
Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Full-Length Histidine-Tagged PPAR Construction
2.4. PPARα Ligand-Binding Domain Mutagenesis
2.5. Purification of PPAR Proteins
2.6. General Autofluoresce Assay Setup
2.7. Statistics
3. Results
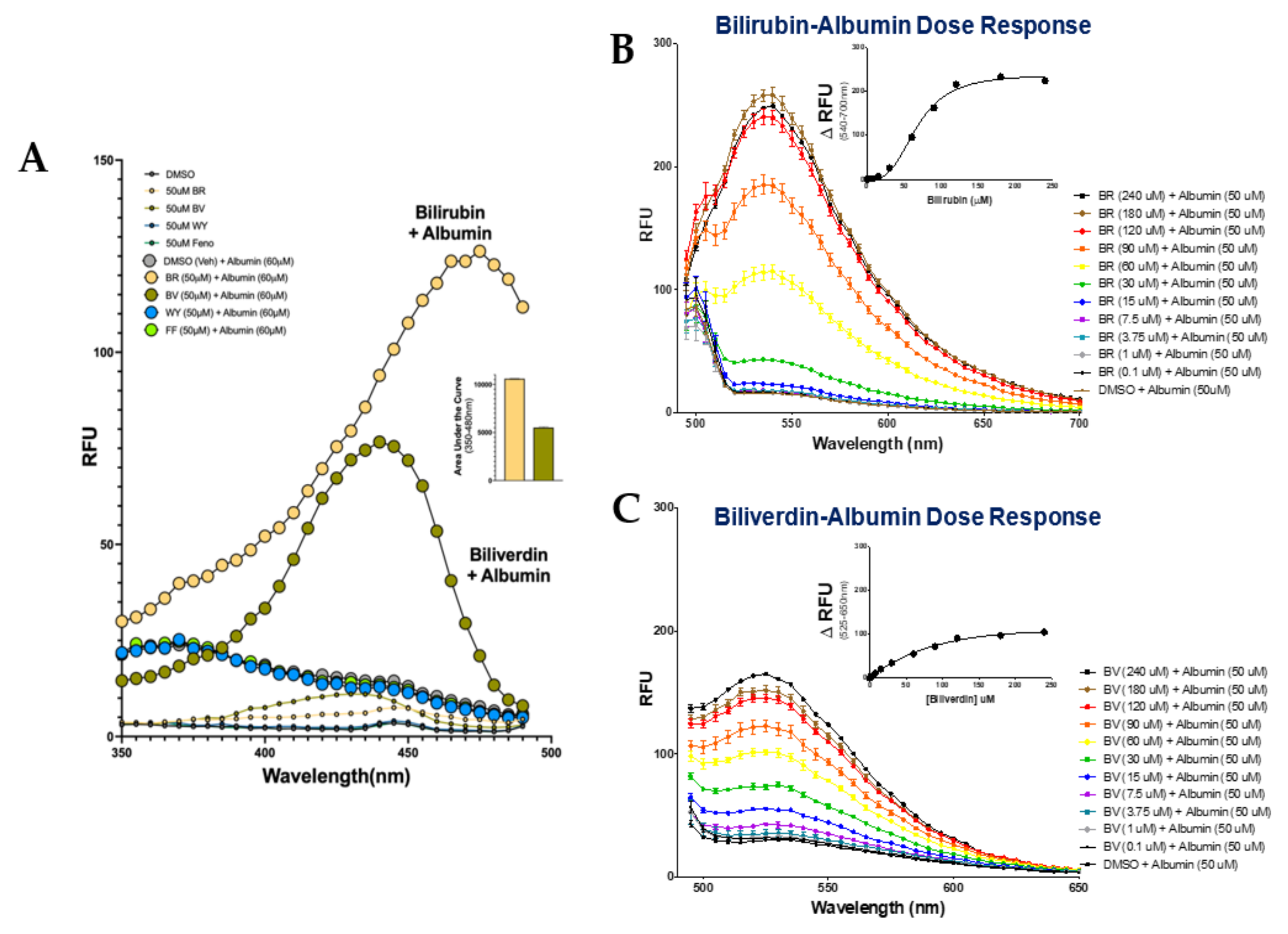
3.1. Autofluorescent Properties of Bilirubin and Biliverdin When Bound to Albumin
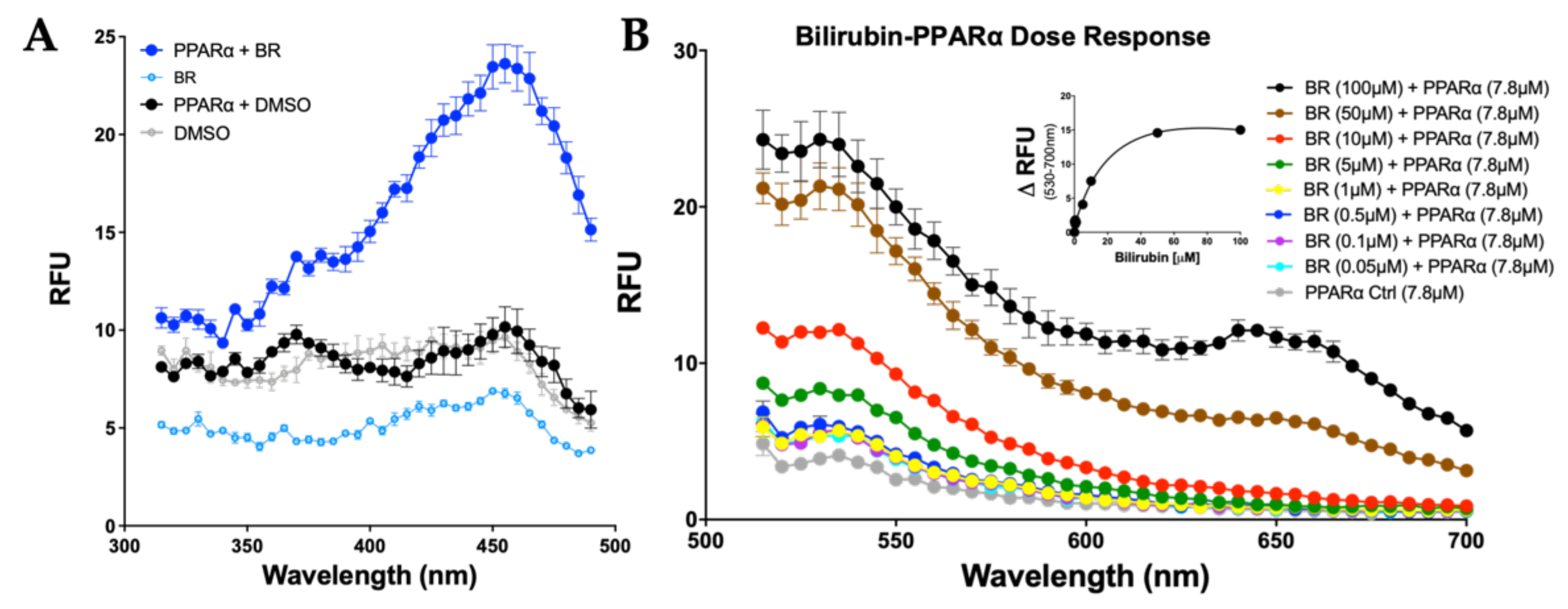
3.2. Bilirubin Fluoresces when Bound to PPARα
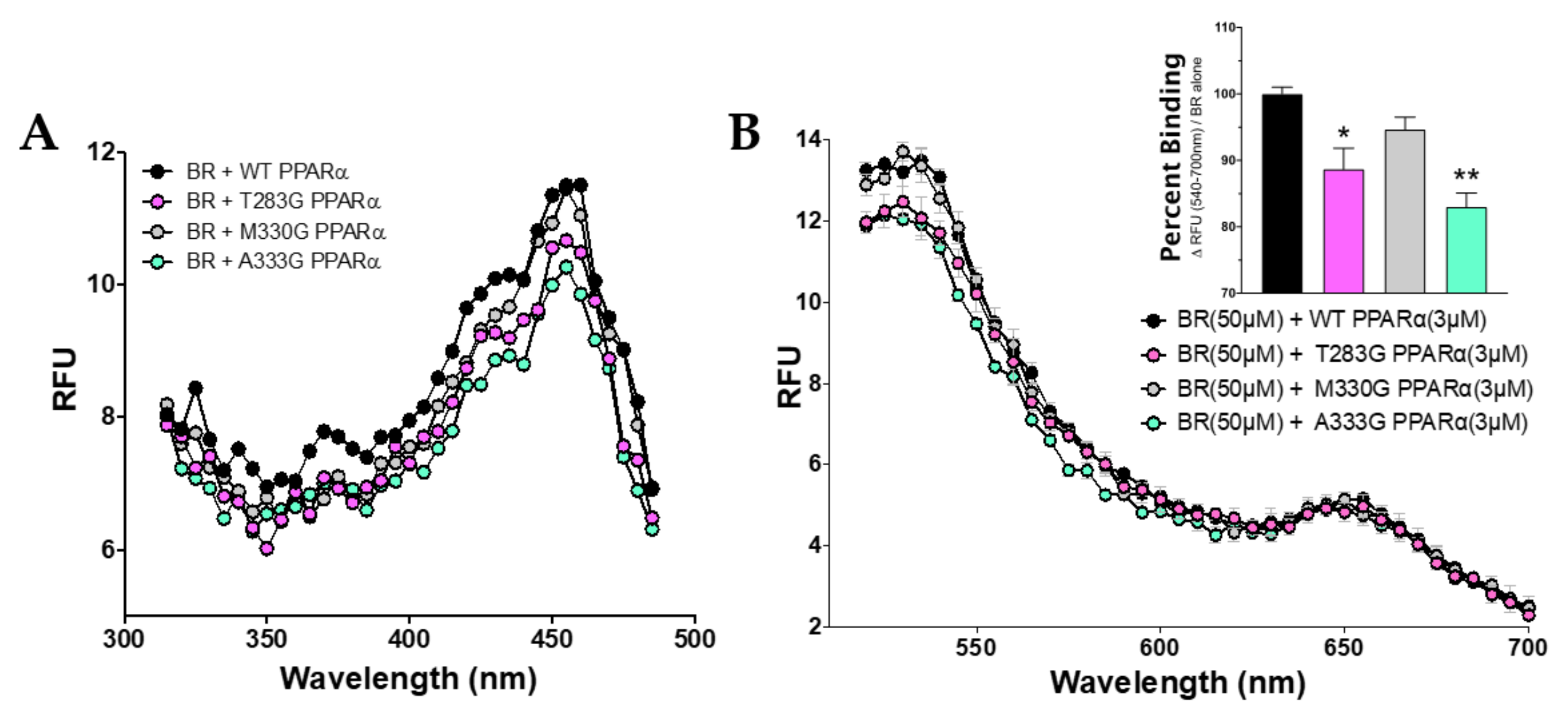
3.3. Bilirubin Requires Distinct Amino Acids to Maximize Binding to PPARα
3.4. Specific Binding of Bilirubin to PPARs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Menendez-Gutierrez, M.P.; Roszer, T.; Ricote, M. Biology and therapeutic applications of peroxisome proliferator- activated receptors. Curr. Top. Med. Chem. 2012, 12, 548–584. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—a review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [Green Version]
- Chiu, M.; McBeth, L.; Sindhwani, P.; Hinds, T.D. Deciphering the Roles of Thiazolidinediones and PPARγin Bladder Cancer. PPAR Res. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D.; Stechschulte, L.A.; Cash, H.A.; Whisler, D.; Banerjee, A.; Yong, W.; Khuder, S.S.; Kaw, M.K.; Shou, W.; Najjar, S.M.; et al. Protein Phosphatase 5 Mediates Lipid Metabolism through Reciprocal Control of Glucocorticoid Receptor and Peroxisome Proliferator-activated Receptor-γ (PPARγ). J. Biol. Chem. 2011, 286, 42911–42922. [Google Scholar] [CrossRef] [Green Version]
- Hinds, T.D.J.; Creeden, J.F.; Gordon, D.M.; Stec, D.F.; Donald, M.C.; Stec, D.E. Bilirubin Nanoparticles Reduce Diet-Induced Hepatic Steatosis, Improve Fat Utilization, and Increase Plasma β-Hydroxybutyrate. Front. Pharmacol. 2020, 11, 594574. [Google Scholar] [CrossRef] [PubMed]
