
Benchmarking Catalysts for Formic Acid/Formate Electrooxidation

 , and
, and 
Abstract
:1. Introduction
Methods for Comparing Electrocatalysts for FA Electrooxidation
2. Platinum for Formic Acid/Formate Electrooxidation
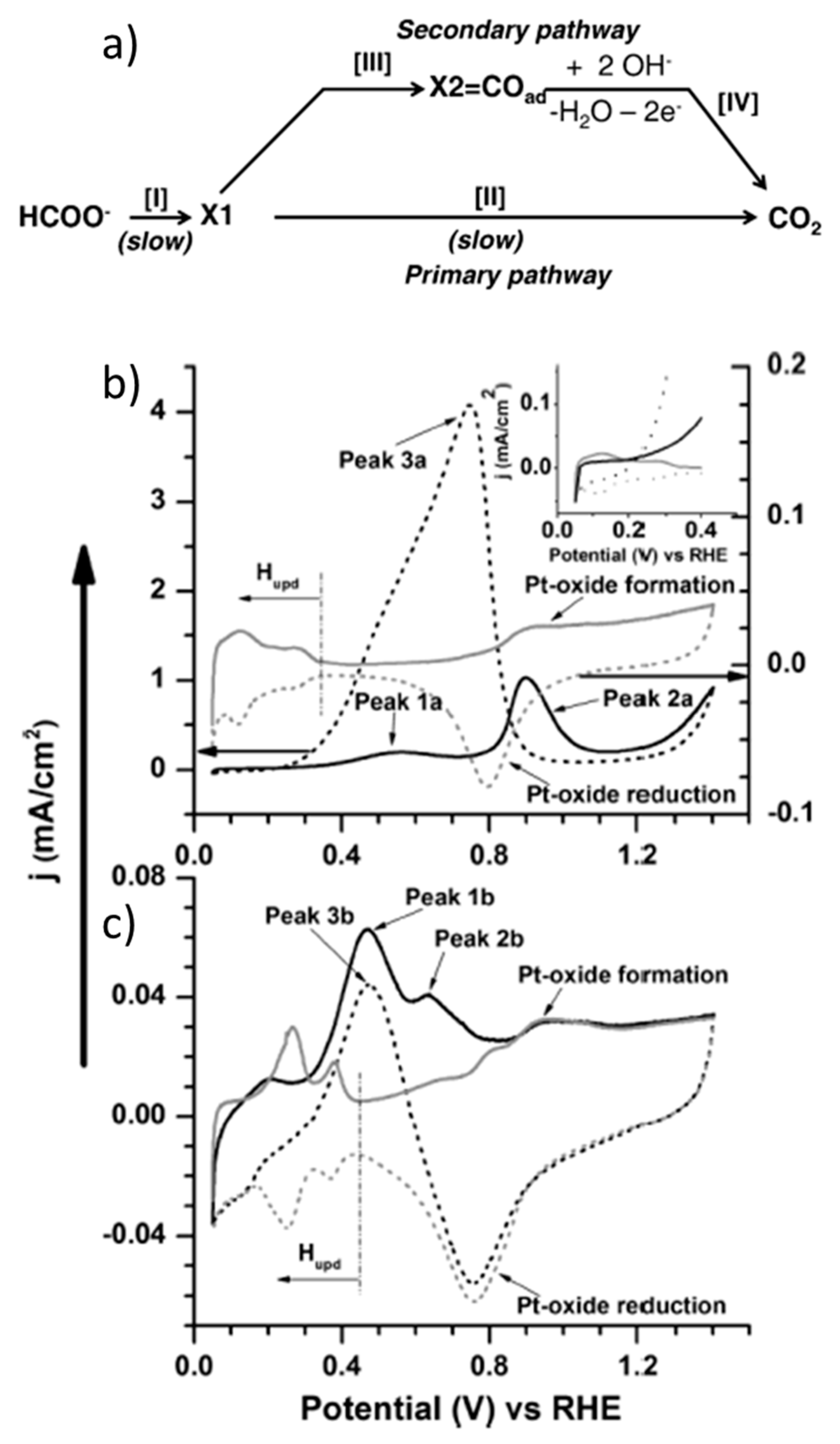
2.1. General Features of Formic Acid/Formate Electrooxidation on Pt
2.2. Monometallic Pt Catalysts
2.3. Bimetallic and Trimetallic Pt-Based Catalysts
2.4. Benchmarking Pt Formic Acid/Formate Electrooxidation Catalysts
3. Palladium for Formic Acid/Formate Electrooxidation
3.1. General Features on Formic Acid/Formate Electrooxidation on Pd
3.2. Monometallic Pd Catalysts
3.3. Bimetallic and Trimetallic Pd-Based Catalysts
3.4. Benchmarking Pd Formic Acid/Formate Electrooxidation Catalysts
4. Pt and Pd-Free Materials for Formic Acid/Formate Electrooxidation
4.1. Bulk Materials
4.1.1. Metals
4.1.2. Metal Oxides
4.1.3. Tungsten Carbide
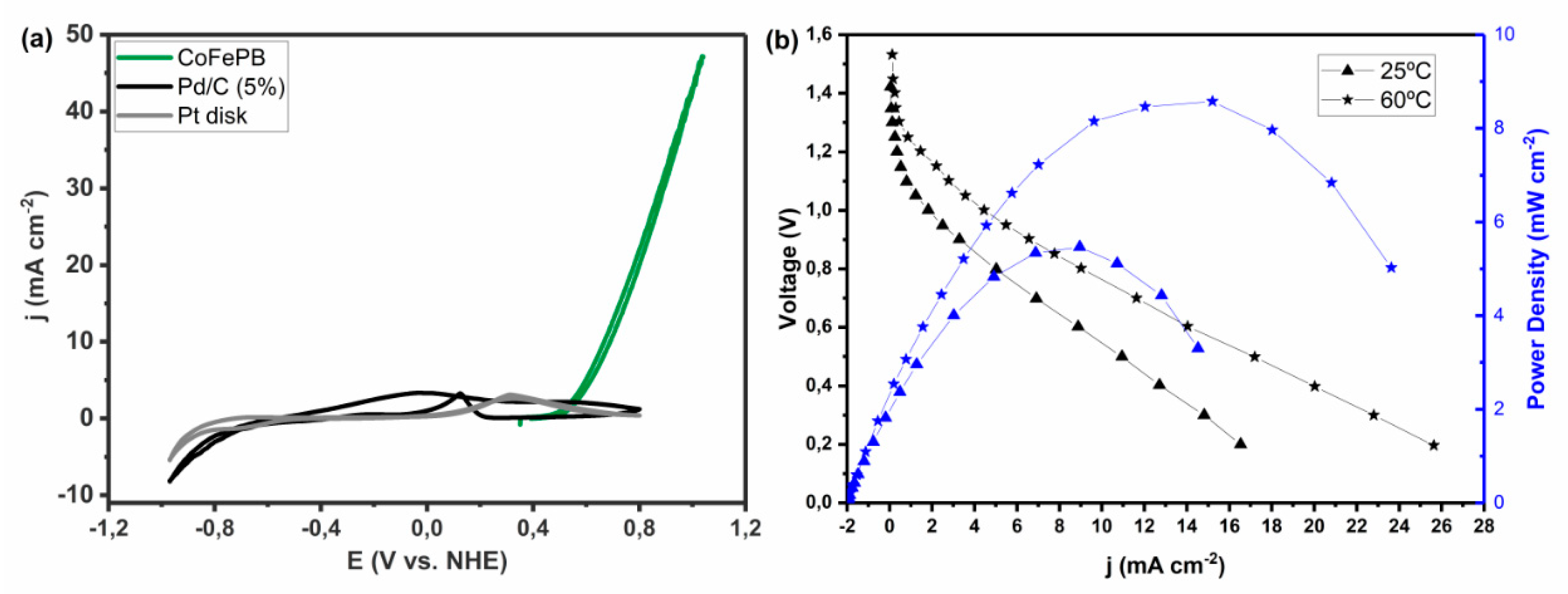
4.1.4. Co-Fe Prussian Blue
4.2. New Trends in Formate Oxidation Catalysts
4.2.1. Nanoparticles
4.2.2. Polymer Composites
4.2.3. Single Atom Catalysts
4.3. Benchmarking Pd and Pt-free Formic Acid/Formate Electrooxidation Catalysts
5. Conclusions
Author Contributions
Funding
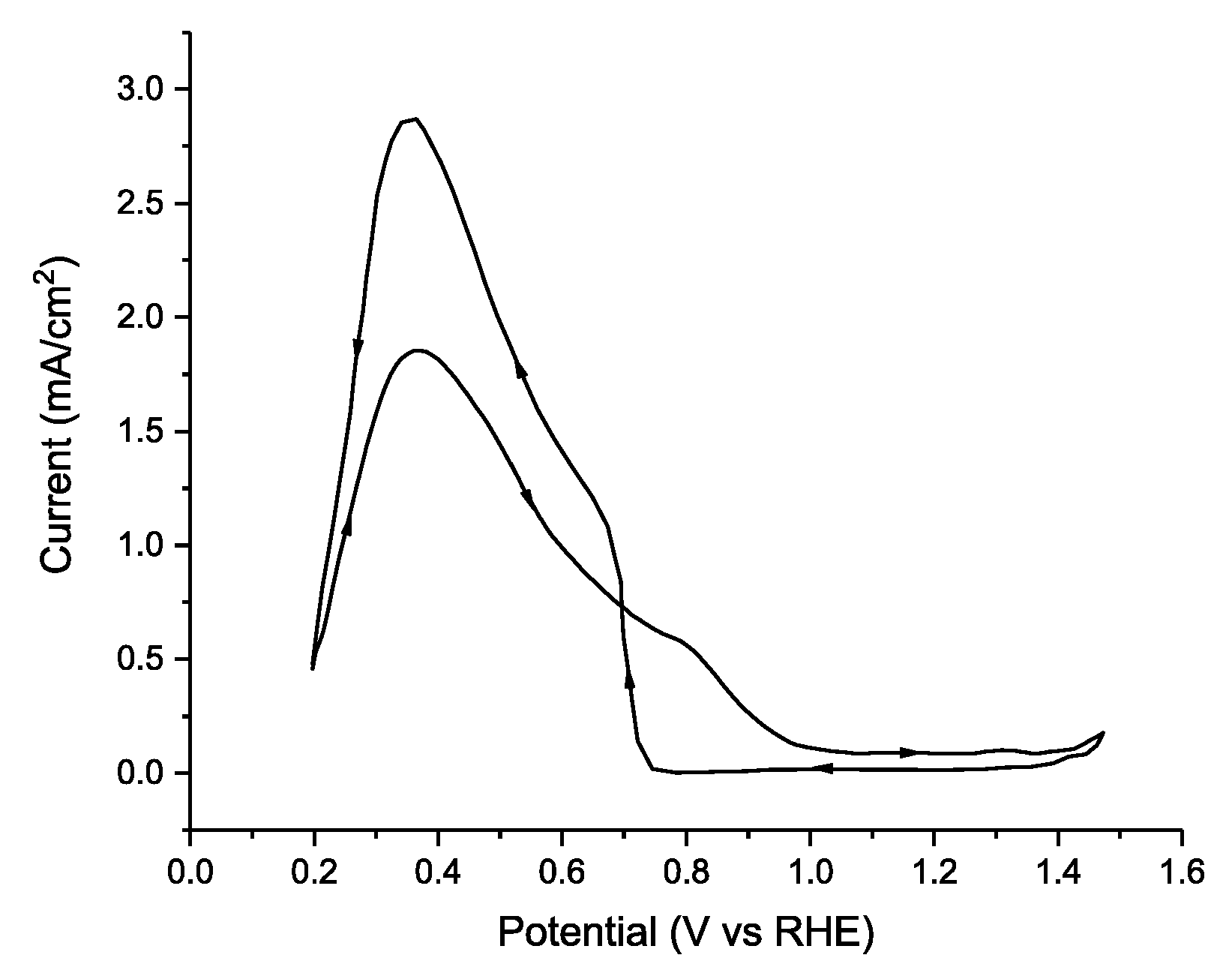
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jiang, S.P.; Li, Q. Introduction to Fuel Cells, 1st ed.; Springer: Singapore, 2021; ISBN 978-981-10-7625-1. [Google Scholar]
- U.S. Department of Energy. DOE–Hydrogen and Fuel Cell Program. 2020. Available online: www.hydrogen.energy.gov (accessed on 2 August 2021).
