
Challenges in the Highly Selective [3 + 1]-Cycloaddition of an Enoldiazoacetamide to Form a Donor–Acceptor Cis-Cyclobutenecarboxamide †


Abstract
:
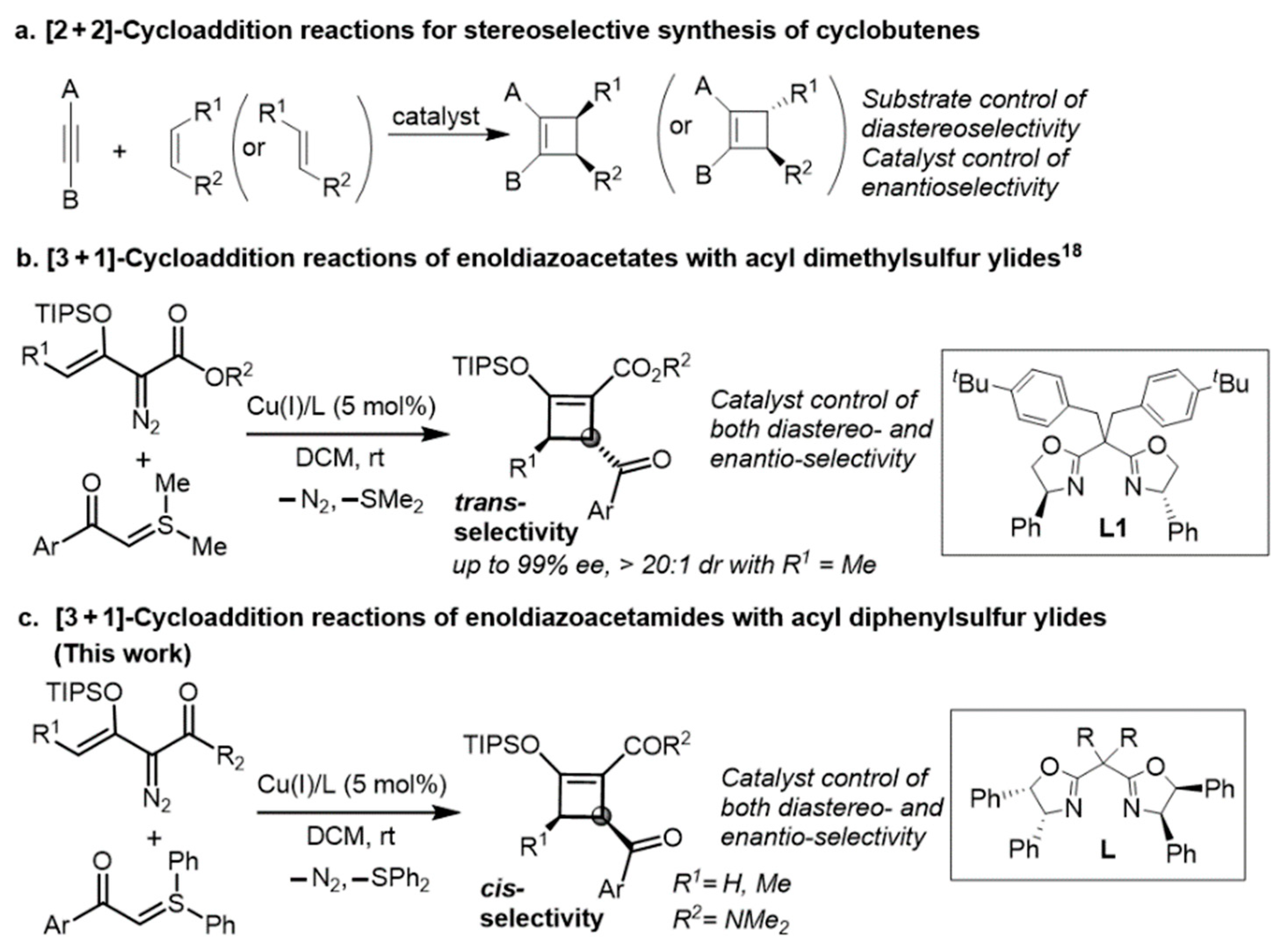
1. Introduction
2. Results and Discussion
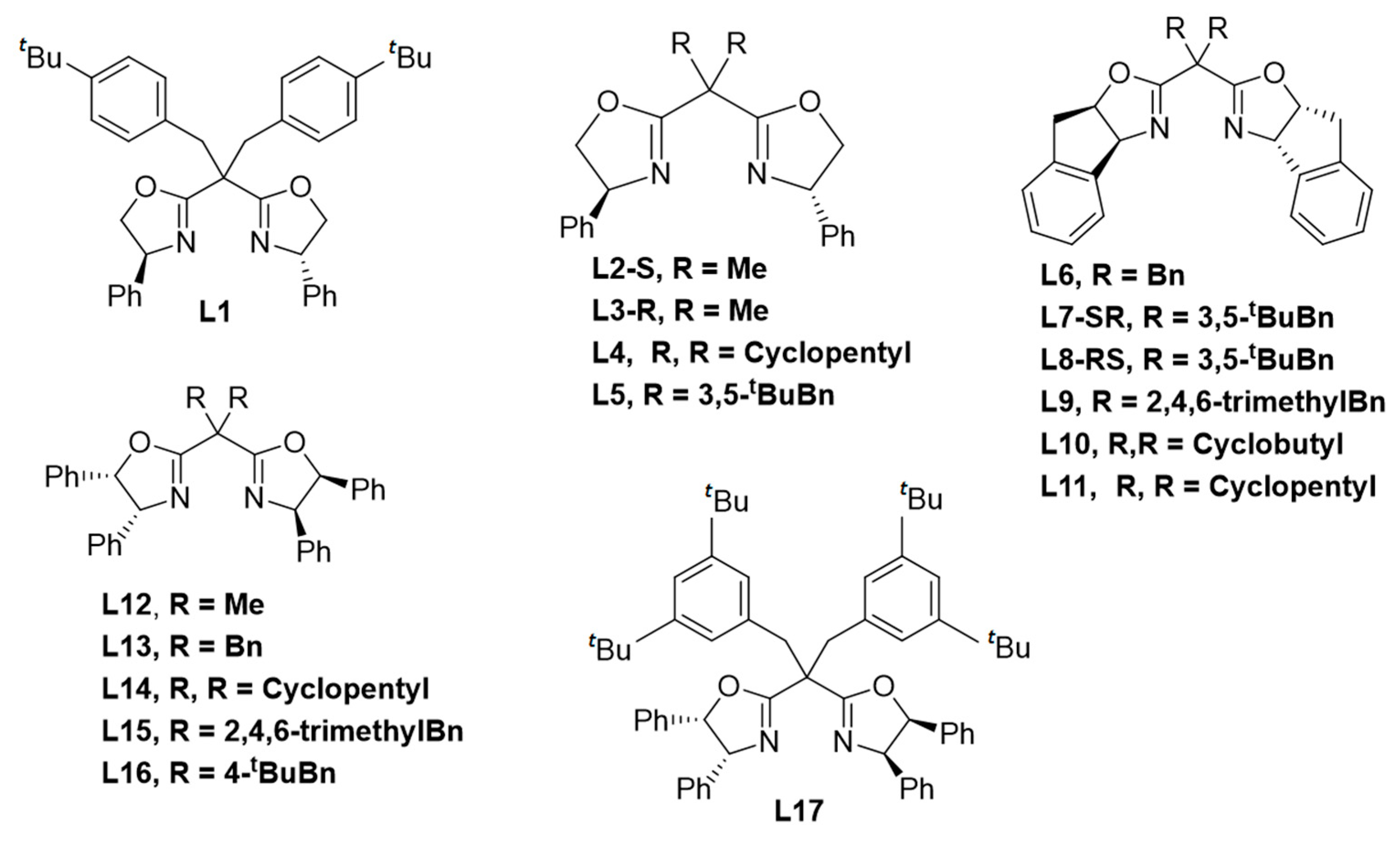
2.1. Ligand Control of Enantioselectivity in the [3 + 1]-Cycloaddition of 2-Diazo-N,N-dimethyl-3-(triisopropylsiloxy)but-3-enamide
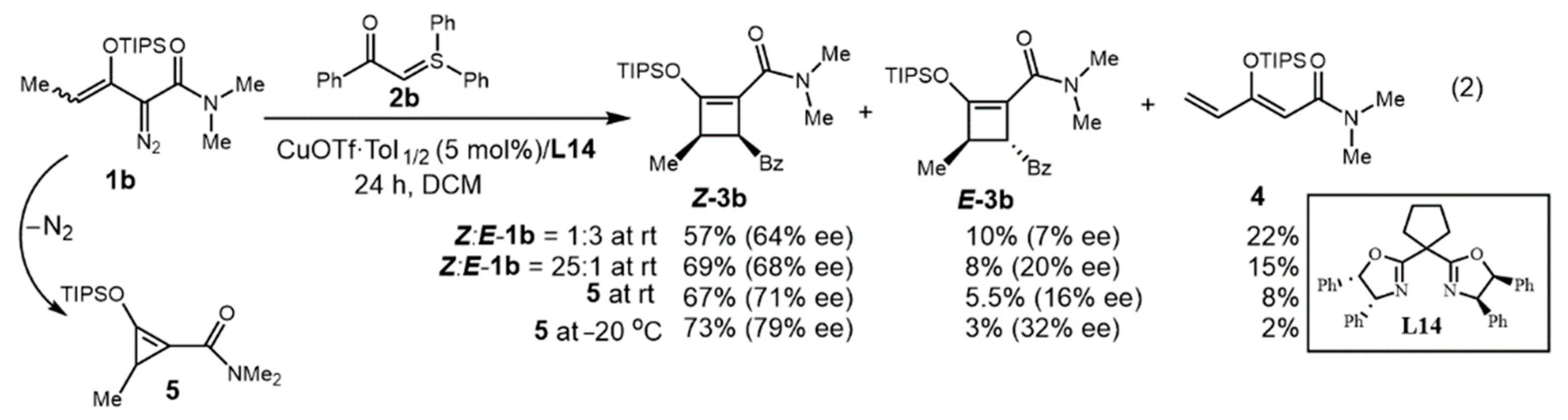
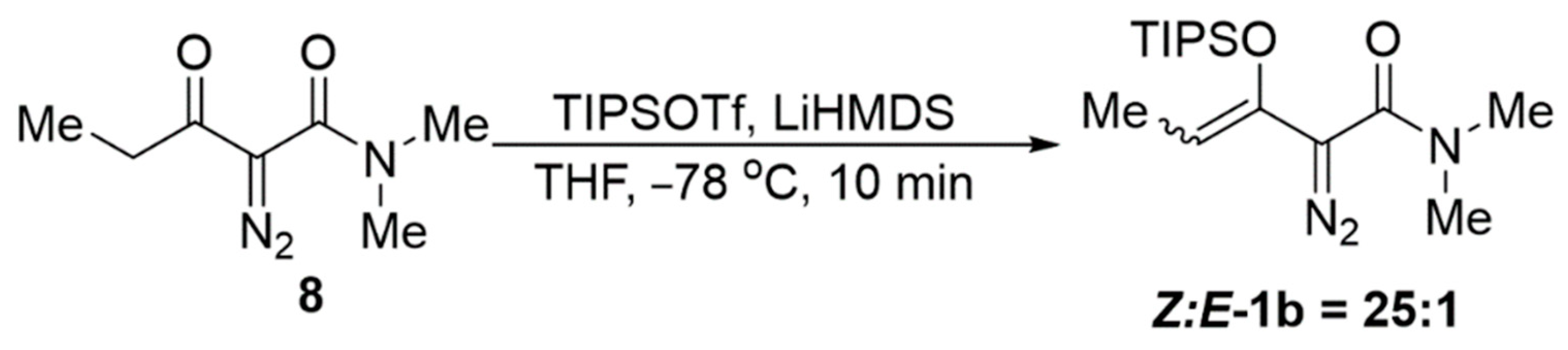
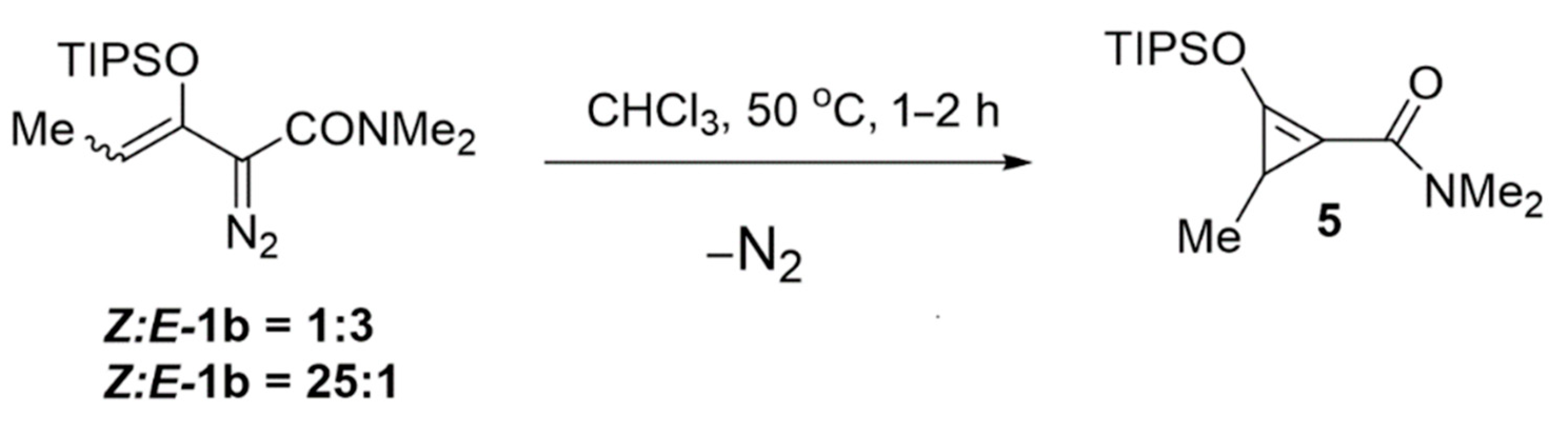
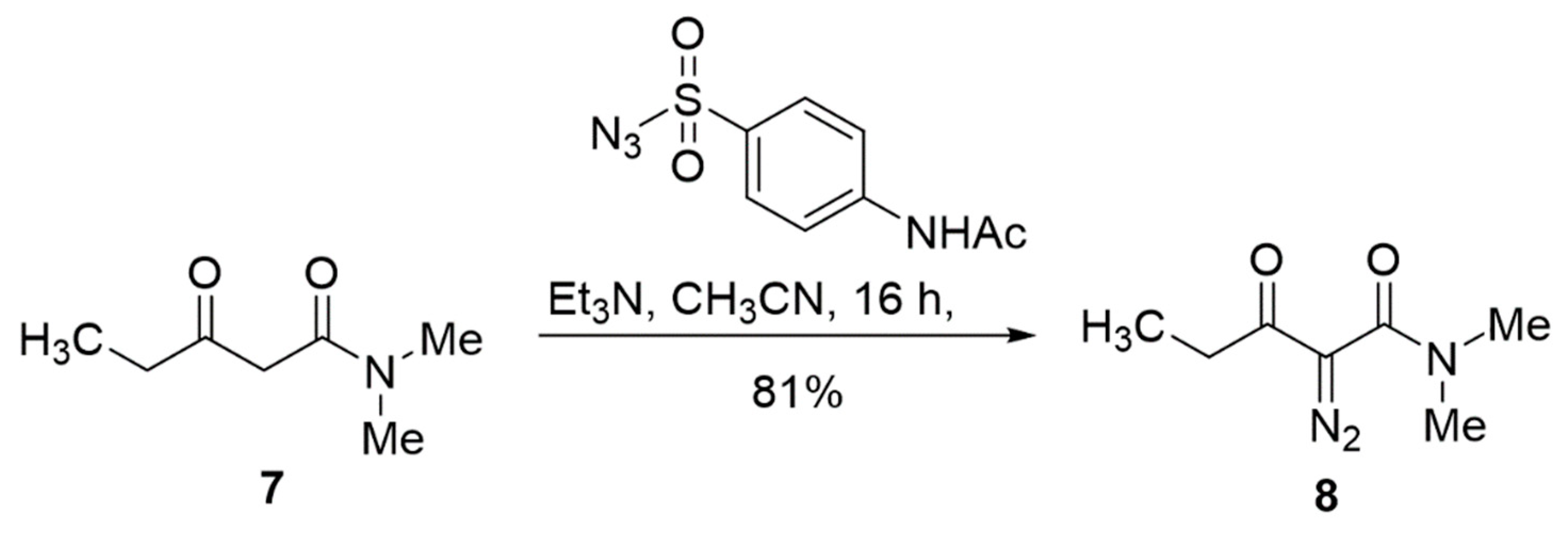
2.2. Ligand Control of Diastereoselectivity and Enantioselectivity in the [3 + 1]-Cycloaddition of 2-Diazo-N,N-dimethyl-3-(triisopropylsiloxy)pent-3-enamide
2.3. Ligand Control of Enantioselectivity in the [3 + 1]-Cycloaddition of 2-Diazo-N,N-dimethyl-3-methyl-2-(triisopropylsiloxy)-1-cyclopropenecarboxamide
3. Materials and Methods
3.1. General Information
3.2. Abbreviations
3.3. Materials
3.4. General Procedure for Asymmetric Catalytic [3 + 1]-Cycloaddition to Prepare (R)-4-Benzoyl-N,N-dimethyl-2-(triisopropylsiloxy)cyclobut-1-ene-1-Carboxamide (3a)
3.5. Preparation of 4-Benzoyl-N,N-Dimethyl-2-(triisopropylsiloxy)cyclobut-1-ene-1-carboxamide (Racemic 3a)
3.6. Preparation of 2-Diazo-N,N-dimethyl-3-(triisopropylsiloxy)but-3-enamide (1a)
3.7. General Procedure for Catalytic [3 + 1]-Cycloaddition Reaction of Sulfur Ylides (2b) with 1b or Cyclopropenecarboxamides (5)