- Lamola, A.A.; Russo, M. Fluorescence excitation spectrum of bilirubin in blood: A model for the action spectrum for phototherapy of neonatal jaundice. Photochem. Photobiol. 2013, 90, 294–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonagh, A.F.; Palma, L.A.; Lightner, D.A. Phototherapy for neonatal jaundice. Stereospecific and regioselective photoisomerization of bilirubin bound to human serum albumin and NMR characterization of intramolecularly cyclized photoproducts. J. Am. Chem. Soc. 1982, 104, 6867–6869. [Google Scholar] [CrossRef]
- Croce, A.C.; Ferrigno, A.; Santin, G.; Vairetti, M.; Bottiroli, G. Bilirubin: An autofluorescence bile biomarker for liver functionality monitoring. J. Biophotonics 2013, 7, 810–817. [Google Scholar] [CrossRef]
- Petersen, C.E.; Ha, C.-E.; Harohalli, K.; Feix, J.B.; Bhagavan, N.V. A Dynamic Model for Bilirubin Binding to Human Serum Albumin. J. Biol. Chem. 2000, 275, 20985–20995. [Google Scholar] [CrossRef] [Green Version]
- Lightner, D.A.; Wooldridge, T.A.; McDonagh, A.F. Photobilirubin: An early bilirubin photoproduct detected by absorbance difference spectroscopy. Proc. Natl. Acad. Sci. USA 1979, 76, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Greene, B.I.; Lamola, A.A.; Shank, C.V. Picosecond primary photoprocesses of bilirubin bound to human serum albumin. Proc. Natl. Acad. Sci. USA 1981, 78, 2008–2012. [Google Scholar] [CrossRef] [Green Version]
- Plavskii, V.Y.; Mostovnikov, V.A.; Mostovnikova, G.R.; Tret’Yakova, A.I. Spectral fluorescence and polarization characteristics of Z,Z-bilirubin IXα. J. Appl. Spectrosc. 2007, 74, 120–132. [Google Scholar] [CrossRef]
- Hostetler, H.A.; Petrescu, A.D.; Kier, A.B.; Schroeder, F. Peroxisome Proliferator-activated Receptor α Interacts with High Affinity and Is Conformationally Responsive to Endogenous Ligands. J. Biol. Chem. 2005, 280, 18667–18682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Sundseth, S.S.; Lehmann, J.M.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors and. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Ruuska, S.E.; Shaw, N.S.; Dong, D.; Noy, N. Ligand Selectivity of the Peroxisome Proliferator-Activated Receptor α†. Biochemistry 1999, 38, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A Bilirubin-Inducible Fluorescent Protein from Eel Muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef] [Green Version]
- Adeosun, S.O.; Moore, K.H.; Lang, D.M.; Nwaneri, A.C.; Hinds, J.T.; Stec, D.E. A Novel Fluorescence-Based Assay for the Measurement of Biliverdin Reductase Activity. React. Oxyg. Spec. 2018, 5, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creeden, J.F.; Gordon, D.M.; Stec, D.E.; Hinds, T.D. Bilirubin as a metabolic hormone: The physiological relevance of low levels. Am. J. Physiol. Metab. 2021, 320, E191–E207. [Google Scholar] [CrossRef]
- Gordon, D.M.; Neifer, K.L.; Hamoud, A.-R.A.; Hawk, C.F.; Nestor-Kalinoski, A.L.; Miruzzi, S.A.; Morran, M.P.; Adeosun, S.O.; Sarver, J.G.; Erhardt, P.W.; et al. Bilirubin remodels murine white adipose tissue by reshaping mitochondrial activity and the coregulator profile of peroxisome proliferator–activated receptor α. J. Biol. Chem. 2020, 295, 9804–9822. [Google Scholar] [CrossRef] [PubMed]
- HindsJr, T.D.; Stec, D.E. Bilirubin, a Cardiometabolic Signaling Molecule. Hypertension 2018, 72, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Hamoud, A.-R.; Weaver, L.; Stec, D.E.; Hinds, T.D. Bilirubin in the Liver–Gut Signaling Axis. Trends Endocrinol. Metab. 2018, 29, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D.; Stec, D.E. Bilirubin Safeguards Cardiorenal and Metabolic Diseases: A Protective Role in Health. Curr. Hypertens. Rep. 2019, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Yun, K.; Choi, H. Relationships between serum total bilirubin levels and metabolic syndrome in Korean adults. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 31–37. [Google Scholar] [CrossRef]