- Ogungbemi, E.; Wilberforce, T.; Ijaodola, O.; Thompson, J.; Olabi, A.G. Review of operating condition, design parameters and material properties for proton exchange membrane fuel cells. Int. J. Energy Res. 2021, 45, 1227–1245. [Google Scholar] [CrossRef]
- Baharuddin, N.A.; Wan Yusoff, W.N.A.; Abd Aziz, A.J.; Mohd Tahir, N.N. Hydrogen fuel cells for sustainable energy: Development and progress in selected developed countries. In Proceedings of the International Postgraduate Conference on Mechanical Engineering (IPCME 2021), Pekan, Malaysia, 19–20 January 2021; Volume 1078, p. 012011. [Google Scholar]
- Shaari, N.; Kamarudin, S.K.; Bahru, R.; Osman, S.H.; Md Ishak, N.A.I. Progress and challenges: Review for direct liquid fuel cell. Int. J. Energy Res. 2021, 45, 6644–6688. [Google Scholar] [CrossRef]
- Ma, Z.; Legrand, U.; Pahija, E.; Tavares, J.R.; Boffito, D.C. From CO2 to Formic Acid Fuel Cells. Ind. Eng. Chem. Res. 2021, 60, 803–815. [Google Scholar] [CrossRef]
- Pan, H.; Heagy, M.D. Photons to formate—A review on photocatalytic reduction of CO2 to formic acid. Nanomaterials 2020, 10, 2422. [Google Scholar] [CrossRef] [PubMed]
- Mardini, N.; Bicer, Y. Direct synthesis of formic acid as hydrogen carrierfrom CO2 for cleaner power generation throughdirect formic acid fuel cell. Int. J. Hydrogen Energy 2021, 46, 13050–13060. [Google Scholar] [CrossRef]
- Vo, T.; Purohit, K.; Nguyen, C.; Biggs, B.; Mayoral, S.; Haan, J. Formate: An Energy Storage and Transport Bridge between Carbon Dioxide and a Formate Fuel Cell in a Single Device. ChemSusChem 2015, 8, 3853–3858. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wu, Y.; Yuan, X.; Wang, H. An Integrated CO2 Electrolyzer and Formate Fuel Cell Enabled by a Reversibly Restructuring Pb–Pd Bimetallic Catalyst. Angew. Chem. Int. Ed. 2019, 58, 4031–4035. [Google Scholar] [CrossRef]
- Xiang, H.; Miller, H.A.; Bellini, M.; Christensen, H.; Scott, K.; Rasul, S.; Yu, E.H. Production of formate by CO2 electrochemical reduction and its application in energy storage. Sustain. Energy Fuels 2019, 4, 277–284. [Google Scholar] [CrossRef]
- Eppinger, J.; Huang, K.W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Chen, R. Direct formate fuel cells: A review. J. Power Source 2016, 320, 127–139. [Google Scholar] [CrossRef]
- Li, Y.; Li, Q.; Wang, H.; Zhang, L.; Wilkinson, D.; Zhang, J. Recent Progresses in Oxygen Reduction Reaction Electrocatalysts for Electrochemical Energy Applications. Electrochem. Energy Rev. 2019, 24, 518–538. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, D.A.; Kirtland, J.D.; Mota, N.D.; Stroock, A.D.; Abruña, H.D. Alternative Oxidants for High-Power Fuel Cells Studied by Rotating Disk Electrode (RDE) Voltammetry at Pt, Au, and Glassy Carbon Electrodes. J. Phys. Chem. C. 2011, 115, 6073–6084. [Google Scholar] [CrossRef]
- Noyes, A.A.; Garner, C.S. Strong Oxidizing Agents in Nitric Acid Solution. I. Oxidation Potential of Cerous—Ceric Salts. J. Am. Chem. Soc. 2002, 58, 1265–1268. [Google Scholar] [CrossRef]
- Nair, V.; Deepthi, A. Cerium(IV) ammonium nitrate—A versatile single-electron oxidant. Chem. Rev. 2007, 107, 1862–1891. [Google Scholar] [CrossRef] [PubMed]
- Vanýsek, P. Table of Standard Electrode Potentials. In CRC Handbook of Chemistry and Physics, 89th ed.; CRC Press: Boca Raton, FL, USA, 1978; Volume 18, pp. 1–21. [Google Scholar]
- Mota, N.; Finkelstein, D.; Kirtland, J.; Rodriguez, C.; Stroock, A.; Abruña, H. Membraneless, Room-Temperature, Direct Borohydride/Cerium Fuel Cell with Power Density of Over 0.25 W/cm2. J. Am. Chem. Soc. 2012, 134, 6076–6079. [Google Scholar] [CrossRef]
- Finkelstein, D.A.; Abruña, H.D. Rediscovering Cr2O72−, an Oxidant with Unrivaled Power and Energy Density, for Affordable, Next-Generation Energy Storage and Conversion. ACS Energy Lett. 2017, 2, 1439–1443. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Yang, W. A high-performance direct formate-peroxide fuel cell with palladium–gold alloy coated foam electrodes. J. Power Source 2015, 278, 569–573. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; He, Y.; Liu, Y.; Jin, L. Performance of direct formate-peroxide fuel cells. J. Power Source 2015, 287, 75–80. [Google Scholar] [CrossRef]
- Han, L.; González-Cobos, J.; Sánchez-Molina, I.; Giancola, S.; Folkmann, S.; Vidal-Ferrán, A.; Galán-Mascarós, J.R. A low temperature aqueous formate fuel cell using cobalt hexacyanoferrate as a non-noble metal oxidation catalyst. Sustain. Energy Fuels 2020, 4, 6227–6233. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, S.; Kumar, A. Hydrogen energy future with formic acid: A renewable chemical hydrogen storage system. Catal. Sci. Technol. 2015, 6, 12–40. [Google Scholar] [CrossRef]
- Yuan, X.Z.; Wang, H. PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications; PEM Fuel Cell Fundamentals; Spinger: London, UK, 2008; pp. 1–87. [Google Scholar]
- Ferrin, P.; Nilekar, A.U.; Greeley, J.; Mavrikakis, M.; Rossmeisl, J. Reactivity descriptors for direct methanol fuel cell anode catalysts. Surf. Sci. 2008, 602, 3424–3431. [Google Scholar] [CrossRef]
- Rejal, S.Z.; Masdar, M.S.; Kamarudin, S.K. A parametric study of the direct formic acid fuel cell (DFAFC) performance and fuel crossover. Int. J. Hydrogen Energy 2014, 39, 10267–10274. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G. Recent advances in direct formic acid fuel cells (DFAFC). J. Power Source 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Davis, D.G. The effect of platinum oxide films on reaction kinetics at platinum electrodes. Talanta 1960, 3, 335–345. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The oxidation of formic acid on noble metal electrodes. II. A comparison of the behaviour of pure electrodes. J. Electroanal. Chem. 1973, 44, 239–254. [Google Scholar] [CrossRef]
- Fang, Z.; Chen, W. Recent advances in formic acid electro-oxidation: From the fundamental mechanism to electrocatalysts. Nanoscale Adv. 2021, 3, 94–105. [Google Scholar] [CrossRef]
- Shen, T.; Zhang, J.; Chen, K.; Deng, S.; Wang, D. Recent Progress of Palladium-Based Electrocatalysts for the FormicAcid Oxidation Reaction. Energy Fuels 2020, 34, 9137–9153. [Google Scholar] [CrossRef]
- Bligaard, T.; Bullock, R.M.; Campbell, C.T.; Chen, J.G.; Gates, B.C.; Gorte, R.J.; Jones, C.W.; Jones, W.D.; Kitchin, J.R.; Scott, S.L. Toward Benchmarking in Catalysis Science: Best Practices, Challenges, and Opportunities. ACS Catal. 2016, 6, 2590–2602. [Google Scholar] [CrossRef]