3.7.1. General Procedure for Asymmetric Catalytic CuOTf-Catalyzed [3 + 1]-Cycloaddition Reactions to Prepare Z-3b and E-3b from 1b (Z:E = 1:3) or 1b (Z:E = 25:1)
3.7.2. General Procedure for Asymmetric Catalytic [3 + 1]-Cycloaddition to Prepare Z-3b and E-3b from 5
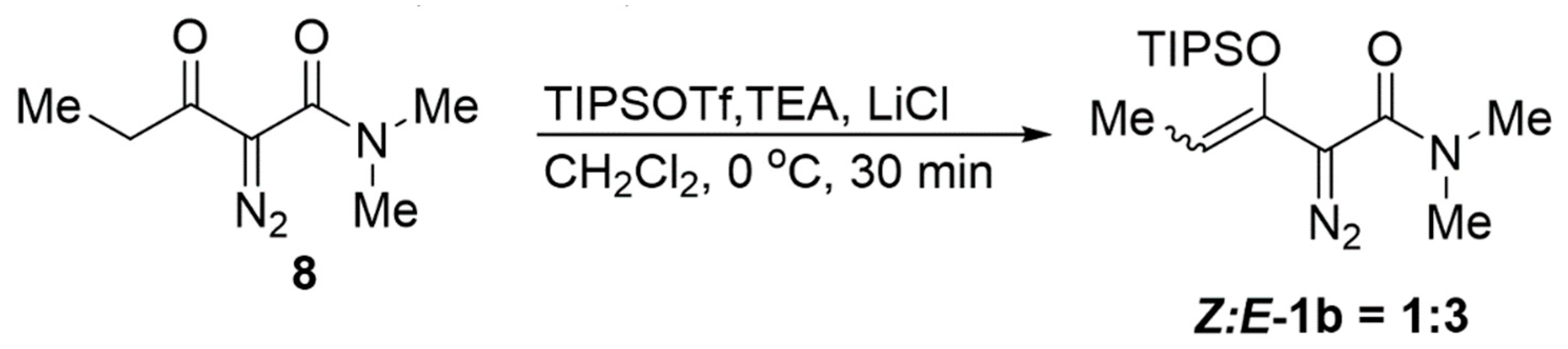
3.8. Preparation of 2-Diazo-N,N-dimethyl-3-oxopentanamide (8)
3.9. Preparation of 2-Diazo-N,N-dimethyl-3-((triisopropylsiloxy)pent-3-enamide, 1b (Z:E-1b = 1:3)
3.10. Preparation of 2-Diazo-N,N-dimethyl-3-(triisopropylsiloxy)pent-3-enamide, 1b (Z:E-1b = 25:1)
3.11. Preparation of N,N,3-Trimethyl-2-(triisopropylsiloxy)cycloprop-1-ene-1-carboxamide (5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Antonsen, S.; Østby, R.B.; Stenstrøm, Y. Chapter 1—Naturally Occurring Cyclobutanes: Their Biological Significance and Synthesis. Stud. Nat. Prod. Chem. 2018, 57, 1–40. [Google Scholar]
- Dembitsky, V.M. Naturally occurring bioactive Cyclobutane-containing (CBC) alkaloids in fungi, fungal endophytes, and plants. Phytomedicine 2014, 21, 1559–1581. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Bioactive cyclobutane-containing alkaloids. J. Nat. Med. 2008, 62, 1–33. [Google Scholar] [CrossRef]
- Namyslo, J.C.; Kaufmann, D.E. The Application of Cyclobutane Derivatives in Organic Synthesis. Chem. Rev. 2003, 103, 1485–1538. [Google Scholar] [CrossRef]
- Lee-Ruff, E.; Mladenova, G. Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev. 2003, 103, 1449–1484. [Google Scholar] [CrossRef]
