 | −119.58 ± 7.11 |
| Ligand4 | 7.86 ± 4.83 | −807.77 ± 33.78 | 680.01 ± 29.12 | −1.43 ± 0.09 | −121.33 ± 9.13 |
| Ligand5 | −2.20 ± 5.73 | −686.56 ± 28.76 | 583.50 ± 27.10 | −2.67 ± 0.13 | −107.93 ± 8.92 |
| Ligand6 | −20.27 ± 4.39 | −533.33 ± 19.94 | 456.46 ± 16.33 | −3.84 ± 0.11 | −100.98 ± 7.20 |
| GTP | −10.86 ± 6.45 | −1287.03 ± 35.53 | 1123.46 ± 29.55 | 3.49 ± 0.17 | −178.09 ± 10.82 |
| Ligand | RMSD | Difference between GTP and Ligand |
|---|---|---|
| Ligand1 | 2.261 ± 0.365 | 0.284 |
| Ligand2 | 2.189 ± 0.336 | 0.212 |
| Ligand3 | 2.165 ± 0.411 | 0.188 |
| Ligand4 | 2.480 ± 0.255 | 0.503 |
| Ligand5 | 2.506 ± 0.350 | 0.529 |
| Ligand6 | 2.305 ± 0.389 | 0.328 |
| GTP | 1.977 ± 0.289 | 0 |
| Ligand | Residues Involve in the H-Bond Formation with the Major Contribution |
|---|---|
| Ligand1 | LYS441 (54.20%), LYS331 (35.88%), PHE330 (7.11%), PHE329 (2.80%) |
| Ligand2 | LYS441 (85.28%), PHE329 (9.37%), PHE330 (4.97%) |
| Ligand3 | LYS441 (38.70%), PHE329 (24.17%), PHE330 (19.74%), LYS331 (17.39%) |
| Ligand4 | LYS441 (52.68%), LYS331 (24.05%), PHE330 (19.28%), PHE329 (3.99%) |
| Ligand5 | LYS441 (54.31%), PHE330 (35.45%), LYS331(9.31%) |
| Ligand6 | LYS441 (36.13%), LYS331 (23.48%), PHE330 (17.81%), ASN334 (17.03%), LYS332 (4.77%) |
| GTP | ARG445 (39.05%), LYS331 (34.63%), LYS441 (17.35%), PHE330 (8.12%) |
| Ligand | Residues Involved in the H-Bond Formation with the Major Contribution |
|---|---|
| PleD–Ligand4 | PHE330 (9.7%), LYS331 (6.01%), LYS441 (5.69%) |
| PleD–GTP | ARG445 (41.44%), LYS331 (15.14%), LYS441 (12.07%), PHE330 (11.95%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pestana-Nobles, R.; Leyva-Rojas, J.A.; Yosa, J. Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation. Molecules 2020, 25, 5334. https://doi.org/10.3390/molecules25225334
Pestana-Nobles R, Leyva-Rojas JA, Yosa J. Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation. Molecules. 2020; 25(22):5334. https://doi.org/10.3390/molecules25225334
Chicago/Turabian StylePestana-Nobles, Roberto, Jorge A. Leyva-Rojas, and Juvenal Yosa. 2020. "Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation" Molecules 25, no. 22: 5334. https://doi.org/10.3390/molecules25225334
APA StylePestana-Nobles, R., Leyva-Rojas, J. A., & Yosa, J. (2020). Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation. Molecules, 25(22), 5334. https://doi.org/10.3390/molecules25225334