- Vítek, L.; Jirsa, M.; Brodanová, M.; Kaláb, M.; Mareček, Z.; Danzig, V.; Novotný, L.; Kotal, P. Gilbert syndrome and ischemic heart disease: A protective effect of elevated bilirubin levels. Atherosclerosis 2002, 160, 449–456. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Takei, K.; Arulmozhiraja, S.; Sladek, V.; Matsuo, N.; Han, S.-I.; Matsuzaka, T.; Sekiya, M.; Tokiwa, T.; Shoji, M.; et al. Molecular association model of PPARα and its new specific and efficient ligand, pemafibrate: Structural basis for SPPARMα. Biochem. Biophys. Res. Commun. 2018, 499, 239–245. [Google Scholar] [CrossRef]
- Dvořák, A.; Pospíšilová, K.; Žížalová, K.; Capková, N.; Muchová, L.; Vecka, M.; Vrzáčková, N.; Křížová, J.; Zelenka, J.; Vítek, L. The Effects of Bilirubin and Lumirubin on Metabolic and Oxidative Stress Markers. Front. Pharmacol. 2021, 12, 567001. [Google Scholar] [CrossRef]
- Gordon, D.M.; Blomquist, T.M.; Miruzzi, S.A.; McCullumsmith, R.; Stec, D.E.; Hinds, T.D. RNA sequencing in human HepG2 hepatocytes reveals PPAR-α mediates transcriptome responsiveness of bilirubin. Physiol. Genom. 2019, 51, 234–240. [Google Scholar] [CrossRef]
- Stec, D.E.; Gordon, D.M.; Hipp, J.A.; Hong, S.; Mitchell, Z.L.; Franco, N.R.; Robison, J.W.; Anderson, C.D.; Stec, D.F.; Hinds, T.D., Jr. The loss of hepatic PPARalpha promotes inflammation and serum hyperlipidemia in diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 317, R733–R745. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Sodhi, K.; Meadows, C.; Fedorova, L.; Puri, N.; Kim, D.H.; Peterson, S.J.; I Shapiro, J.; Abraham, N.G.; Kappas, A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity 2014, 22, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.M.; Adeosun, S.O.; Ngwudike, S.I.; Anderson, C.D.; Hall, J.E.; Hinds, T.D.; Stec, D.E. CRISPR Cas9-mediated deletion of biliverdin reductase A (BVRA) in mouse liver cells induces oxidative stress and lipid accumulation. Arch. Biochem. Biophys. 