- Ha, S.; Lee, Y.; Kwak, Y.; Mishra, A.; Yu, E.; Ryou, B.; Park, C.-M. Alkyne–Alkene [2 + 2] cycloaddition based on visible light photocatalysis. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Bai, Y.-B.; Luo, Z.; Wang, Y.; Gao, J.-M.; Zhang, L. Au-Catalyzed Intermolecular [2 + 2] Cycloadditions between Chloroalkynes and Unactivated Alkenes. J. Am. Chem. Soc. 2018, 140, 5860–5865. [Google Scholar] [CrossRef]
- Ogoshi, S.; Kumar, R.; Tamai, E.; Ohnishi, A.; Nishimura, A.; Hoshimoto, Y.; Ohashi, M. Nickel-Catalyzed Enantioselective Synthesis of Cyclobutenes via [2 + 2] Cycloaddition of α,β-Unsaturated Carbonyls with 1,3-Enynes. Synthesis 2016, 48, 2789–2794. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Zhao, K.; Doitomi, K.; Ganguly, R.; Li, Y.-X.; Shen, Z.-L.; Hirao, H.; Loh, T.-P. Lewis acid-catalyzed selective [2 + 2]-cycloaddition anddearomatizing cascade reaction of aryl alkynes with acrylates. J. Am. Chem. Soc. 2017, 139, 13570–13578. [Google Scholar] [CrossRef]
- Yoon, T.P. Visible Light Photocatalysis: The Development of Photocatalytic Radical Ion Cycloadditions. ACS Catal. 2013, 3, 895–902. [Google Scholar] [CrossRef]
- Sakai, K.; Kochi, T.; Kakiuchi, F. Rhodium-catalyzed intermolecular [2 + 2] cycloaddition of terminal alkynes with electron-deficient alkenes. Org. Lett. 2013, 15, 1024–1027. [Google Scholar] [CrossRef]
- Nishimura, A.; Ohashi, M.; Ogoshi, S. Nickel-catalyzed intermolecular [2 + 2] cycloaddition of conjugated enynes with alkenes. J. Am. Chem. Soc. 2012, 134, 15692–15695. [Google Scholar] [CrossRef]
