
Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates



Abstract
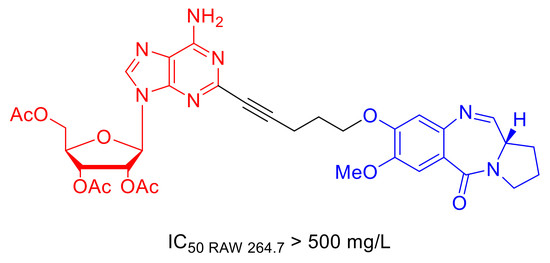
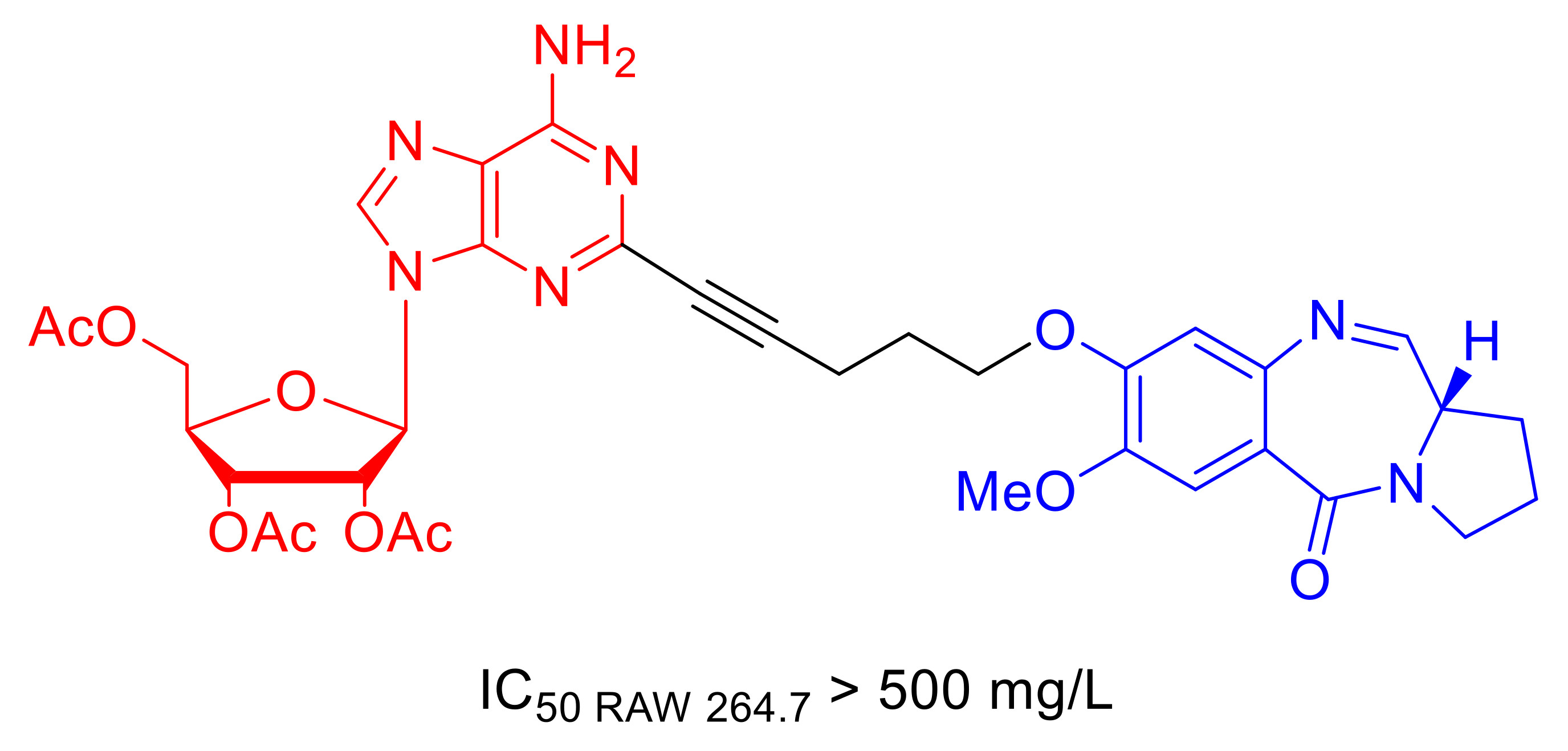
1. Introduction
2. Chemical and Biological Experiments
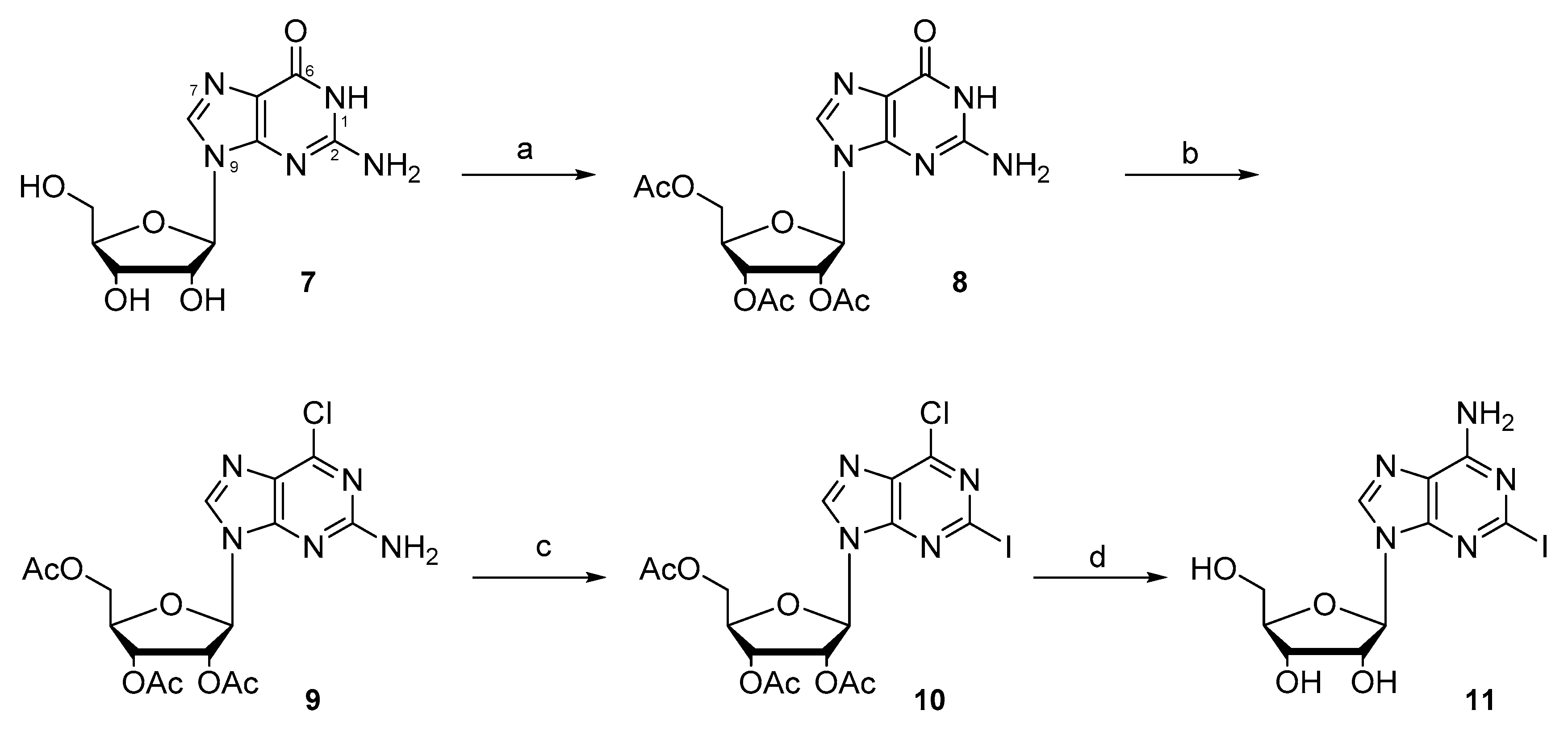
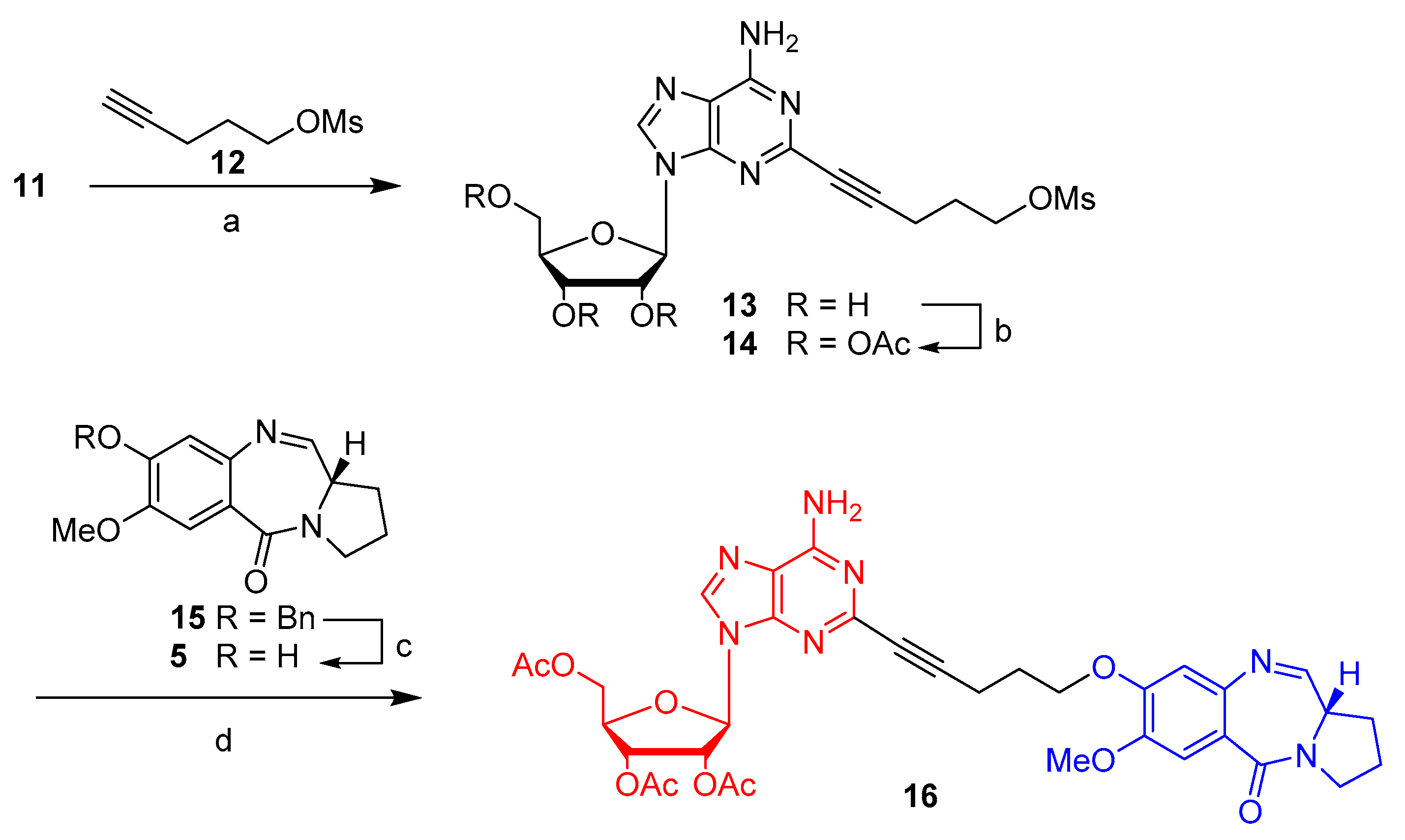
2.1. Synthesis
2.2. Cytotoxicity Vs. Antimycobacterial Activity
2.3. Footprinting Studies
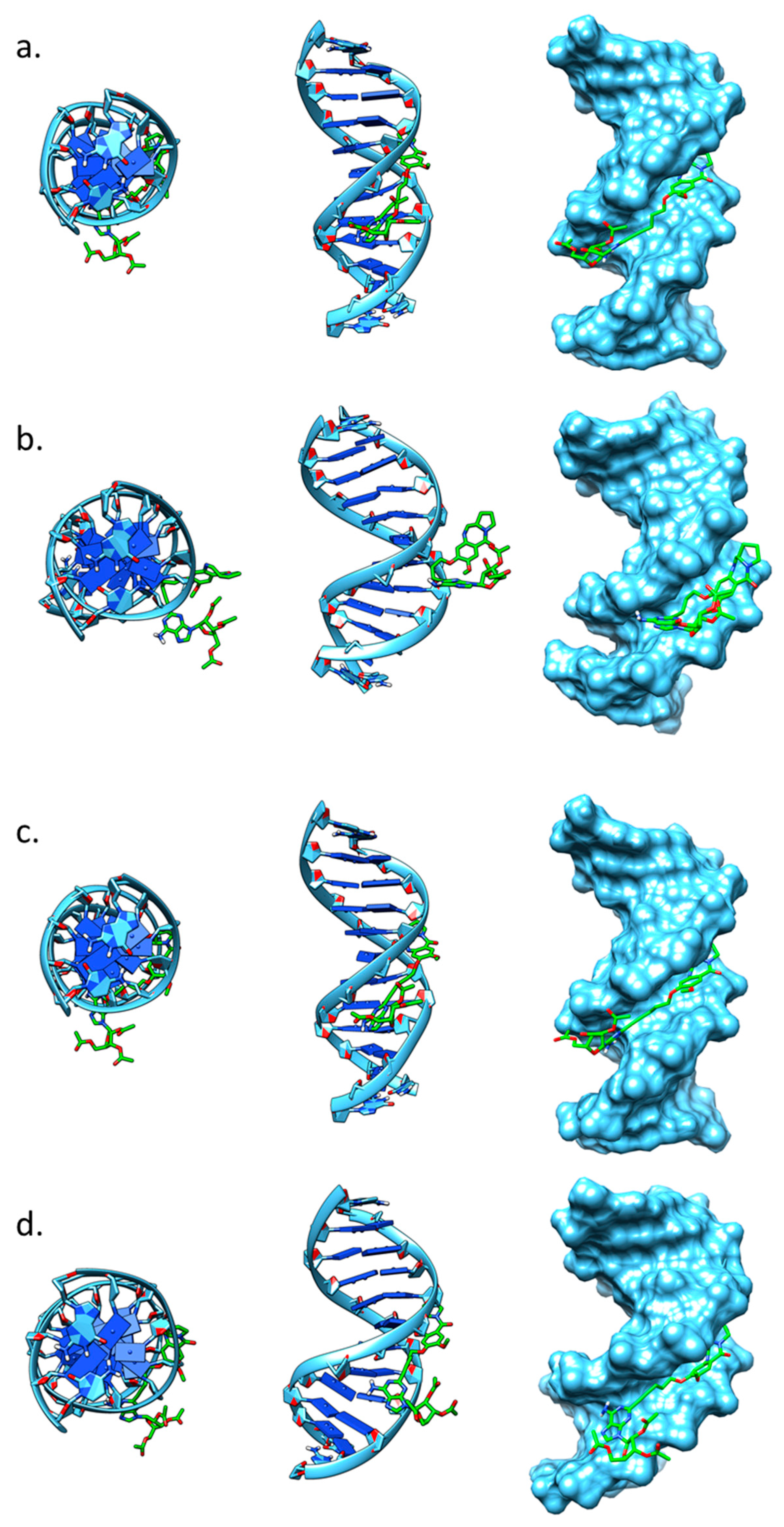
2.4. Molecular Modelling
3. Conclusions
4. Materials and Methods
4.1. General Chemistry Reactions Information
4.2. DNAse I Footprinting Assay
4.3. Microbiology and Cytotoxicity Experiments
4.3.1. Mycobacteria
4.3.2. Preparation of Microbiological Media
4.3.3. Growth, Maintenance and Cryopreservation of M. aurum
4.3.4. Optical Density of Bacterial Culture
Growth Curve
4.3.5. MIC Determination Using Solid Agar Method (Spot Culture Growth Inhibition Assay)
Preparation of the Master Plate
Preparation of Spot Culture Growth Inhibition Assay
4.3.6. Resazurin Cytotoxicity Assay
4.4. Molecular Dynamics Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leimgruber, W.; Stefanović, V.; Schenker, F.; Karr, A.; Berger, J. Isolation and Characterization of Anthramycin, a New Antitumor Antibiotic. J. Am. Chem. Soc. 1965, 87, 5791–5793. [Google Scholar] [CrossRef]
- Hurley, L.H.; Reck, T.; Thurston, D.E.; Langley, D.R.; Holden, K.G.; Hertzberg, R.P.; Hoover, J.R.; Gallagher, G., Jr.; Faucette, L.F. Pyrrolo[1,4]benzodiazepine antitumor antibiotics: Relationship of DNA alkylation and sequence specificity to the biological activity of natural and synthetic compounds. Chem. Res. Toxicol. 1988, 1, 258–268. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer. 2002, 2, 188. [Google Scholar] [CrossRef]