- Wei, C.; Rao, R.; Peng, J.; Huang, B.; Stephens, I.; Risch, M.; Xu, Z.; Shao-Horn, Y. Recommended Practices and Benchmark Activity for Hydrogen and Oxygen Electrocatalysis in Water Splitting and Fuel Cells. Adv. Mater. 2019, 31, 1806296. [Google Scholar] [CrossRef]
- Buck, R.P.; Griffith, L.R. Voltammetric and Chronopotentiometric Study of the Anodic Oxidation of Methanol, Formaldehyde, and Formic Acid. J. Electrochem. Soc. 1962, 109, 1005. [Google Scholar] [CrossRef]
- Kutschker, A.; Vielstich, W. Zum mechanismus der eletrochemischen ameisensäureoxidation in saurem leitelektrolyten. Electrochim. Acta 1963, 8, 985–989. [Google Scholar] [CrossRef]
- Breiter, M.W. Anodic oxidation of formic acid on platinum-I. Adsorption of formic acid, oxygen, and hydrogen in perchloric acid solutions. Electrochim. Acta 1963, 8, 447–456. [Google Scholar] [CrossRef]
- Juliard, A.L.; Shalit, H. Application of Cyclic Voltammetry to the Kinetic Study of Electro-Oxidation of Organic Compounds. J. Electrochem. Soc. 1963, 110, 1002. [Google Scholar] [CrossRef]
- Schmidt, H.; Vielstich, W. Einfluß von Edelmetall-Mischkatalysatoren auf die anodische Oxydation von Methanol und Formiat. Fresenius’ Z. Anal. Chem. 1966, 224, 84–93. [Google Scholar] [CrossRef]
- John, J.; Wang, H.; Rus, E.D.; Abruña, H.D. Mechanistic studies of formate oxidation on platinum in alkaline medium. J. Phys. Chem. C 2012, 116, 5810–5820. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Electrochemical Hydrogen Adsorption and Absorption. Part 1: Under-potential Deposition of Hydrogen. Electrocatalysis 2010, 1, 179–199. [Google Scholar] [CrossRef]
- Spendelow, J.S.; Goodpaster, J.D.; Kenis, P.J.A.; Wieckowski, A. Mechanism of CO oxidation on Pt(111) in alkaline media. J. Phys. Chem. B 2006, 110, 9545–9555. [Google Scholar] [CrossRef]
- Couto, A.; Rincón, A.; Pérez, M.C.; Gutiérrez, C. Adsorption and electrooxidation of carbon monoxide on polycrystalline platinum at pH 0.3–13. Electrochim. Acta 2001, 46, 1285–1296. [Google Scholar] [CrossRef]
- Marković, N.M.; Sarraf, S.T.; Gasteiger, H.A.; Ross, P.N. Hydrogen electrochemistry on platinum low-index single-crystal surfaces in alkaline solution. J. Chem. Soc. Faraday Trans. 1996, 92, 3719–3725. [Google Scholar] [CrossRef]
- García, G.; Koper, M.T.M. Stripping voltammetry of carbon monoxide oxidation on stepped platinum single-crystal electrodes in alkaline solution. Phys. Chem. Chem. Phys. 2008, 10, 3802–3811. [Google Scholar] [CrossRef]
- Joo, J.; Uchida, T.; Cuesta, A.; Koper, M.T.M.; Osawa, M. The effect of pH on the electrocatalytic oxidation of formic acid/formate on platinum: A mechanistic study by surface-enhanced infrared spectroscopy coupled with cyclic voltammetry. Electrochim. Acta 2014, 129, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Miki, A.; Ye, S.; Osawa, M. Surface-enhanced IR absorption on platinum nanoparticles: An application to real-time monitoring of electrocatalytic reactions. Chem. Commun. 2002, 2, 1500–1501. [Google Scholar] [CrossRef]
- Samjeské, G.; Osawa, M. Current oscillations during formic acid oxidation on a Pt electrode: Insight into the mechanism by time-resolved IR spectroscopy. Angew. Chem. Int. Ed. 2005, 44, 5694–5698. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X.; Heinen, M.; Jusys, Z.; Behm, R.J. Kinetics and mechanism of the electrooxidation of formic acid–Spectroelectrochemical studies in a flow cell. Angew. Chem. Int. Ed. 2006, 45, 981–985. [Google Scholar] [CrossRef]
- Chen, Y.X.; Heinen, M.; Jusys, Z.; Behm, R.J. Bridge-bonded formate: Active intermediate or spectator species in formic acid oxidation on a Pt film electrode? Langmuir 2006, 22, 10399–10408. [Google Scholar] [CrossRef]
- Kumar, M.K.; Jha, N.S.; Mohan, S.; Jha, S.K. Reduced graphene oxide-supported nickel oxide catalyst with improved CO tolerance for formic acid electrooxidation. Int. J. Hydrogen Energy 2014, 39, 12572–12577. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, Y.; Dong, T.; Yuan, X.; Lv, L.; Wei, X.; Wang, J. Formate: A possible replacement for formic acid in fuel cells. Aust. J. Chem. 2017, 70, 757–763. [Google Scholar] [CrossRef]
- Han, L.; González-Cobos, J.; Sánchez-Molina, I.; Giancola, S.; Folkman, S.; Tang, P.; Heggen, M.; Dunin-Borkowski, R.; Arbiol, J.; Giménez, S.; et al. Cobalt Hexacyanoferrate as a Selective and High Current Density Formate Oxidation Electrocatalyst. ACS Appl. Energy Mater. 2020, 3, 9198–9207. [Google Scholar] [CrossRef]
- Jeong, S.; Shin, W. Triple-pulse Method for Monitoring Formate in CO2 Conversion Process. Electroanalysis 2016, 28, 1437–1440. [Google Scholar] [CrossRef]
- Perales-Rondón, J.V.; Brimaud, S.; Solla-Gullón, J.; Herrero, E.; Jürgen-Behm, R.; Feliu, J. Further Insights into the Formic Acid Oxidation Mechanism on Platinum: pH and Anion Adsorption Effects. Electrochim. Acta 2015, 180, 479–485. [Google Scholar] [CrossRef]
- Joo, J.; Choun, M.; Jeong, J.; Lee, J. Influence of Solution pH on Pt Anode Catalyst in Direct Formic Acid Fuel Cells. ACS Catal. 2015, 5, 6848–6851. [Google Scholar] [CrossRef]
- Okamoto, H.; Kon, W.; Mukouyama, Y. Stationary Voltammogram for Oxidation of Formic Acid on Polycrystalline Platinum. J. Phys. Chem. B 2004, 108, 4432–4438. [Google Scholar] [CrossRef]
- Brimaud, S.; Solla-Gullón, J.; Weber, I.; Feliu, J.M.; Behm, R.J. Formic Acid Electrooxidation on Noble-Metal Electrodes: Role and Mechanistic Implications of pH, Surface Structure, and Anion Adsorption. ChemElectroChem 2014, 1, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Busó-Rogero, C.; Ferre-Vilaplana, A.; Herrero, E.; Feliu, J.M. The role of formic acid/formate equilibria in the oxidation of formic acid on Pt(111). Electrochem. Commun. 2019, 98, 10–14. [Google Scholar] [CrossRef]
- Abdelrahman, A.; Hermann, J.M.; Kibler, L.A. Electrocatalytic Oxidation of Formate and Formic Acid on Platinum and Gold: Study of pH Dependence with Phosphate Buffers. Electrocatalysis 2017, 8, 509–517. [Google Scholar] [CrossRef]
- Perales-Rondón, J.V.; Herrero, E.; Feliu, J.M. On the activation energy of the formic acid oxidation reaction on platinum electrodes. J. Electroanal. Chem. 2015, 742, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Habibi, B.; Imanzadeh, H.; Haghighi Shishavan, Y.; Amiri, M. Effect of Carbon Support on the Electrocatalytic Performance of the Pt Nanoparticles Toward Oxidation of Formic Acid. Catal. Lett. 2020, 150, 312–321. [Google Scholar] [CrossRef]
- Bisht, A.; Pentyala, P.; Deshpande, P.A.; Sharma, S. La0.80Sr0.20CoO3 as a noble-metal-free catalyst for the direct oxidation of formic acid under zero applied potential. Electrochem. Commun. 2019, 99, 1–4. [Google Scholar] [CrossRef]