- Fan, B.-M.; Li, X.-J.; Peng, F.-Z.; Zhang, H.-B.; Chan, A.S.C.; Shao, Z.-H. Ligand-controlled enantioselective [2 + 2] cycloaddition of oxabicyclic alkenes with terminal alkynes using chiraliridium catalyst. Org. Lett. 2010, 12, 304–306. [Google Scholar] [CrossRef]
- Lopez-Carrillo, V.; Echavarren, A.M. ChemInform Abstract: Gold(I)-Catalyzed Intermolecular [2 + 2] Cycloaddition of Alkynes with Alkenes. J. Am. Chem. Soc. 2010, 41, 9292–9294. [Google Scholar] [CrossRef] [PubMed]
- Treutwein, J.; Hilt, G. Cobalt-catalyzed [2 + 2] cycloaddition. Angew. Chem. Int. Ed. 2008, 47, 6811–6813. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Conner, M.L.; Brown, M.K. Cyclobutane and Cyclobutene Synthesis: Catalytic Enantioselective [2 + 2] Cycloadditions. Angew. Chem. Int. Ed. 2015, 54, 11918–11928. [Google Scholar] [CrossRef] [PubMed]
- Parsutkar, M.M.; Pagar, V.V.; Rajanbabu, T.V. Catalytic Enantioselective Synthesis of Cyclobutenes from Alkynes and Alkenyl Derivatives. J. Am. Chem. Soc. 2019, 141, 15367–15377. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Massey, L.A.; Zavalij, P.; Doyle, M.P. Catalytic Asymmetric [3+1]-Cycloaddition Reaction of Ylides with Electrophilic Metallo-enolcarbene Intermediates. Angew. Chem. Int. Ed. 2017, 56, 7479–7483. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.Q.; Yedoyan, J.; Arman, H.; Doyle, M.P. Dirhodium(II)-Catalyzed Annulation of Enoldiazoacetamides with α-Diazoketones: An Efficient and Highly Selective Approach to Fused and Bridged Ring System. Angew. Chem. Int. Ed. 