- Jenkins, T.C.; Hurley, L.H.; Neidle, S.; Thurston, D.E. Structure of a covalent DNA minor groove adduct with a pyrrolobenzodiazepine dimer: Evidence for sequence-specific interstrand crosslinking. J. Med. Chem. 1994, 37, 4529–4537. [Google Scholar] [CrossRef]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Thurston, D.E.; Bose, D.S. Synthesis of DNA-Interactive Pyrrolo [2,1-c][1,4] benzodiazepines. Chem. Rev. 1994, 94, 433–465. [Google Scholar] [CrossRef]
- Hertzberg, R.P.; Hecht, S.M.; Reynolds, V.L.; Molineux, I.J.; Hurley, L.H. DNA sequence specificity of the pyrrolo[1,4]benzodiazepine antitumor antibiotics. Methidiumpropyl-EDTA-iron(II) footprinting analysis of DNA binding sites for anthramycin and related drugs. Biochemistry 1986, 25, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Puvvada, M.S.; Forrow, S.A.; Hartley, J.A.; Stephenson, P.; Gibson, I.; Jenkins, T.C.; Thurston, D.E. Inhibition of Bacteriophage T7 RNA Polymerase in Vitro Transcription by DNA-Binding Pyrrolo[2,1-c][1,4]benzodiazepines. Biochemistry 1997, 36, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Mantaj, J.; Jackson, P.J.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef]