- Jiang, J.; Scott, J.; Wieckowski, A. Direct evidence of a triple-path mechanism of formate electrooxidation on Pt black in alkaline media at varying temperature. Part I: The electrochemical studies. Electrochim. Acta 2013, 104, 124–133. [Google Scholar] [CrossRef]
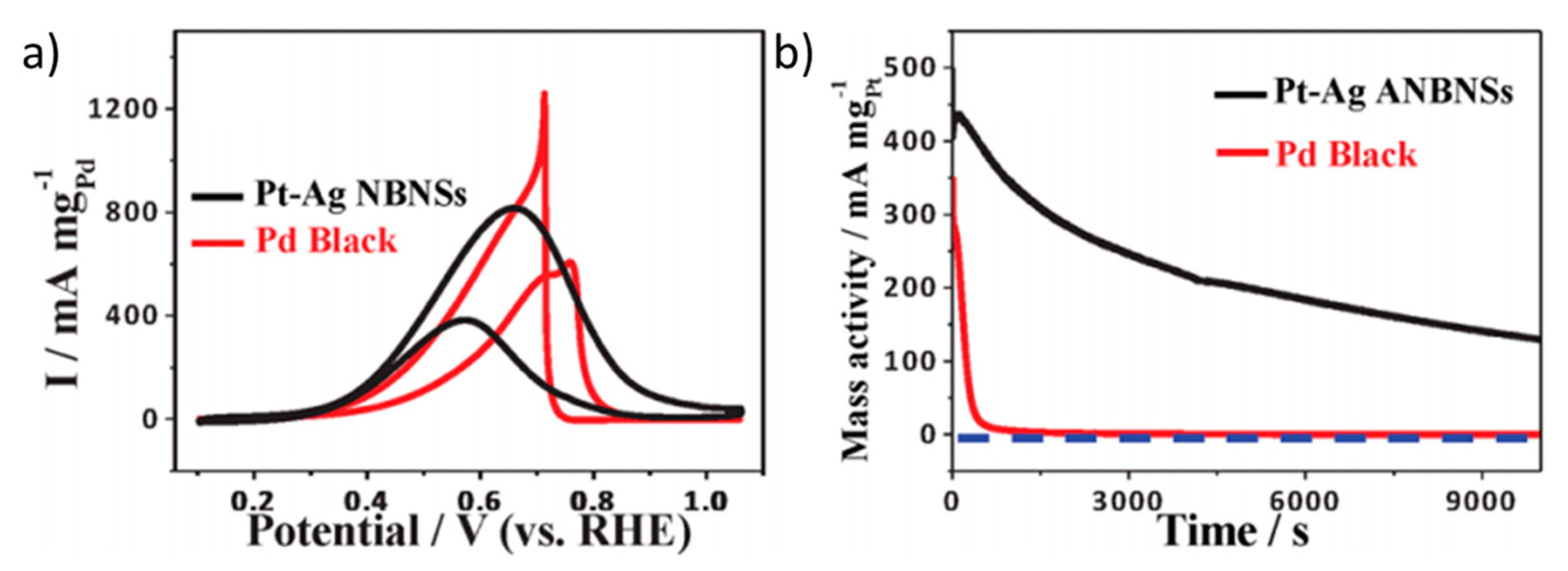
- Han, S.; Liu, H.; Bai, J.; Tian, X.; Xia, B.; Zeng, J.; Jiang, J.; Chen, Y. Platinum-Silver Alloy Nanoballoon Nanoassemblies with Super Catalytic Activity for the Formate Electrooxidation. ACS Appl. Energy Mater. 2018, 1, 1252–1258. [Google Scholar] [CrossRef]
- Lashkenari, M.; Ghorbani, M.; Safabakhsh, M.; Shahrokhi, B.; Fallah, J.; Rezaei, S. Fabrication of polyaniline/SBA-15-supported platinum/cobalt nanocomposites as promising electrocatalyst for formic acid oxidation. J. Appl. Electrochem. 2020, 50, 523–534. [Google Scholar] [CrossRef]
- Shi, H.; Liao, F.; Zhu, W.; Shao, C.; Shao, M. Effective PtAu nanowire network catalysts with ultralow Pt content for formic acid oxidation and methanol oxidation. Int. J. Hydrogen Energy 2020, 45, 16071–16079. [Google Scholar] [CrossRef]
- Menshikov, V.; Novomlinsky, I.; Belenov, S.; Alekseenko, A.; Safronenko, O.; Guterman, V. Methanol, Ethanol, and Formic Acid Oxidation on New Platinum-Containing Catalysts. Catalysts 2021, 11, 158. [Google Scholar] [CrossRef]
- Wang, C.; Yu, Z.; Li, G.; Song, Q.; Li, G.; Luo, C.; Yin, S.; Lu, B.; Xiao, C.; Xu, B.; et al. Intermetallic PtBi Nanoplates with High Catalytic Activity towards Electro-oxidation of Formic Acid and Glycerol. ChemElectroChem 2020, 7, 239–245. [Google Scholar] [CrossRef]
- Yu, X.; Manthiram, A. Catalyst-selective, scalable membraneless alkaline direct formate fuel cells. Appl. Catal. B Environ. 2015, 165, 63–67. [Google Scholar] [CrossRef]
- Chen, Y.; Niu, H.; Feng, Y.; Wu, J.; Wang, A.; Huang, H.; Feng, J. Three-dimensional hierarchical urchin-like PdCuPt nanoassembles with zigzag branches: A highly efficient and durable electrocatalyst for formic acid oxidation reaction. Appl. Surf. Sci. 2020, 510, 145480. [Google Scholar] [CrossRef]
- Hsieh, C.; Hsiao, H.; Tzou, D.; Yu, P.; Chen, P.; Jang, B. Electro-oxidation of methanol and formic acid on platinum nanoparticles with different oxidation levels. Mater. Chem. Phys. 2015, 149, 359–367. [Google Scholar] [CrossRef]
- Pisarek, M.; Kedzierzawski, P.; Andrzejczuk, M.; Holdynsky, M.; Mikolajkzuk-Zichora, A.; Borodzinsky, A.; Janik-Czachor, M. TiO2 nanotubes with pt and pd nanoparticles as catalysts for electro-oxidation of formic acid. Materials 2020, 13, 1195. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Song, L.; Elnabawy, A.; Marinkovic, M.; Mavrikakis, M.; Adzic, R. Platinum and Palladium Monolayer Electrocatalysts for Formic Acid Oxidation. Top. Catal. 2020, 63, 742–749. [Google Scholar] [CrossRef]
- Adić, R.R.; Spasojević, M.D.; Despić, A.R. Electrocatalysis by foreign metal monolayers. Oxidation of formic acid on palladium. J. Electroanal. Chem. 1978, 92, 31–43. [Google Scholar] [CrossRef]
- Chen, W.; Xu, L.P.; Chen, S. Enhanced electrocatalytic oxidation of formic acid by platinum deposition on ruthenium nanoparticle surfaces. J. Electroanal. Chem. 2009, 631, 36–42. [Google Scholar] [CrossRef]
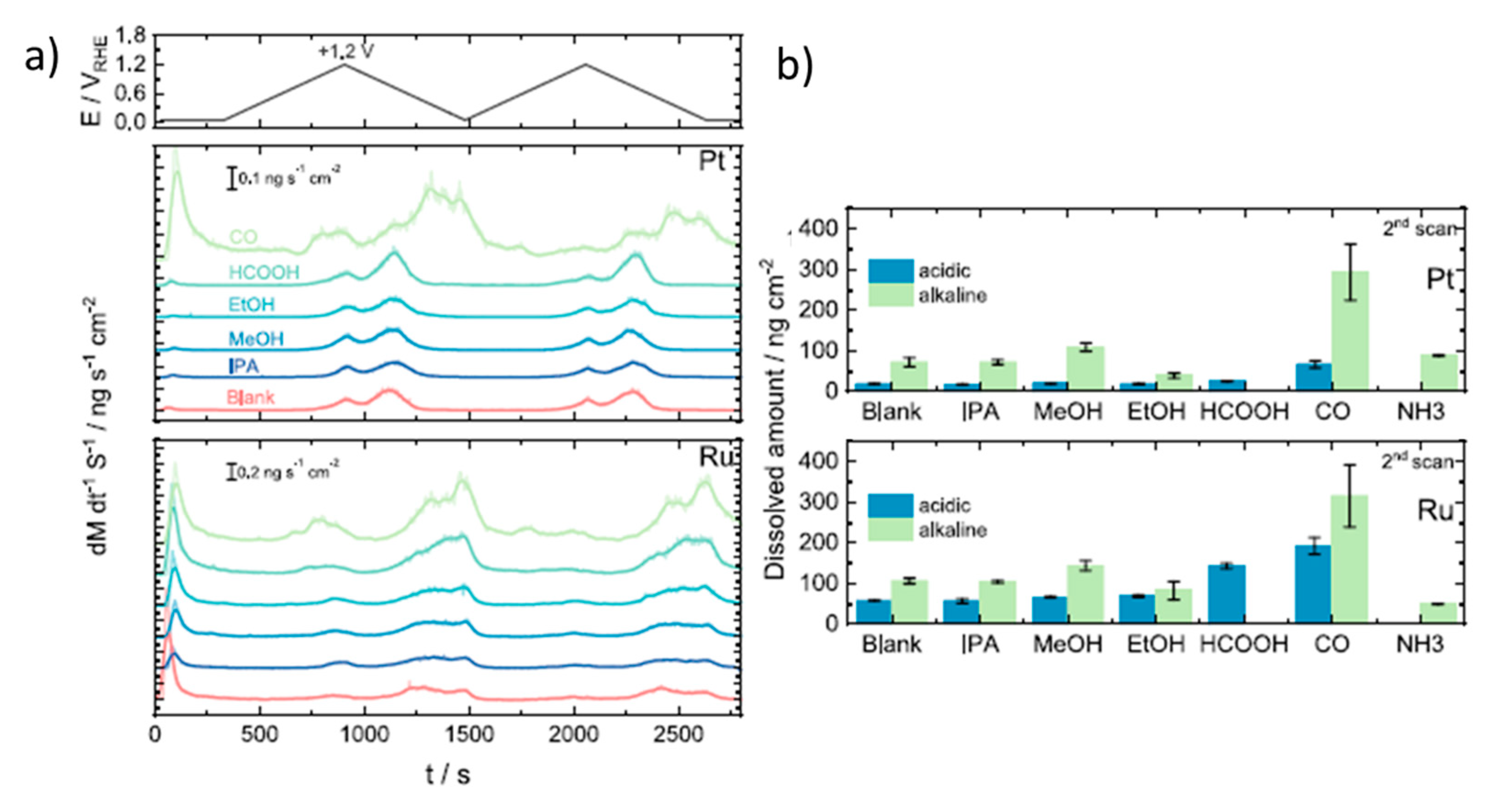
- Kormányos, A.; Speck, F.D.; Mayrhofer, K.J.J.; Cherevko, S. Influence of Fuels and pH on the Dissolution Stability of Bifunctional PtRu/C Alloy Electrocatalysts. ACS Catal. 2020, 10, 10858–10870. [Google Scholar] [CrossRef]
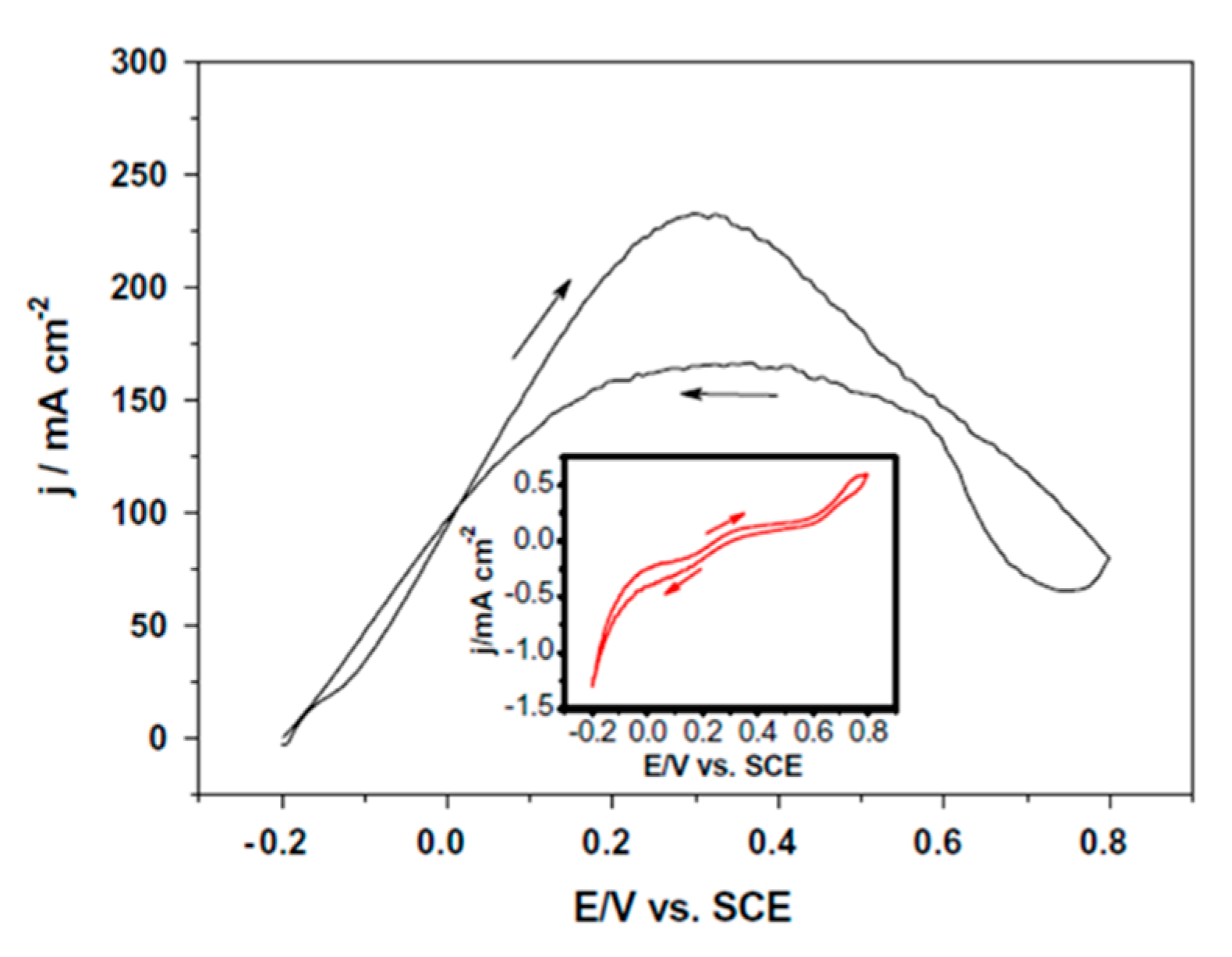
- Park, I.S.; Lee, K.S.; Choi, J.H.; Park, H.Y.; Sung, Y.E. Surface Structure of Pt-Modified Au Nanoparticles and Electrocatalytic Activity in Formic Acid Electro-Oxidation. J. Phys. Chem. C 2007, 111, 19126–19133. [Google Scholar] [CrossRef]
- Chen, W.; Kim, J.; Sun, S.; Chen, S. Electro-oxidation of formic acid catalyzed by FePt nanoparticles. Phys. Chem. Chem. Phys. 2006, 8, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
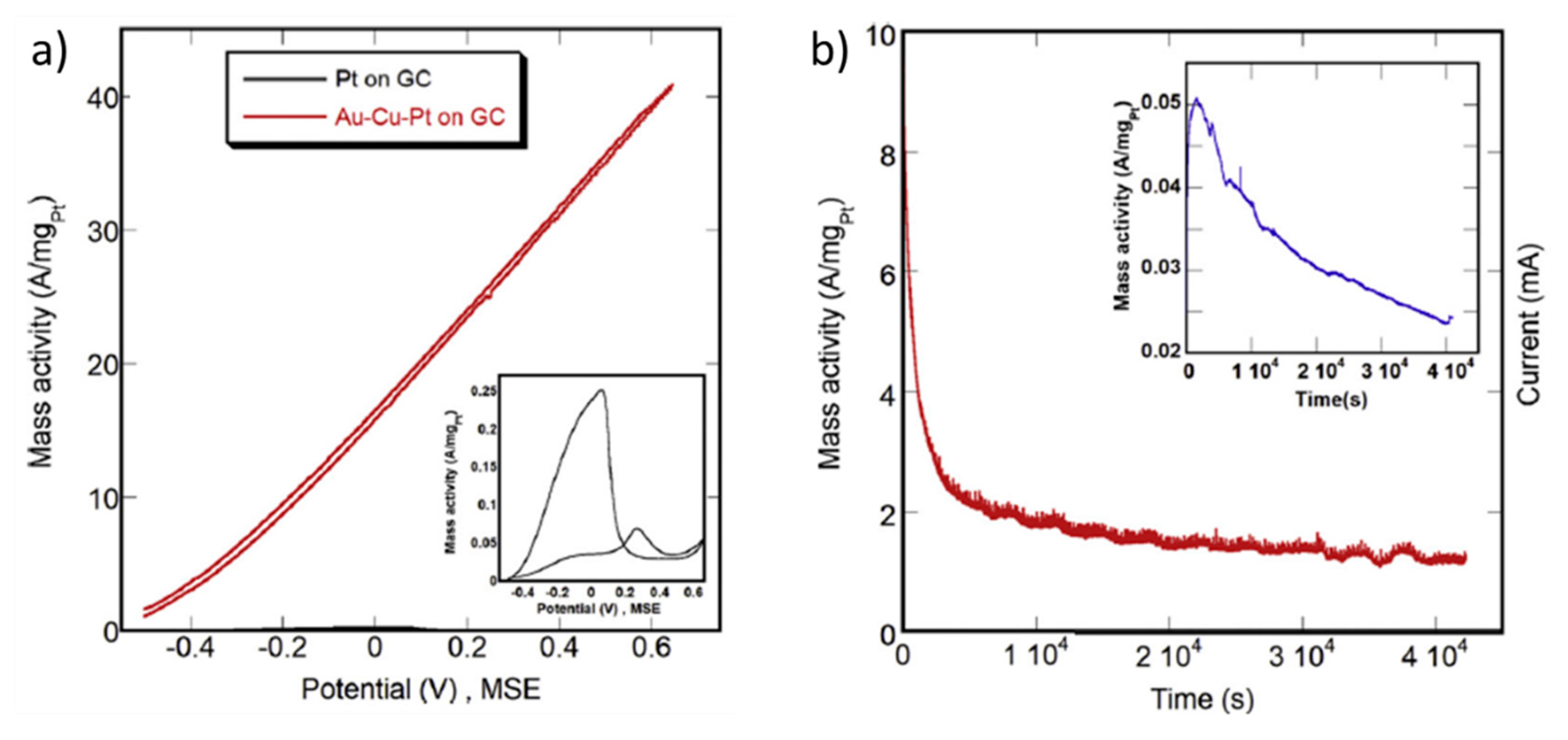
- Xie, Y.; Dimitrov, N. Ultralow Pt loading nanoporous Au-Cu-Pt thin film as highly active and durable catalyst for formic acid oxidation. Appl. Catal. B Environ. 2020, 263, 118366. [Google Scholar] [CrossRef]
- Ferre-Vilaplana, A.; Perales-Rondón, J.V.; Buso-Rogero, C.; Feliu, J.M.; Herrero, E. Formic acid oxidation on platinum electrodes: A detailed mechanism supported by experiments and calculations on well-defined surfaces. J. Mater. Chem. A 2017, 5, 21773–21784. [Google Scholar] [CrossRef] [Green Version]
- Aslam, N.M.; Masdar, M.S.; Kamarudin, S.K.; Daud, W.R.W. Overview on Direct Formic Acid Fuel Cells (DFAFCs) as an Energy Sources. Apcbee Procedia 2012, 3, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Nogalska, A.; Navarro, A.B.; Garcia-Valls, R. MEA preparation for direct formate/formic acid fuel cell—Comparison of palladium black and palladium supported on activated carbon performance on power generation in passive fuel cell. Membranes 2020, 10, 355. [Google Scholar] [CrossRef] [PubMed]
- Petrii, O.A. Pt-Ru electrocatalysts for fuel cells: A representative review. J. Solid State Electrochem. 2008, 12, 609–642. [Google Scholar] [CrossRef]
- Marković, N.; Gasteiger, H.; Ross, P.; Jiang, X.; Villegas, I.; Weaver, M. Electro-oxidation mechanisms of methanol and formic acid on Pt-Ru alloy surfaces. Electrochim. Acta 1995, 40, 91–98. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Marković, N.; Ross, P.N.; Cairns, E.J. Electro-oxidation of small organic molecules on well-characterized Pt-Ru alloys. Electrochim. Acta 1994, 39, 1825–1832. [Google Scholar] [CrossRef]
- Neurock, M.; Janik, M.; Wieckowski, A. A first principles comparison of the mechanism and site requirements for the electrocatalytic oxidation of methanol and formic acid over Pt. Faraday Discuss. 2008, 140, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Schalenbach, M.; Kasian, O.; Ledendecker, M.; Speck, F.; Mingers, A.; Mayrhofer, K.; Cherevko, S. The Electrochemical Dissolution of Noble Metals in Alkaline Media. Electrocatalysis 2018, 9, 153–161. [Google Scholar] [CrossRef]
- Cherevko, S.; Zeradjanin, A.R.; Keeley, G.P.; Mayrhofer, K.J.J. A Comparative Study on Gold and Platinum Dissolution in Acidic and Alkaline Media. J. Electrochem. Soc. 2014, 161, H822–H830. [Google Scholar] [CrossRef]
- da Silva, S.G.; Silva, J.C.M.; Buzzo, G.S.; Neto, A.O.; Assumpção, M.H.M.T. Use of PtAu/C electrocatalysts toward formate oxidation: Electrochemical and fuel cell considerations. Mater. Renew. Sustain. Energy 2016, 54, 15. [Google Scholar] [CrossRef] [Green Version]