2016, 55, 5573–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padwa, A.; Austin, D.J.; Hornbuckle, S.F.; Semones, M.A.; Doyle, M.P.; Protopopova, M.N. Control of Chemoselectivity in Catalytic Carbenoid Reactions. J. Am. Chem. Soc. 1992, 114, 1874–1876. [Google Scholar] [CrossRef]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratts, K.W.; Yao, A.N. Stable Sulfonium Ylids. J. Org. Chem. 1966, 31, 1185–1188. [Google Scholar] [CrossRef]
- Seshadri, R.; Pegg, W.J.; Israel, M. Diethyl oxomalonate. An improved synthesis. J. Org. Chem. 1981, 46, 2596–2598. [Google Scholar] [CrossRef]
- Nozaki, H.; Takaku, M.; Kondô, K. Stable sulphonium phenacylides: Isolation and reactions. Tetrahedron 1966, 22, 2145–2152. [Google Scholar] [CrossRef]
- Kramer, S.; Skrydstrup, T. Gold-Catalyzed Carbene Transfer to Alkynes: Access to 2,4-Disubstituted Furans. Angew. Chem. Int. Ed. 2012, 51, 4681–4684. [Google Scholar] [CrossRef] [PubMed]
- Shved, A.S.; Tabolin, A.A.; Novikov, R.A.; Nelyubina, Y.V.; Timofeev, V.P.; Ioffe, S.L. Six-Membered Cyclic Nitroso Acetals: Synthesis and Studies of the Nitrogen Inversion Process of N-Silyloxy-3,6-dihydro-2H-1,2-oxazines. Eur. J. Org. Chem. 2016, 2016, 5569–5578. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, G.; Sun, J. Gold-Catalyzed Formal [4+1]/[4+3] Cycloadditions of Diazo Esters with Triazines. Angew. Chem. Int. Ed. 2016, 55, 11867–11871. [Google Scholar] [CrossRef]