- Hartley, J.A.; Flynn, M.J.; Bingham, J.P.; Corbett, S.; Reinert, H.; Tiberghien, A.; Masterson, L.A.; Antonow, D.; Adams, L.; Chowdhury, S.; et al. Pre-clinical pharmacology and mechanism of action of SG3199, the pyrrolobenzodiazepine (PBD) dimer warhead component of antibody-drug conjugate (ADC) payload tesirine. Sci. Rep. 2018, 8, 4. [Google Scholar] [CrossRef]
- Tiberghien, A.C.; Levy, J.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and synthesis of tesirine, a clinical antibody–drug conjugate pyrrolobenzodiazepine dimer payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., III; Robert, F. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef]
- Wells, G.; Martin, C.R.; Howard, P.W.; Sands, Z.A.; Laughton, C.A.; Tiberghien, A.; Woo, C.K.; Masterson, L.A.; Stephenson, M.J.; Hartley, J.A.; et al. Design, Synthesis, and Biophysical and Biological Evaluation of a Series of Pyrrolobenzodiazepine−Poly(N-methylpyrrole) Conjugates. J. Med. Chem. 2006, 49, 5442–5461. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Balboni, G.; Cacciari, B.; Guiotto, A.; Manfredini, S.; Romagnoli, R.; Spalluto, G.; Thurston, D.E.; Howard, P.W.; Bianchi, N.; et al. Synthesis, in Vitro Antiproliferative Activity, and DNA-Binding Properties of Hybrid Molecules Containing Pyrrolo[2,1-c][1,4]benzodiazepine and Minor-Groove-Binding Oligopyrrole Carriers. J. Med. Chem. 1999, 42, 5131–5141. [Google Scholar] [CrossRef]
- Brucoli, F.; Hawkins, R.M.; James, C.H.; Jackson, P.J.; Wells, G.; Jenkins, T.C.; Ellis, T.; Kotecha, M.; Hochhauser, D.; Hartley, J.A.; et al. An extended pyrrolobenzodiazepine–polyamide conjugate with selectivity for a DNA sequence containing the ICB2 transcription factor binding site. J. Med. Chem. 2013, 56, 6339–6351. [Google Scholar] [CrossRef]