- Capon, A.; Parsons, R. The oxidation of formic acid at noble metal electrodes Part 4. Platinum + palladium alloys. J. Electroanal. Chem. 1975, 65, 285–305. [Google Scholar] [CrossRef]
- Waszczuk, P.; Crown, A.; Mitrovski, S.; Wieckowski, A. Methanol and formic acid oxidation on ad-metal modified electrodes. In Handbook of Fuel Cells; John Wiley and Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Watanabe, M.; Horiuchi, M.; Motoo, S. Electrocatalysis by ad-atoms. Part XXIII. Design of platinum ad-electrodes for formic acid fuel cells with ad-atoms of the IVth and the Vth groups. J. Electroanal. Chem. 1988, 250, 117–125. [Google Scholar] [CrossRef]
- Motoo, S.; Watanabe, M. Electrocatalysis by Sn and Ge AD-atoms. J. Electroanal. Chem. 1976, 69, 429–431. [Google Scholar] [CrossRef]
- Adžić, R.R.; Simić, D.N.; Despić, A.R.; Dražić, D.M. Electrocatalysis by foreign metal monolayers: Oxidation of formic acid on platinum. J. Electroanal. Chem. Interfac. Electrochem. 1975, 65, 587–601. [Google Scholar] [CrossRef]
- Kwon, Y.; Birdja, Y.; Spanos, I.; Rodriguez, P.; Koper, M.T.M. Highly selective electro-oxidation of glycerol to dihydroxyacetone on platinum in the presence of bismuth. ACS Catal. 2012, 2, 759–764. [Google Scholar] [CrossRef]
- González-Cobos, J.; Baranton, S.; Coutanceau, C. A Systematic in Situ Infrared Study of the Electrooxidation of C3 Alcohols on Carbon-Supported Pt and Pt-Bi Catalysts. J. Phys. Chem. C 2016, 120, 7155–7164. [Google Scholar] [CrossRef]
- Tusi, M.M.; Polanco, N.S.O.; Da Silva, S.G.; Spinacé, E.V.; Neto, A.O. The high activity of PtBi/C electrocatalysts for ethanol electro-oxidation in alkaline medium. Electrochem. Commun. 2011, 13, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.; Uchida, T.; Cuesta, A.; Koper, M.T.M.; Osawa, M. Importance of acid-base equilibrium in electrocatalytic oxidation of formic acid on platinum. J. Am. Chem. Soc. 2013, 135, 9991–9994. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chen, W.; Wei, Z.; Xu, M.; He, Z.; Cai, J.; Chen, Y.; Santos, E. Mechanistic Implication of the pH Effect and H/D Kinetic Isotope Effect on HCOOH/HCOO–Oxidation at Pt Electrodes: A Study by Computer Simulation. ACS Catal. 2021, 11, 6920–6930. [Google Scholar] [CrossRef]
- Haan, J.L.; Masel, R.I. The influence of solution pH on rates of an electrocatalytic reaction: Formic acid electrooxidation on platinum and palladium. Electrochim. Acta 2009, 54, 4073–4078. [Google Scholar] [CrossRef]
- Rees, N.V.; Compton, R.G. Sustainable energy: A review of formic acid electrochemical fuel cells. J. Solid State Electrochem. 2011, 15, 2095–2100. [Google Scholar] [CrossRef]
- Beletskaya, I.P. The cross-coupling reactions of organic halides with organic derivatives of tin, mercury and copper catalyzed by palladium. J. Organomet. Chem. 1983, 250, 551–564. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Li, J.J. An introduction to palladium catalysis. In Tetrahedron Organic Chemistry Series; Elsevier: Amsterdam, The Netherlands, 2007; Volume 26, pp. 1–35. [Google Scholar]
- Conway, B.E.; Bockris, J.O. The d-Band Character of Metals and the Rate and Mechanism of the Electrolytic Hydrogen Evolution Reaction. Nature 1956, 178, 488–489. [Google Scholar] [CrossRef]
- Pentland, N.; Bockris, J.O.; Sheldon, E. Hydrogen Evolution Reaction on Copper, Gold, Molybdenum, Palladium, Rhodium, and Iron. J. Electrochem. Soc. 1957, 104, 182. [Google Scholar] [CrossRef]
- Savadogo, O.; Lee, K.; Oishi, K.; Mitsushima, S.; Kamiya, N.; Ota, K. New palladium alloys catalyst for the oxygen reduction reaction in an acid medium. Electrochem. Commun. 2004, 6, 105–109. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, H.-X.; Jiang, K.; Cai, W.-B. From HCOOH to CO at Pd Electrodes: A Surface-Enhanced Infrared Spectroscopy Study. J. Am. Chem. Soc. 2011, 133, 14876–14879. [Google Scholar] [CrossRef]
- Arenz, M.; Stamenkovic, V.; Schmidt, T.; Wandelt, K.; Ross, P.; Markovic, N. The electro-oxidation of formic acid on Pt-Pd single crystal bimetallic surfaces. Phys. Chem. Chem. Phys. 2003, 5, 4242–4251. [Google Scholar] [CrossRef]
- Solis, V.; Iwasita, T.; Pavese, A.; Vielstich, W. Investigation of formic acid oxidation on palladium in acidic solutions by on-line mass spectroscopy. J. Electroanal. Chem. 1988, 255, 155–162. [Google Scholar] [CrossRef]
- Hoshi, N.; Kida, K.; Nakamura, M.; Nakada, M.; Osada, K. Structural Effects of Electrochemical Oxidation of Formic Acid on Single Crystal Electrodes of Palladium. J. Phys. Chem B 2006, 110, 12480–12484. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Herron, J.; Scaranto, J.; Huang, H.; Wang, Y.; Xia, X.; Lv, T.; Park, J.; Peng, H.; Mavrikakis, M.; et al. A Comprehensive Study of Formic Acid Oxidation on Palladium Nanocrystals with Different Types of Facets and Twin Defects. ChemCatChem 2015, 7, 2077–2084. [Google Scholar] [CrossRef]
- Wang, H.; Qian, X.; Lui, S.; Yin, S.; Xu, Y.; Li, X.; Wang, Z.; Wang, L. Boron-Doped PdCuAu Nanospine Assembly as an Efficient Electrocatalyst toward Formic Acid Oxidation. Chem. Eur. J. 2020, 26, 2493–2498. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Qi, Z.; Zhao, C.; Ji, H.; Zhang, Z. High catalytic activity of ultrafine nanoporous palladium for electro-oxidation of methanol, ethanol, and formic acid. Electrochem. Commun. 2009, 11, 1896–1899. [Google Scholar] [CrossRef]
- Wang, J.; Chen, F.; Jin, Y.; Guo, L.; Gong, X.; Wang, X.; Johnston, R. In situ high-potential-driven surface restructuring of ternary AgPd-Ptdilute aerogels with record-high performance improvement for formate oxidation electrocatalysis. Nanoscale 2019, 11, 14174–14185. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Hwang, H.; Kim, J.W.; Lee, J. The Effect of Synthesis Temperature on Pd-H Catalyst Structure for Alkaline Direct Formate Fuel Cells. ECS Trans. 2018, 85, 149–158. [Google Scholar] [CrossRef]
- Galvan, V.; Glass, D.E.; Baxter, A.F.; Surya Prakash, G.K. Reduced Graphene Oxide Supported Palladium Nanoparticles for Enhanced Electrocatalytic Activity toward Formate Electrooxidation in an Alkaline Medium. ACS Appl. Energy Mater. 2019, 2, 7104–7111. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, F.; Jin, T.; Guo, L.; Wang, Q.; Liu, H. Alloying in inverse CeO2/Pd nanoparticles to enhance the electrocatalytic activity for the formate oxidation reaction. J. Mater. Chem. A 2019, 7, 22996–23007. [Google Scholar] [CrossRef]
- Choun, M.; Ham, K.; Shin, D.; Lee, J.K.; Lee, J. Catalytically active highly metallic palladium on carbon support for oxidation of HCOO−. Catal. Today 2017, 295, 26–31. [Google Scholar] [CrossRef]