- Nocquet, P.; Opatz, T. Total Synthesis of (±)-Scopolamine: Challenges of the Tropane Ring. Eur. J. Org. Chem. 2016, 2016, 1156–1164. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Zavalij, P.Y.; Doyle, M.P. Straightforward Access to the [3.2.2] Nonatriene Structural Framework via Intramolecular Cyclopropenation/Buchner Reaction/Cope Rearrangement Cascade. Org. Lett. 2015, 17, 790–793. [Google Scholar] [CrossRef]
- Dong, K.; Marichev, K.O.; Xu, X.; Doyle, M.P. High Stereocontrol in the Preparation of Silyl-Protected γ-Substituted Enoldiazoacetates. Synlett 2019, 30, 1457–1461. [Google Scholar] [CrossRef]
- Dong, K.; Marichev, K.O.; Doyle, M.P. The Role of Donor-Acceptor Cyclopropenes in Metal Carbene Reactions. Conversion of E-Substituted Enoldiazoacetates to Z-Substituted Metallo-Enolcarbenes. Organometal 2019, 38, 4043–4050. [Google Scholar] [CrossRef]
- Deng, Y.; Jing, C.; Doyle, M.P. Dinitrogen extrusion from enoldiazo compounds under thermal conditions: synthesis of donor–acceptor cyclopropenes. Chem. Commun. 2015, 51, 12924–12927. [Google Scholar] [CrossRef]
- Liu, Q.; Rovis, T. Enantio- and Diastereoselective Intermolecular Stetter Reaction of Glyoxamide and Alkylidene Ketoamides. Org. Lett. 2009, 11, 2856–2859. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.-S.; Wang, Z.-L.; An, X.-L.; Luo, X.; Deng, W.-P. Direct synthesis of polysubstituted 2-aminothiophenes by Cu(ii)-catalyzed addition/oxidative cyclization of alkynoates with thioamides. Org. Biomol. Chem. 2014, 12, 8473–8479. [Google Scholar] [CrossRef] [PubMed]