- Rahman, K.M.; Jackson, P.J.; James, C.H.; Basu, B.P.; Hartley, J.A.; de la Fuente, M.; Schatzlein, A.; Robson, M.; Pedley, R.B.; Pepper, C.; et al. GC-targeted C8-linked pyrrolobenzodiazepine–biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. [Google Scholar] [CrossRef]
- Corcoran, D.B.; Lewis, T.; Nahar, K.S.; Jamshidi, S.; Fegan, C.; Pepper, C.; Thurston, D.E.; Rahman, K.M. Effects of systematic shortening of noncovalent C8 side chain on the cytotoxicity and NF-κB inhibitory capacity of pyrrolobenzodiazepines (PBDs). J. Med. Chem. 2019, 62, 2127–2139. [Google Scholar] [CrossRef]
- Kamal, A.; Vijaya Bharathi, E.; Janaki Ramaiah, M.; Dastagiri, D.; Surendranadha Reddy, J.; Viswanath, A.; Sultana, F.; Pushpavalli, S.N.; Pal-Bhadra, M.; Srivastava, H.K.; et al. Quinazolinone linked pyrrolo[2,1-c][1,4]benzodiazepine (PBD) conjugates: Design, synthesis and biological evaluation as potential anticancer agents. Bioorg. Med. Chem. 2010, 18, 526–542. [Google Scholar] [CrossRef]
- Andriollo, P.; Hind, C.K.; Picconi, P.; Nahar, K.S.; Jamshidi, S.; Varsha, A.; Clifford, M.; Sutton, J.M.; Rahman, K.M. C8-linked pyrrolobenzodiazepine monomers with inverted building blocks show selective activity against multidrug resistant Gram-positive bacteria. ACS Infect Dis. 2018, 4, 158–174. [Google Scholar] [CrossRef]