2019, 672, 108072. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. 2015, 26, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Heuvel, J.P.V.; Stec, D.E. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; Gordon, D.M.; Nestor-Kalinoski, A.L.; Donald, M.C.; Mitchell, Z.L.; Creeden, J.F.; Hinds, J.T.D. Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice. Biomolecules 2020, 10, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, D.E.; Hinds, J.T.D. Natural Product Heme Oxygenase Inducers as Treatment for Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef] [PubMed]
- Hinds, J.T.D.; Creeden, J.F.; Gordon, D.M.; Spegele, A.C.; Britton, S.L.; Koch, L.G.; Stec, D.E. Rats Genetically Selected for High Aerobic Exercise Capacity Have Elevated Plasma Bilirubin by Upregulation of Hepatic Biliverdin Reductase-A (BVRA) and Suppression of UGT1A1. Antioxidants 2020, 9, 889. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Hosick, P.A.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert’s Syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon, D.M.; Hong, S.H.; Kipp, Z.A.; Hinds, T.D., Jr. Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules 2021, 26, 2975. https://doi.org/10.3390/molecules26102975
Gordon DM, Hong SH, Kipp ZA, Hinds TD Jr. Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules. 2021; 26(10):2975. https://doi.org/10.3390/molecules26102975
Chicago/Turabian StyleGordon, Darren M., Stephen H. Hong, Zachary A. Kipp, and Terry D. Hinds, Jr. 2021. "Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha)" Molecules 26, no. 10: 2975. https://doi.org/10.3390/molecules26102975
APA StyleGordon, D. M., Hong, S. H., Kipp, Z. A., & Hinds, T. D., Jr. (2021). Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARalpha). Molecules, 26(10), 2975. https://doi.org/10.3390/molecules26102975