- Sankar, S.; Anilkumar, G.M.; Tamaki, T.; Yamaguchi, T. Binary Pd–Ni Nanoalloy Particles over Carbon Support with Superior Alkaline Formate Fuel Electrooxidation Performance. ChemCatChem 2019, 11, 4731–4737. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, J.; Yang, C.; Yang, Y.; Zhang, Y.; Yang, F.; Ma, R.; Yang, L.; He, H.; Huang, H. Pd nanocrystals anchored on 3D hybrid architectures constructed from nitrogen-doped graphene and low-defect carbon nanotube as high-performance multifunctional electrocatalysts for formic acid and methanol oxidation. Mater. Today Energy 2020, 16, 100409–100417. [Google Scholar] [CrossRef]
- Wang, H.; Chen, H.; Wang, H.; Ou, C.; Li, R.; Liu, H. Synthesis of ultrafine low loading Pd–Cu alloy catalysts supported on graphene with excellent electrocatalytic performance for formic acid oxidation. Int. J. Hydrogen Energy 2020, 45, 10735–10744. [Google Scholar] [CrossRef]
- Yépez, O.; Scharifker, B.R. Oxidation of formate on hydrogen-loaded palladium. Int. J. Hydrogen Energy 2002, 27, 99–105. [Google Scholar] [CrossRef]
- Gharib, A.; Arab, A. Electrodeposited Pd, PdCd, and PdBi nanostructures: Preparation, characterization, corrosion behavior, and their electrocatalytic activities for formic acid oxidation. J. Electroanal. Chem. 2020, 866, 114166–114176. [Google Scholar] [CrossRef]
- Hwang, E.; Park, H.; Kim, H.; Ahn, S.H.; Kim, S.K. Electrochemically Fabricated Pd–In Catalysts for Carbon Dioxide-Formate/Formic Acid Inter-Conversion. Bull. Korean Chem. Soc. 2017, 38, 607–613. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, F.; Tang, Q.; Guo, L.; Gebremarian, T.; Jin, T.; Liu, H.; Kou, B.; Li, Z.; Bian, W. Transition from core-shell to janus segregation pattern in AgPd nanoalloy by Ni doping for the formate oxidation. Appl. Catal. B Env. 2020, 270, 118861. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, W. Nanoneedle-covered Pd-Ag nanotubes: High electrocatalytic activity for formic acid oxidation. J. Phys. Chem. C 2010, 114, 21190–21200. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, F.; Guo, L.; Jin, T.; Liu, H.; Wang, X.; Gong, X.; Liu, Y. Nanoalloying effects on the catalytic activity of the formate oxidation reaction over AgPd and AgCuPd aerogels. J. Mater. Chem. A 2019, 7, 16122–16135. [Google Scholar] [CrossRef]
- Douk, A.; Farsadrooh, M.; Damanigol, F.; Moghaddam, A.; Saravani, H.; Noroozifar, N. Porous three-dimensional network of Pd-Cu aerogel toward formic acid oxidation. RSC Adv. 2018, 8, 23539–23545. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Chung, S.; Park, J.; Hwang, J.; Lee, C.; Uhm, S.; Bong, S.; Lee, J. Contribution of Interstitial Boron in a Boron-Incorporated Palladium Catalyst Toward Formate Oxidation in an Alkaline Direct Formate Fuel Cell. ACS Catal. 2021, 11, 4722–4729. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The oxidation of formic acid at noble metal electrodes Part I. Review of Previous Work. Electroanal. Chem. Interfaclal Electrochem. 1973, 44, 1–7. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The oxidation of formic acid at noble metal electrodes Part III. Intermediates and mechanism on platinum electrodes. Electroanal. Chem. Interfac. Electrochem. 1973, 45, 205–231. [Google Scholar] [CrossRef]
- Sankar, S.; Anilkumar, G.M.; Tamaki, T.; Yamaguchi, T. Cobalt-Modified Palladium Bimetallic Catalyst: A Multifunctional Electrocatalyst with Enhanced Efficiency and Stability toward the Oxidation of Ethanol and Formate in Alkaline Medium. ACS Appl. Energy Mater. 2018, 1, 4140–4149. [Google Scholar] [CrossRef]
- Xi, Z.; Li, J.; Su, D.; Muzzio, M.; Yi, C.; Li, Q.; Sun, S. Stabilizing CuPd Nanoparticles via CuPd Coupling to WO2.72 Nanorods in Electrochemical Oxidation of Formic Acid. J. Am. Chem. Soc. 2017, 139, 15191–15196. [Google Scholar] [CrossRef]
- Rettenmaier, C.; Arán-Ais, R.; Timoshenko, J.; Rizo, R.; Jeon, H.; Külh, S.; Chee, S.; Bergmann, A.; Cuenya, B. Enhanced Formic Acid Oxidation over SnO2-decorated Pd Nanocubes. ACS Catal. 2020, 10, 14540. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.M.; Mattausch, Y.; Weiß, A.; Jacob, T.; Kibler, L.A. Enhanced Electrocatalytic Oxidation of Formic Acid on Au(111) in the Presence of Pyridine. J. Electrochem. Soc. 2018, 165, J3192–J3198. [Google Scholar] [CrossRef]
- Burke, L.D.; O’Dwyer, K.J. Application of the hydrous oxide mediation model of electrocatalysis to reactions at noble metal anodes in base-I.; Pt, Pd and Rh. Electrochim. Acta 1990, 35, 1821–1827. [Google Scholar] [CrossRef]
- Sathe, B.R.; Balan, B.K.; Pillai, V.K. Enhanced electrocatalytic performance of interconnected Rh nano-chains towards formic acid oxidation. Energy Environ. Sci. 2011, 4, 1029–1036. [Google Scholar] [CrossRef]
- Motoo, S.; Furuya, N. Electrochemistry of iridium single crystal surfaces. Part, I. Structural effect on formic acid oxidation and poison formation on Ir(111), (100) and (110). J. Electroanal. Chem. 1986, 197, 209–218. [Google Scholar] [CrossRef]
- Orozco, G.; Gutiérrez, C. Adsorption and electro-oxidation of carbon monoxide, methanol, ethanol and formic acid on osmium electrodeposited on glassy carbon. J. Electroanal. Chem. 2000, 484, 64–72. [Google Scholar] [CrossRef]
- Ouattara, L.; Fierro, S.; Frey, O.; Koudelka, M.; Comninellis, C. Electrochemical comparison of IrO2 prepared by anodic oxidation of pure iridium and IrO2 prepared by thermal decomposition of H2IrCl6 precursor solution. J. Appl. Electrochem. 2009, 39, 1361–1367. [Google Scholar] [CrossRef]
- Lindner, V.E.; Vitzthum, I.L.G.; Baresel, V.D.; Gellcrt, W.; Heidemeyer, J. Bindungsisomerie bei Sulfinato-Komplexen von Obergangsmetallen Wolframcarbid als Anodenmaterial fur Brenn-stoffzellen. Angew. Chem. 1971, 83, 213–214. [Google Scholar] [CrossRef]
- Palanker, V.S.; Gajyev, R.A.; Sokolsky, D.V. On adsorption and electro-oxidation of some compounds on tungsten carbide; their effect on hydrogen electro-oxidation. Electrochim. Acta 1977, 22, 133–136. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Zhao, F.; Quaas, M.; Wulff, H.; Schröder, U.; Scholz, F. Evaluation of catalytic properties of tungsten carbide for the anode of microbial fuel cells. Appl. Catal. B Environ. 2007, 74, 261–269. [Google Scholar] [CrossRef]
- Böhm, H. New non-noble metal anode catalysts for acid fuel cells. Nature 1970, 227, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.N.; Noor Azam, A.M.I.; Isahak, W.N.R.W.; Mohd Zainoodin, A.; Masdar, M.S. Improving the electrocatalytic activity for formic acid oxidation of bimetallic Ir-Zn nanoparticles decorated on graphene nanoplatelets. Mater. Res. Express 2020, 7, 015095. [Google Scholar] [CrossRef]