- Rosado, H.; Rahman, K.M.; Feuerbaum, E.A.; Hinds, J.; Thurston, D.E.; Taylor, P.W. The minor groove-binding agent ELB-21 forms multiple interstrand and intrastrand covalent cross-links with duplex DNA and displays potent bactericidal activity against methicillin-resistant Staphylococcus Aureus. J. Antimicrob. Chemother. 2011, 66, 985–996. [Google Scholar] [CrossRef]
- Brucoli, F.; Guzman, J.D.; Basher, M.A.; Evangelopoulos, D.; McMahon, E.; Munshi, T.; McHugh, T.D.; Fox, K.R.; Bhakta, S. DNA sequence-selective C8-linked pyrrolobenzodiazepine–heterocyclic polyamide conjugates show anti-tubercular-specific activities. J. Antibiot. (Tokyo) 2016, 69, 843–849. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iacobino, A.; Giannoni, F.; Fattorini, L.; Brucoli, F. Activity of DNA-targeted C8-linked pyrrolobenzodiazepine–heterocyclic polyamide conjugates against aerobically and hypoxically grown Mycobacterium tuberculosis under acidic and neutral conditions. J. Antibiot. (Tokyo) 2018, 71, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Picconi, P.; Jeeves, R.; Moon, C.; Jamshidi, S.; Nahar, K.S.; Laws, M.; Bacon, J.; Rahman, K.M. Noncytotoxic Pyrrolobenzodiazepine–Ciprofloxacin Conjugate with Activity against Mycobacterium Tuberculosis. ACS Omega 2019, 4, 20873–22088. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Bovero, A.; Fruttarolo, F.; Preti, D.; Tabrizi, M.A.; Pavani, M.G.; Romagnoli, R. DNA minor groove binders as potential antitumor and antimicrobial agents. Med. Res. Rev. 2004, 24, 475–528. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F. DNA-Minor Groove Binding Agents as Anti-Tubercular Probes. Old Tools for a New Challenge? Anti-Infect. Agents 2018, 16, 71–79. [Google Scholar] [CrossRef]