- Nolin, A.; Ahn, R.W.; Kobayashi, Y.; Kennedy-Smith, J.J.; Toste, F.D. Enantioselective Reduction of Ketones and Imines Catalyzed by (CN-Box)ReV–Oxo Complexes. Chem. Eur. J. 2010, 16, 9555–9562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.-B.; Zhao, Y.-M.; Gu, P. Asymmetric Intramolecular Desymmetrization of meso-α,α′-Diazido Alcohols with Aryldiazo-acetates: Assembly of Chiral C3 Fragments with Three Continuous Stereocenters. Org. Lett. 2016, 18, 1984–1987. [Google Scholar] [CrossRef] [PubMed]
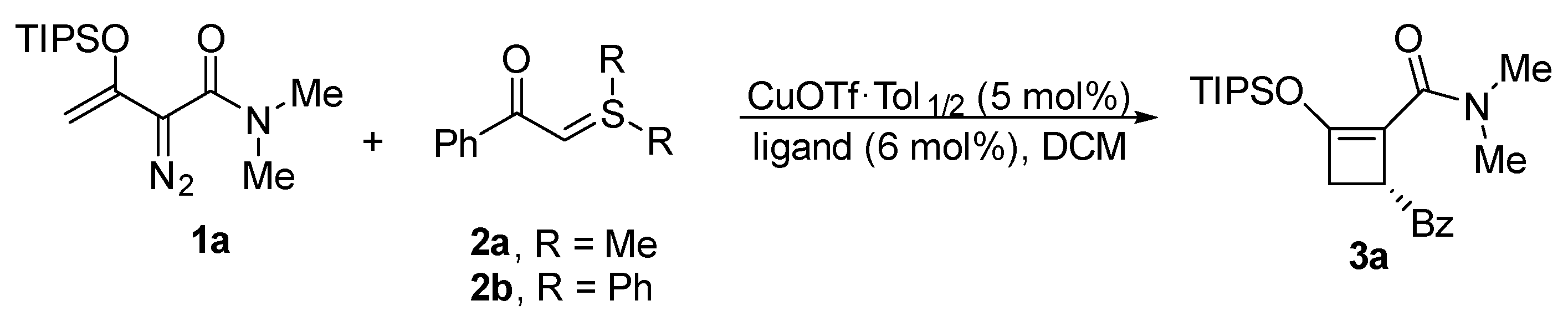

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Catalyst | Ligand | Ylide | t/h | T/°C | Yield [%] b | ee [%] c |
|---|---|---|---|---|---|---|---|
| 1 | CuOTf·Tol1/2 | L1 | 2a | 18 | rt | 58 | 0 |
| 2 | CuOTf·Tol1/2 | L5 | 2b | 24 | rt | 74 | 64 |
| 3 | CuOTf·Tol1/2 | L9 | 2b | 16 | rt | 71 | 32 |
| 4 | CuOTf·Tol1/2 | L10 | 2b | 16 | rt | 76 | 45 |
| 5 | CuOTf·Tol1/2 | L12 | 2b | 16 | rt | 90 | 38 |
| 6 | CuOTf·Tol1/2 | L13 | 2b | 16 | rt | 78 | 44 |
| 7 | CuOTf·Tol1/2 | L14 | 2b | 16 | rt | 54 | 34 |
| 8 | CuOTf·Tol1/2 | L14 | 2b | 24 | 0 | 60 | 38 |
| 9 | CuOTf·Tol1/2 | L16 | 2b | 24 | rt | 80 | 58 |
| 10 | CuOTf·Tol1/2 | L17 | 2b | 16 | rt | 79 | 60 |
| 11 | CuOTf·Tol1/2 | L17 | 2b | 24 | 0 | 82 | 64 |
| 12 d | CuOTf·Tol1/2 | L17 | 2b | 24 | −20 | 74 | 68 |
| 13 | CuBF4(CH3CN)4 | L17 | 2b | 16 | rt | 79 | 59 |
| 14 | CuBF4(CH3CN)4 | L17 | 2b | 16 | 0 | 75 | 65 |
| 15 d | CuBF4(CH3CN)4 | L17 | 2b | 24 | −20 | 82 | 67 |