- Kumar, A.; Joshi, L.; Prakash, R. Electrocatalytic performance of interfacially synthesized Au-polyindole composite toward formic acid oxidation. Ind. Eng. Chem. Res. 2013, 52, 9374–9380. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Suresh, P.; Viva, F.; Olah, G.A. Novel single step electrochemical route to γ-MnO2 nanoparticle-coated polyaniline nanofibers: Thermal stability and formic acid oxidation on the resulting nanocomposites. J. Power Source 2008, 181, 79–84. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, A.C.; Prakash, R. Electro-oxidation of formic acid using polyindole-SnO2 nanocomposite. Catal. Sci. Technol. 2012, 2, 2533–2538. [Google Scholar] [CrossRef]
- Li, Z.; Chen, Y.; Ji, S.; Tang, Y.; Chen, W.; Li, A.; Zhao, J.; Xiong, W.; Wu, Y.; Gong, Y.; et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host–guest strategy. Nat. Chem. 2020, 12, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Dong, J.; Huang, Z.; Xin, P.; Chen, W.; Wang, Y.; Li, Z.; Yin, Z.; Xin, W.; Zhuang, Z.; et al. Single-atom Rh/N-doped carbon electrocatalyst for formic acid oxidation. Nat. Nanotechnol. 2020, 15, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Crépy, G.; Lamy, C.; Maximovitch, S. Oxydation de l’acide formique sur électrode d’or. J. Electroanal. Chem. 1974, 54, 161–179. [Google Scholar] [CrossRef]
- Cuesta, A.; Cabello, G.; Hartl, F.; Escudero-Escribano, M.; Vazquez-Dominguez, C.; Kibler, L.; Osawa, M.; Gutierrez, C. Electrooxidation of formic acid on gold: An ATR-SEIRAS study of the role of adsorbed formate. Catal. Today 2013, 202, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Kibler, L.A.; Al-Shakran, M. Adsorption of Formate on Au(111) in Acid Solution: Relevance for Electro-Oxidation of Formic Acid. J. Phys. Chem. C 2016, 120, 16238–16245. [Google Scholar] [CrossRef]
- Fierro, S.; Ouattara, L.; Calderon, E.; Passas-Lagos, E.; Baltruschat, H.; Comninellis, C. Investigation of formic acid oxidation on Ti/IrO2 electrodes. Electrochim. Acta 2009, 54, 2053–2061. [Google Scholar] [CrossRef]
- Ma, C.A.; Zhang, W.K.; Cheng, D.H.; Zhou, B.X. Preparation and electrocatalytic properties of tungsten carbide electrocatalysts. Trans. Nonferrous Met. Soc. China 2002, 12, 1015–1019. [Google Scholar]
- Baresel, D.; Gellert, W.; Heidemeyer, J.; Scharner, P. Wolframcarbid als Anodenmaterial für Brennstoffzellen. Angew. Chem. 1971, 83, 213–214. [Google Scholar] [CrossRef]
- Kim, D.; Cargnello, M. Formic acid oxidation boosted by Rh single atoms. Nat. Nanotechnol. 2020, 15, 346–347. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidant | Reaction | Thermodynamic Potential, E0 vs. NHE (V) i | Theoretical Fuel Cell Voltage (V) ii | |
|---|---|---|---|---|
| With Acid Anode (HCOOH Oxidation at pH 0) | With Alkaline Anode (HCOO− Oxidation at pH 14) | |||
| O2/Air | O2 + 4H+ + 4e− → 2H2O | 1.229 | 1.449 | 2.275 |
| K2CrO7 | Cr2O72− + 14H+ + 6e− → 2Cr3+ + 7H2O | 1.360 | 1.580 | 2.406 |
| HClO | HClO + H+ + 2e− → Cl− + H2O | 1.482 | 1.702 | 2.528 |
| HClO + H+ + e− → 0.5Cl2 + H2O | 1.611 | 1.831 | 2.657 | |
| KMnO4 | MnO4− + 8H+ + 5e− → Mn2+ + 4H2O | 1.507 | 1.727 | 2.553 |
| MnO4− + 4H+ + 3e− → MnO2 + 2H2O | 1.679 | 1.899 | 2.725 | |
| Ce(NH4)2(NO3)6 | Ce4+ + e− → Ce3+ | 1.720 | 1.940 | 2.766 |
| H2O2 | H2O2 + 2H+ + 2e− → 2H2O | 1.776 | 1.996 | 2.822 |
| K2S2O8 | S2O82− + 2H+ + 2e− → 2HSO4− | 2.123 | 2.343 | 3.169 |
| Ref. | Catalyst | Experimental Conditions i | Eonset (V vs. RHE) ii | jmax (mA cm−2) at Epeak (V vs. RHE) ii | j (mA cm−2) ii at | |||
|---|---|---|---|---|---|---|---|---|
| 1.00 V vs. RHE | 1.20 V vs. RHE | 1.40 V vs. RHE | 1.60 V vs. RHE | |||||
| [40] | Pt disk | 0.2 M HCOOH + 1 M HClO4 (pH ≈ 0) 20 mV s−1 | 0.2 | 1.0 mA cm−2 (0.9 V vs. RHE) | 0.4 | 0.2 | 0.9 | - |
| 0.2 M HCOOH + 1 M NaOH (pH ≈ 14.0) 20 mV s−1 | 0.3 | 0.1 mA cm−2 (0.5 V vs. RHE) | <0.1 | <0.1 | <0.1 | - | ||
| [51] | Pt disk | 0.3 M HCOOH (pH 3.5) 100 mV s−1 | 0.4 | 1.8 mA cm−2 (1.0 V) | 1.7 | - | - | - |
| [52] | Pt disk | 1 M HCOONa + 0.1 M H2SO4 (pH 4.5) 50 mV s−1 | 0.2 | 2.5 mA cm−2 (0.6 V vs. RHE) | 1.9 | 1.8 | - | - |
| [53] | Pt disk | 0.4 M HCOOH + 1 M KNO3 (pH 5) 5 mV s−1 | 0.3 | 0.5 mA cm−2 (1.4 V vs. RHE) | 0.3 | 0.3 | 0.4 | 0.3 |
| 0.4 M HCOOH + 1 M KNO3 (pH 13) 5 mV s−1 | 0.5 | 3.1 mA cm−2 (1.1 V vs. RHE) | 2.1 | 2.0 | 0.9 | - | ||
| [54] | Pt disk | 0.1 M HCOOK + 0.2 M K2SO4 (pH ≈ 8.4) 50 mV s−1 | 0.5 | 4.3 mA cm−2 (0.9 V vs. RHE) | 2.7 | 1.5 | - | - |
| [55] | Pt rotating disk | 0.1 M HCOOH + 0.2 M KPi (pH 3.7) 50 mV s−1, 1000 rpm | 0.5 | 2.9 mA cm−2 (0.9 V vs. RHE) | 0.9 | 0.4 | 0.3 | - |
| [56] | Pt rotating disk | 0.02 M HCOONa + 0.2 M KPi (pH 4.2) 20 mV s−1, 1000 rpm | 0.3 | 8.5 mA cm−2 (0.9 V vs. RHE) | 7.7 | 4.5 | - | - |
| [57] | Pt net | 0.1 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 100 mV s−1 | 0.3 | 3.1 mA cm−2 (0.9 V vs. RHE) | 1.5 | 0.4 | 2.9 | - |
| [58] | Pt bead | 0.1 M HCOOH + 0.5 M Na2SO4 (pH 3.6) 50 mV s−1 | 0.5 | 5.4 mA cm−2 (0.9 V vs. RHE) | 2.4 | 1.0 | 0.6 | - |
| [59] | Pt(111) | 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.3 | 1.5 mA cm−2 (0.6 V vs. RHE) | <0.1 | - | - | - |
| [60] | Pt(111) | 0.05 M HCOONa + 0.2 M NaPi (pH 5.1) 10 mV s−1 | 0.3 | 2.4 mA cm−2 (0.6 V vs. RHE) | - | - | - | - |
| [61] | Pt(111) | 0.1 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.3 | 2.2 mA cm−2 (0.6 V vs. RHE) | - | - | - | - |
| 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.3 | 2.8 mA cm−2 (0.5 V vs. RHE) | - | - | - | - | ||
| [62] | Pt nanoparticles | 0.5 M HCOOH + 0.1 M H2SO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 10 mA cm−2 (0.8 V vs. RHE) | 3.4 | 6.1 | 8.3 | - |
| [63] | Pt nanoparticles | 2.1 M HCOOH + 0.5 M KNO3 (pH ≈ 1.7) 40 mV s−1 | 0.5 | 55.0 mA cm−2 (1.2 V vs. RHE) | 45.0 | 48.0 | 36.0 | - |
| [64] | Pt nanoparticles (Pt black) | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 0.7 mA cm−2 (0.6 V vs. RHE) | 0.5 | 0.4 | - | - |
| 0.5 M HCOOK + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.2 | 0.2 mA cm−2 (0.5 V vs. RHE) | <0.1 | <0.1 | - | - | ||
| [65] | Pt nanoparticles (Pt black) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.3 | 2.6 mA cm−2 (0.5 V vs. RHE) | <1.0 | - | - | - |
| [52] | Polyaniline/Pt disk | 1 M HCOONa + 0.1 M H2SO4 (pH 4.5) 50 mV s−1 | 0.2 | 23.0 mA cm−2 (0.8 V vs. RHE) | 8.6 | 4.5 | 6.8 | - |
| [66] | Pt nanoparticles/Polyaniline/SBA-15 | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.3 | 33.2 mA cm−2 (1.0 V vs. RHE) | 32.9 | 15.6 | - | - |