- Raju, G.; Srinivas, R.; Reddy, V.S.; Idris, M.M.; Kamal, A.; Nagesh, N. Interaction of Pyrrolobenzodiazepine (PBD) Ligands with Parallel Intermolecular G-Quadruplex Complex Using Spectroscopy and ESI-MS. PLoS ONE 2012, 7, e35920. [Google Scholar] [CrossRef]
- Kamal, A.; Prabhakar, S.; Janaki Ramaiah, M.; Venkat Reddy, P.; Ratna Reddy, C.; Mallareddy, A.; Shankaraiah, N.; Lakshmi Narayan Reddy, T.; Pushpavalli, S.N.; Pal-Bhadra, M. Synthesis and anticancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers. Eur. J. Med. Chem. 2011, 46, 3820–3831. [Google Scholar] [CrossRef]
- Rahman, K.M.; Corcoran, D.B.; Bui, T.T.T.; Jackson, P.J.M.; Thurston, D.E. Pyrrolobenzodiazepines (PBDs) do not bind to DNA G-quadruplexes. PLoS ONE 2014, 9, e105021. [Google Scholar] [CrossRef]
- Ramkumar, V.; Hallam, D.M.; Nie, Z. Adenosine, Oxidative Stress and Cytoprotection. Jpn. J. Pharmacol. 2001, 86, 265–274. [Google Scholar] [CrossRef]
- Matsuda, A.; Shinozaki, M.; Yamaguchi, T.; Homma, H.; Nomoto, R.; Miyasaka, T.; Watanabe, Y.; Abiru, T. Nucleosides and nucleotides. 103. 2-Alkynyladenosines: A novel class of selective adenosine A2 receptor agonists with potent antihypertensive effects. J. Med. Chem. 1992, 35, 241–252. [Google Scholar] [CrossRef]
- Robins, M.J.; Uznański, B. Nucleic acid related compounds. 33. Conversions of adenosine and guanosine to 2, 6-dichloro, 2-amino-6-chloro, and derived purine nucleosides. Can. J. Chem. 1981, 59, 2601–2607. [Google Scholar] [CrossRef]