| Entry a | Ligand | Solvent | T/°C | t/h | dr (Z:E) b | Yield (ee) of Z-3b c | Yield (ee) of E-3b d | Yield (4) e |
|---|---|---|---|---|---|---|---|---|
| 1 | L1 | DCM | rt | 38 | (2:1) | 32% (−12%) | 12% (65%) | 47% |
| 2 | L4 | DCM | rt | 24 | (2.5:1) | 50% (0%) | 18% (0%) | 4% |
| 3 | L5 | DCM | rt | 24 | (8:1) | 28% (−69%) | 3% (62%) | 59% |
| 4 | L11 | DCM | rt | 24 | (5:1) | 40% (62%) | 8% (56%) | 44% |
| 5 | L12 | DCM | rt | 24 | (5:1) | 61% (48%) | 12% (10%) | 16% |
| 6 | L14 | DCM | rt | 24 | (12:1) | 67% (71%) | 5.5% (16%) | 8% |
| 7 f | L14 | DCM | −20 | 48 | (24:1) | 73% (79%) | 3% (32%) | 2% |
| 8 | L16 | DCM | rt | 38 | (10:1) | 57% (59%) | 6% (NA) | 22% |
| 9 | L17 | DCM | rt | 24 | (18:1) | 24% (63%) | 2% (NA) | 51% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joyasawal, S.; Ma, D.; Doyle, M.P. Challenges in the Highly Selective [3 + 1]-Cycloaddition of an Enoldiazoacetamide to Form a Donor–Acceptor Cis-Cyclobutenecarboxamide. Molecules 2021, 26, 3520. https://doi.org/10.3390/molecules26123520
Joyasawal S, Ma D, Doyle MP. Challenges in the Highly Selective [3 + 1]-Cycloaddition of an Enoldiazoacetamide to Form a Donor–Acceptor Cis-Cyclobutenecarboxamide. Molecules. 2021; 26(12):3520. https://doi.org/10.3390/molecules26123520
Chicago/Turabian StyleJoyasawal, Sipak, Donghui Ma, and Michael P. Doyle. 2021. "Challenges in the Highly Selective [3 + 1]-Cycloaddition of an Enoldiazoacetamide to Form a Donor–Acceptor Cis-Cyclobutenecarboxamide" Molecules 26, no. 12: 3520. https://doi.org/10.3390/molecules26123520