- Tercel, M.; Stribbling, S.M.; Sheppard, H.; Siim, B.G.; Wu, K.; Pullen, S.M.; Botting, K.J.; Wilson, W.R.; Denny, W.A.J. Unsymmetrical DNA cross-linking agents: Combination of the CBI and PBD pharmacophores. Med. Chem. 2003, 46, 2132–2151. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bhakta, S.J. An integrated surrogate model for screening of drugs against Mycobacterium tuberculosis. Antimicrob. Chemother. 2012, 67, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Rizi, K.; Murdan, S.; Danquah, C.A.; Faull, J.; Bhakta, S.J. Development of a rapid, reliable and quantitative method—“SPOTi” for testing antifungal efficacy. Microbiol. Methods 2015, 117, 36–40. [Google Scholar] [CrossRef]
- Danquah, C.A.; Maitra, A.; Gibbons, S.; Faull, J.; Bhakta, S. HT-SPOTi: A rapid drug susceptibility test (DST) to evaluate antibiotic resistance profiles and novel chemicals for anti-infective drug discovery. Curr. Protoc. Microbiol. 2016, 40, 12. [Google Scholar]
- Barrow, E.W.; Westbrook, L.; Bansal, N.; Suling, W.J.; Maddry, J.A.; Parker, W.B.; Barrow, W.W. Antimycobacterial activity of 2-methyl-adenosine. J. Antimicrob. Chemother. 2003, 52, 801–808. [Google Scholar] [CrossRef][Green Version]
- Vitali, L.A.; Petrelli, D.; Lambertucci, C.; Prenna, M.; Volpini, R.; Cristalli, G.J. In vitro antibacterial activity of different adenosine analogues. Med. Microbiol. 2012, 61, 525–528. [Google Scholar] [CrossRef]
- Hampshire, A.J.; Fox, K.R. Preferred binding sites for the bifunctional intercalator TANDEM determined using DNA fragments that contain every symmetrical hexanucleotide sequence. Anal. Biochem. 2008, 374, 298–303. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 11 and 15 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
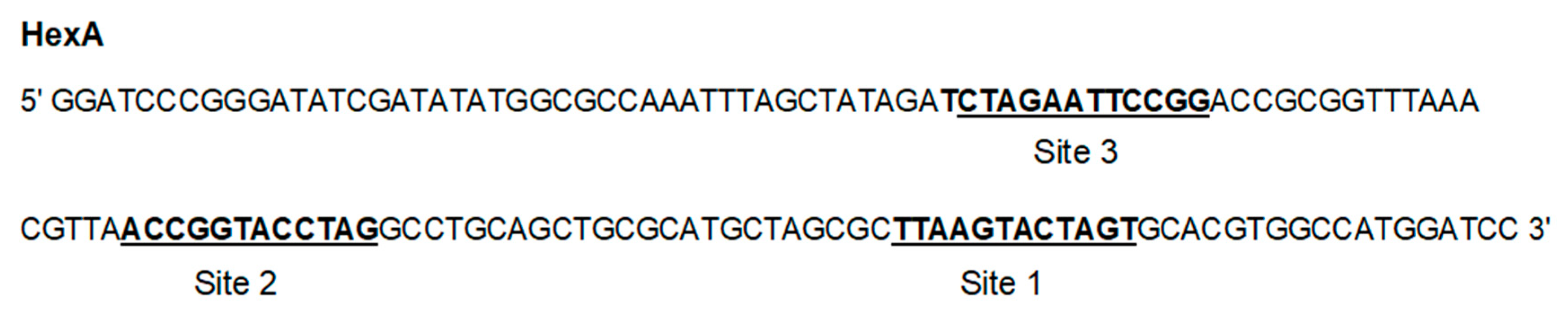{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GIC50 a RAW 264.7 (mg/L) | MIC90 b M. aurum (mg/L) | MIC90 M. bovis BCG (mg/L) | SI c GIC50/MIC90 M. bovis |
|---|---|---|---|---|
| PBD (5) | 0.49 | 62.5 | 31.25 | 0.01 |
| Bn-PBD (15) | 0.48 | 3.91 | 7.89 | 0.06 |
| 2-I-ADN (11) | 250 | 125 | 15.63 | 16 |
| PBD–ADN (16) | 500 | 500 | 250 | 2 |
| INH | 500 | 7.81 | 0.49 | 1020 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferguson, L.; Bhakta, S.; Fox, K.R.; Wells, G.; Brucoli, F. Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates. Molecules 2020, 25, 1243. https://doi.org/10.3390/molecules25051243
Ferguson L, Bhakta S, Fox KR, Wells G, Brucoli F. Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates. Molecules. 2020; 25(5):1243. https://doi.org/10.3390/molecules25051243
Chicago/Turabian StyleFerguson, Lindsay, Sanjib Bhakta, Keith R. Fox, Geoff Wells, and Federico Brucoli. 2020. "Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates" Molecules 25, no. 5: 1243. https://doi.org/10.3390/molecules25051243
APA StyleFerguson, L., Bhakta, S., Fox, K. R., Wells, G., & Brucoli, F. (2020). Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates. Molecules, 25(5), 1243. https://doi.org/10.3390/molecules25051243