| [67] | Pt(20%)/C | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 13.1 mA cm−2 (1.0 V vs. RHE) | 12.0 | 10.0 | - | - |
| [68] | Pt(20%)/C | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1) 20 mV s−1 | 0.3 | 13.5 mA cm−2 (1.0 V vs. RHE) | 9.9 | - | - | - |
| [69] | Pt(20%)/Vulcan carbon | 0.25 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.3 | 4.4 mA cm−2 (0.9 V vs. RHE) | 2.7 | - | - | - |
| [70] | Pt(40%)/Vulcan carbon | 1 M HCOOH + 1 M H2SO4 (pH ≈ 0) 20 mV s−1 | 0.2 | 110.0 mA cm−2 (1.1 V vs. RHE) | 96.0 | 58.0 | 78.0 | - |
| 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | 0.4 | 14.6 mA cm−2 (0.6 V vs. RHE) | 3.4 | 3.4 | 3.7 | 9.2 | ||
| [71] | Pt(50%)/C | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.8 | 33.9 mA cm−2 (1.2 V vs. RHE) | 18.5 | 33.0 | 13.7 | 8.0 |
| [72] | Pt nanoparticles/Vulcan carbon | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.3 | 5.2 mA cm−2 (1.0 V vs. RHE) | 5.2 | 6.4 | - | - |
| [62] | Pt nanoparticles/Carbon nanoparticles | 0.5 M HCOOH + 0.1 M H2SO4 (pH ≈ 1.0) 50 mV s−1 | 0.1 | 38.9 mA cm−2 (0.9 V vs. RHE) | 23.4 | 23.7 | 32.5 | - |
| Pt nanoparticles/Reduced graphene oxide | 0.5 M HCOOH + 0.1 M H2SO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 27.7 mA cm−2 (0.9 V vs. RHE) | 9.7 | 10.4 | 12.7 | - | |
| [73] | Pt nanoparticles/ TiO2 nanotubes | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 10 mV s−1 | 0.8 | 14.0 mA cm−2 (1.0 V vs. RHE) | 14.0 | 4.4 | - | - |
| [74] | Pt monolayer/Ru(0001) | 0.5 M HCOOH + 0.1 M HclO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 1.1 mA cm−2 (0.9 V vs. RHE) | 0.9 | - | - | - |
| Pt monolayer/Rh(111) | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 1.5 mA cm−2 (1.0 V vs. RHE) | 1.4 | - | - | - | |
| Pt monolayer/Pd(111) | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 0.6 mA cm−2 (1.0 V vs. RHE) | 0.4 | - | - | - | |
| Pt monolayer/Au(111) | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 7.3 mA cm−2 (0.6 V vs. RHE) | 4.1 | - | - | - | |
| [75] | Pt disk + 1 × 10−3 M Cd2+ (in electrolyte) | 0.265 M HCOOH + 1 M HClO4 (pH ≈ 0) 50 mV s−1 | 0.2 | 7.2 mA cm−2 (0.5 V vs. RHE) | 1.0 | 0.9 | 3.3 | - |
| Pt disk + 5 × 10−4 M Ti+ (in electrolyte) | 0.265 M HCOOH + 1 M HClO4 (pH ≈ 0) 50 mV s−1 | 0.1 | 20.7 mA cm−2 (0.5 V vs. RHE) | 1.1 | - | - | - | |
| Pt disk + 1 × 10−3 M Bi3+ (in electrolyte) | 0.265 M HCOOH + 1 M HClO4 (pH ≈ 0) 50 mV s−1 | 0.5 | 46.0 mA cm−2 (0.8 V vs. RHE) | 1.1 | 1.0 | 1.9 | - | |
| Pt disk + 1 × 10−3 M Pb2+ (in electrolyte) | 0.265 M HCOOH + 1 M HClO4 (pH ≈ 0) 50 mV s−1 | 0.2 | 70.2 mA cm−2 (0.5 V vs. RHE) | 1.3 | 1.5 | 3.7 | - | |
| [76] | PtRu | 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 100 mV s−1 | −0.1 | 2.2 mA cm−2 (0.6 V vs. RHE) | 2.7 | - | - | - |
| [77] | PtRu | 0.05 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 10 mV s−1 | 0.2 | 5.1 mA cm−2 (0.8 V vs. RHE) | 2.9 | 1.7 | - | - |
| [70] | Pt1Ru1(40%)/Vulcan carbon | 1 M HCOOH + 1 M H2SO4 (pH ≈ 0) 20 mV s−1 | 0.3 | 145.0 mA cm−2 (1.0 V vs. RHE) | 145.0 | 44.0 | 49.0 | - |
| [78] | PtAu/C | 1 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 20 mV s−1 | 0.1 | 4.0 mA cm−2 (0.6 V vs. RHE) | 1.8 | 1.3 | - | - |
| [68] | PtCu/C | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1) 20 mV s−1 | 0.3 | 30.8 mA cm−2 (1.1 V vs. RHE) | 19.0 | - | - | - |
| Pt/SnO2/C | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1) 20 mV s−1 | 0.2 | 32.8 mA cm−2 (1.1 V vs. RHE) | 14.3 | - | - | - | |
| [69] | Pt(20%)Bi/Vulcan carbon | 0.25 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.1 | 46.1 mA cm−2 (0.8 V vs. RHE) | 3.0 | - | - | - |
| [79] | PtFe nanoparticles | 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 100 mV s−1 | 0.2 | 32.3 mA cm−2 (1.3 V vs. RHE) | 14.3 | 22.4 | 30.2 | - |
| [67] | Pt0.05Au nanowires | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 33.5 mA cm−2 (0.6 V vs. RHE) | 10.0 | 11.0 | - | - |
| [66] | PtCo nanoparticles | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.4 | 19.5 mA cm−2 (1.0 V vs. RHE) | 17.4 | 9.1 | - | - |
| PtCo nanoparticles/Polyaniline/SBA–15 | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.3 | 63.8 mA cm−2 (1.1 V vs. RHE) | 46.8 | 39.4 | - | - | |
| [65] | PtAg alloy nanoballoon nanoassembly | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 32.6 mA cm−2 (0.7 V vs. RHE) | <2.0 | - | - | - |
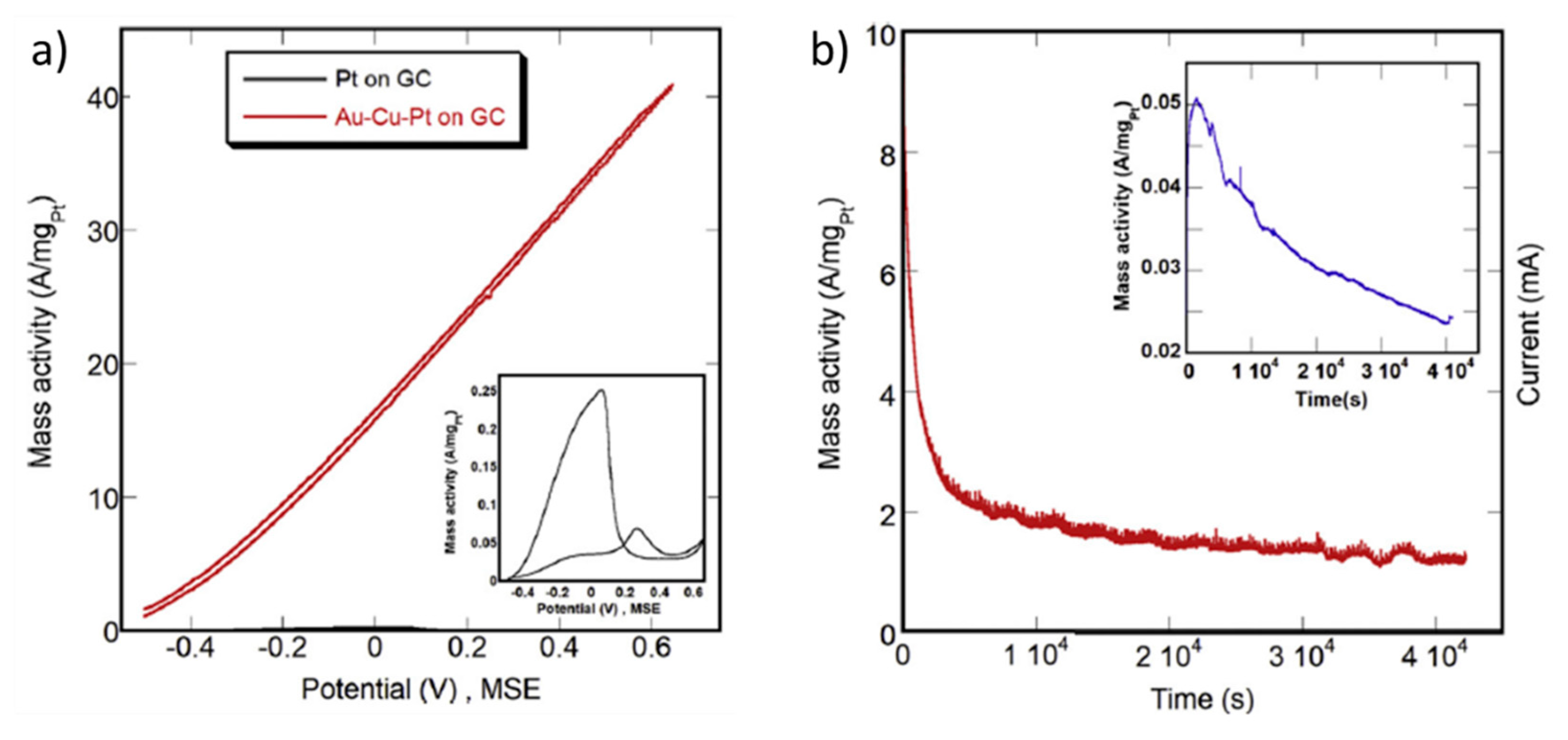
| [80] | Pt(1.3 at.%)AuCu dealloyed | 2 M HCOOH + 0.1 M HClO4 (pH ≈ 0.9) 50 mV s−1 | 0.2 | 54.8 A mgPt−1 (1.1 V vs. RHE) iii | 52.1 A mgPt−1 iii | 1.4 A mgPt−1 iii | - | - |
| Pt(2.6 at.%)AuCu dealloyed | 2 M HCOOH + 0.1 M HClO4 (pH ≈ 0.9) 50 mV s−1 | 0.2 | - | 28.0 iii | 35.8 iii | - | - | |
| Ref. | Catalyst | Experimental Conditions i | Eonset (V vs. RHE) ii | jmax (mA cm) at Epeak (V vs. RHE) ii | j (mA cm−2) ii at | |||
|---|---|---|---|---|---|---|---|---|
| 1.00 V vs. RHE | 1.20 V vs. RHE | 1.40 V vs. RHE | 1.60 V vs. RHE | |||||
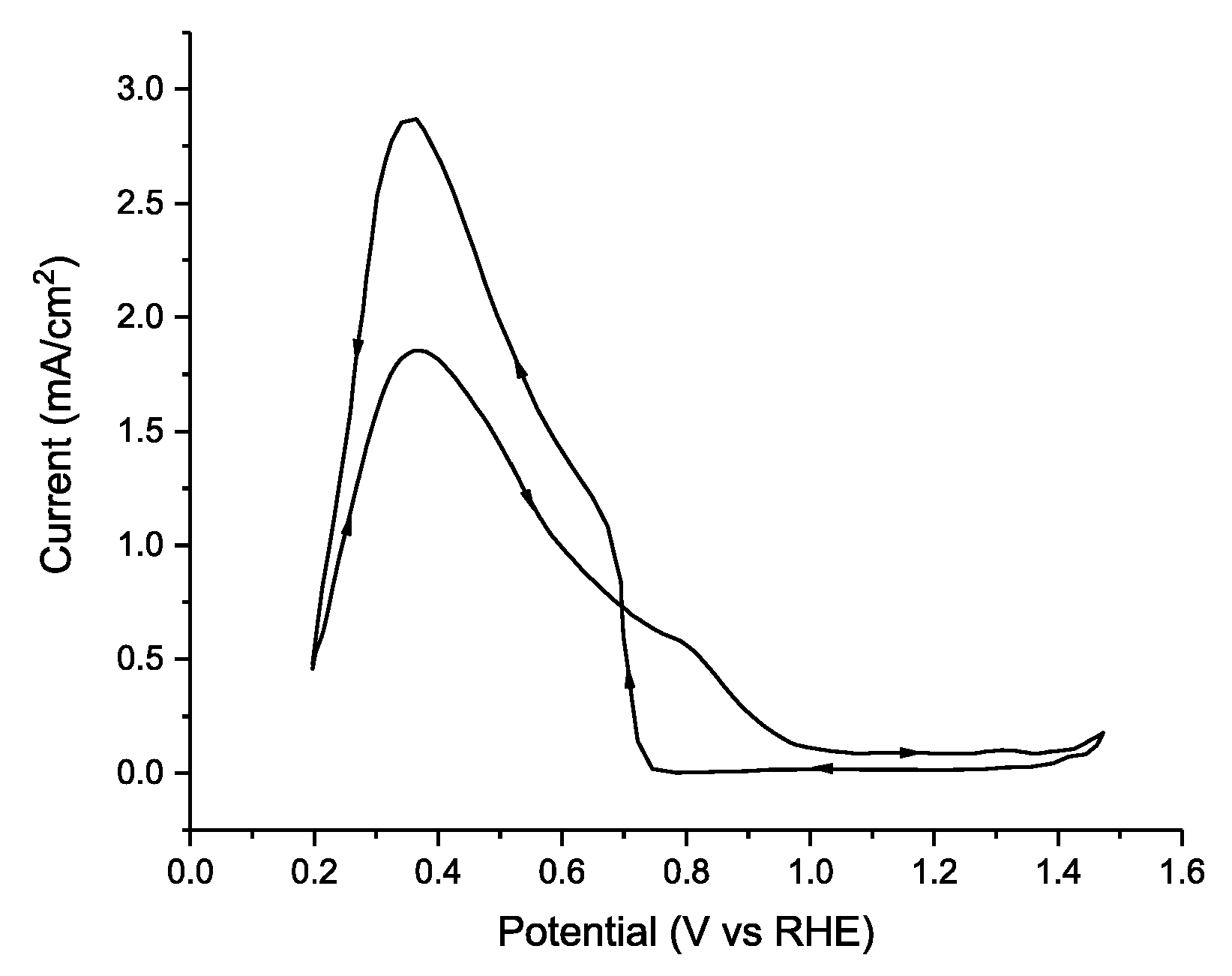
| [30] | Pd disk | 1.0 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 70 mV s−1 | N/A | 2.8 mA/cm−2 (0.36 V vs. RHE) | 0.12 | 0.087 | 0.094 | - |
| [110] | Pd foil | 0.01 M HCOOH + 0.5 M HClO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 3.2 mA/cm (0.3 V vs. RHE) | 0.11 | 0.11 | 0.16 | 1.07 |
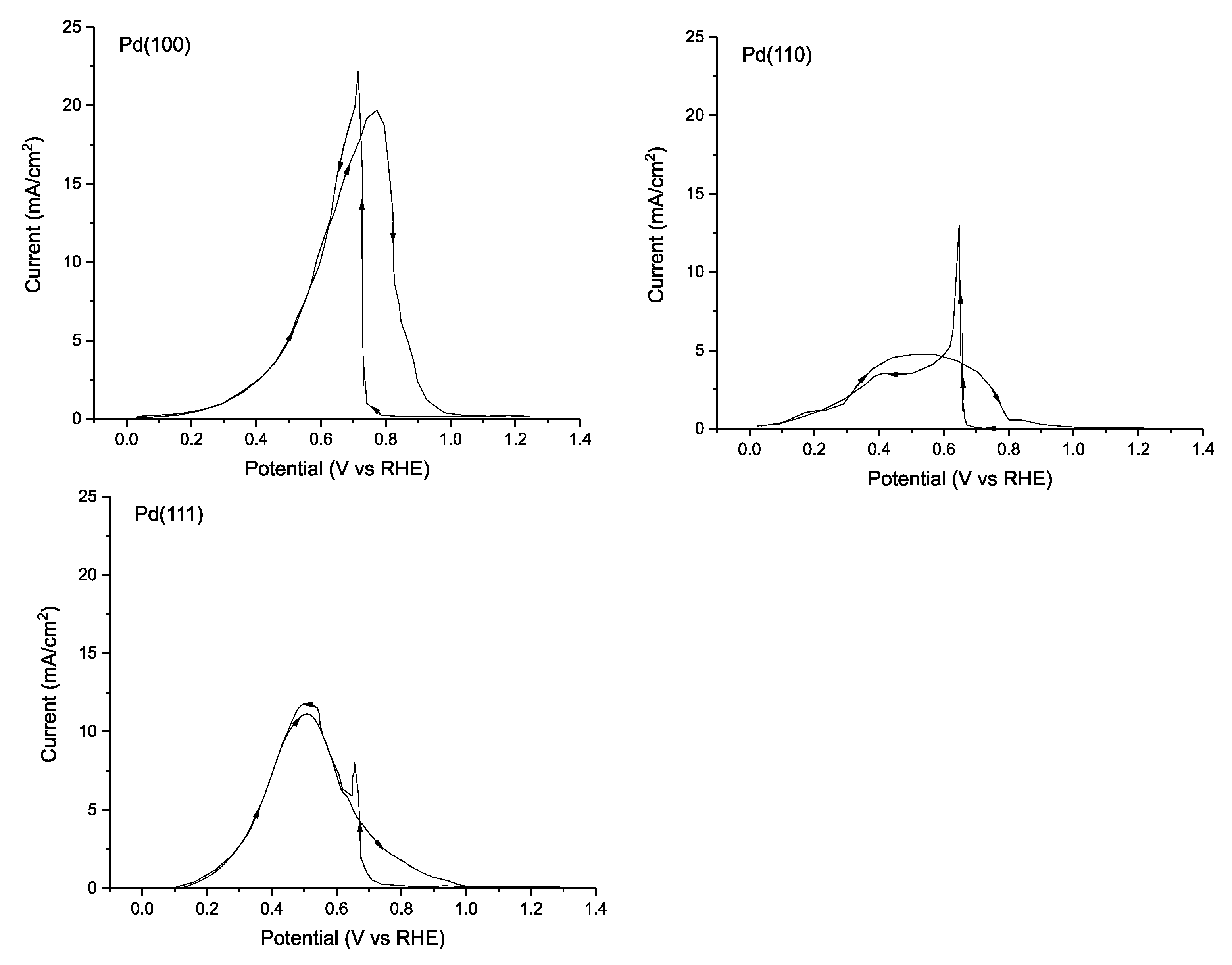
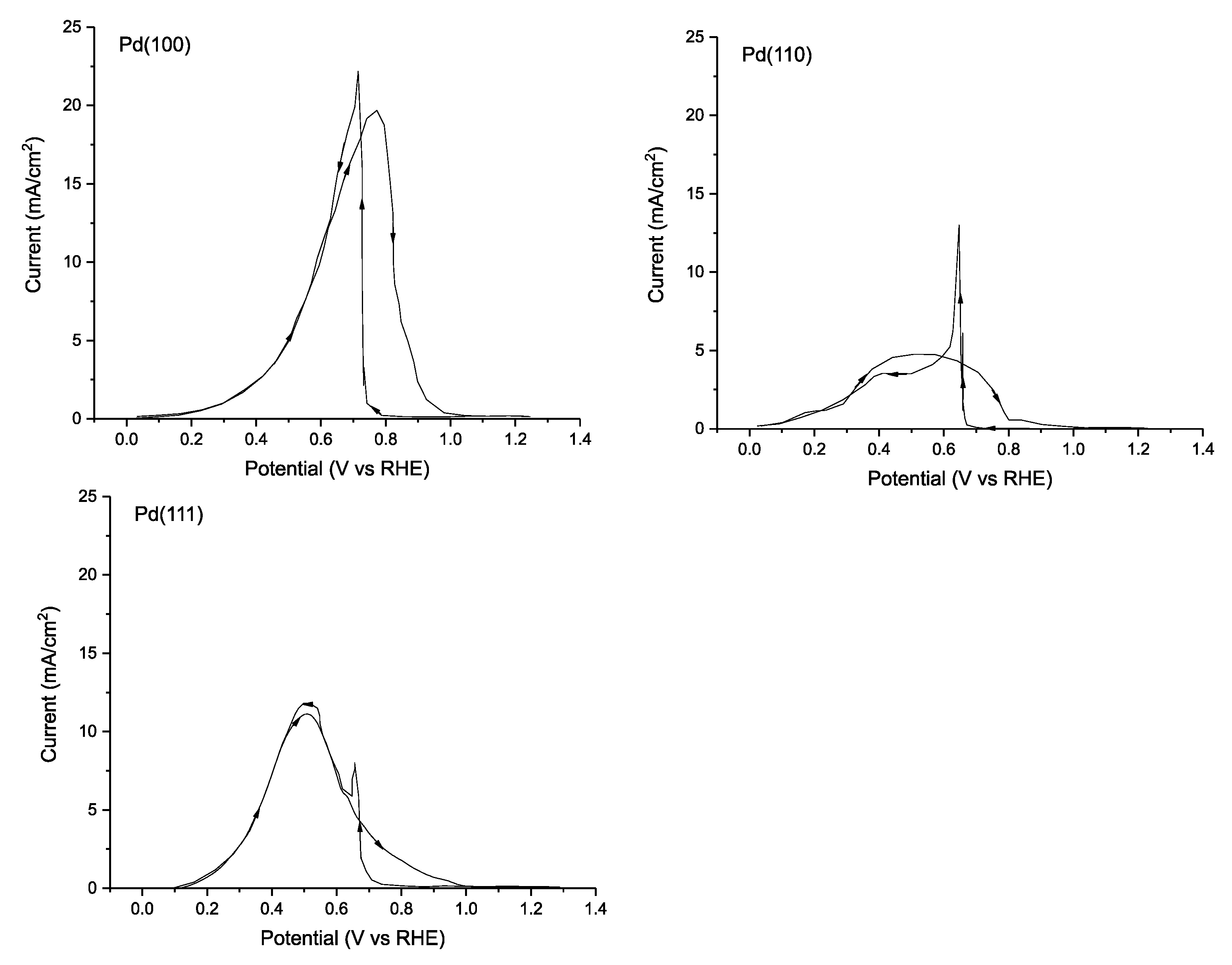
| [111] | Pd(100) | 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 20 mV s−1 | 0.1 | 19.7 mA cm (0.77 V vs. RHE) | <1.0 | <1.0 | - | - |
| Pd(110) | 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 20 mV s−1 | 0.1 | 11.0 mA cm (0.52 V vs. RHE) | <1.0 | <1.0 | - | - | |
| Pd(111) | 0.1 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 20 mV s−1 | 0.2 | 4.8 mA cm (0.5 V vs. RHE) | <1.0 | <1.0 | - | - | |
| [112] | Pd nanocubes | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 10.1 mA cm (0.54 V vs. RHE) | 1.0 | - | - | - |
| Pd nanooctahedra | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 6 mA cm−2 (0.47 V vs. RHE) | 0.45 | - | - | - | |
| Pd nanoicosahedrons | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.2 | 10.4 mA cm−2 (0.46 V vs. RHE) | 0.32 | - | - | - | |
| [71] | Pd black | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.8 | 27.3 mA cm−2 (1.2 V vs. RHE) | 13.6 | 26.4 | 12.5 | 6.0 |
| [113] | Pd black | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 5.4 mA cm−2 (0.5 V vs. RHE) | <1.0 | <1.0 | - | - |
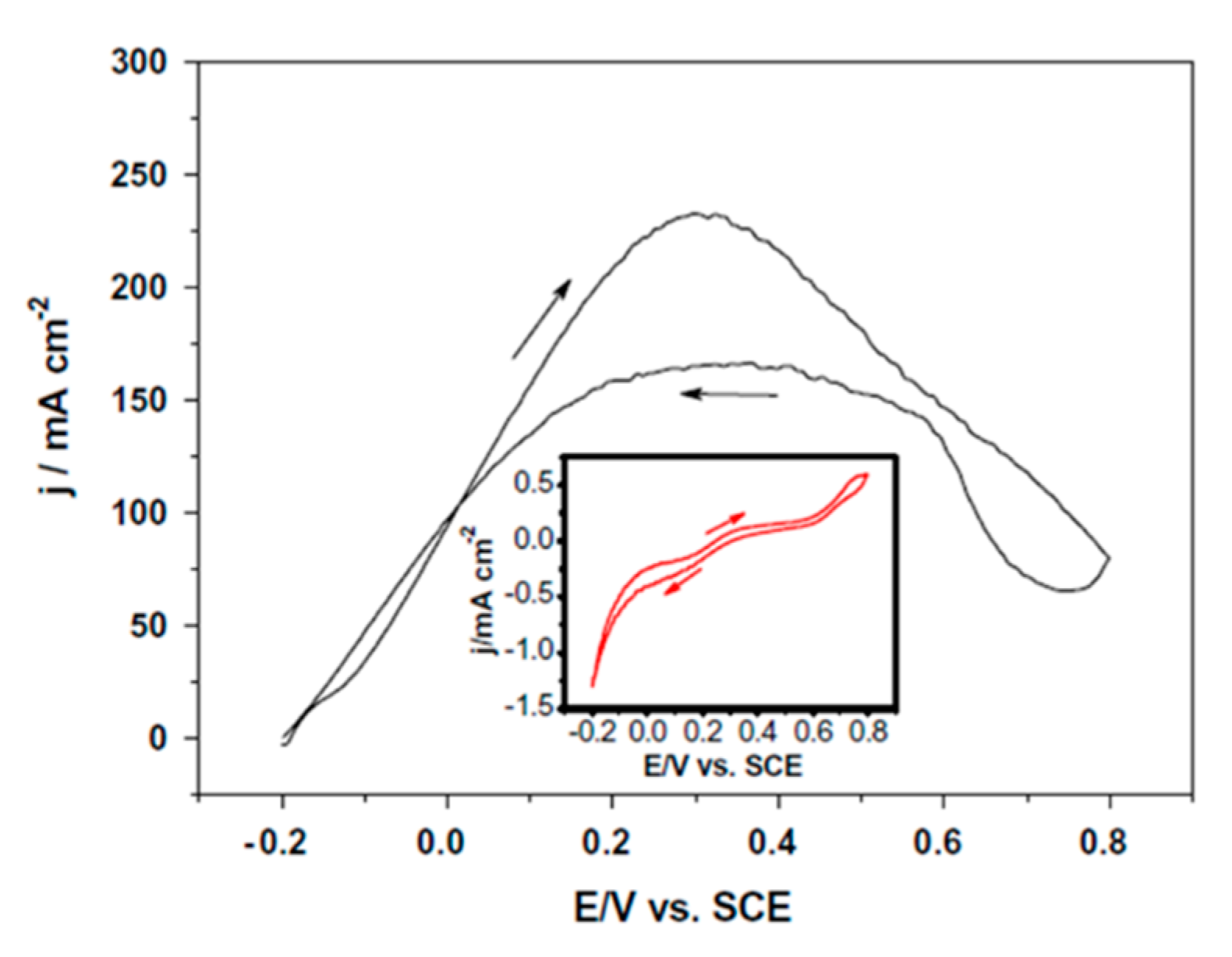
| [114] | Pd (nanoporous) | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 10 mV s−1 | 0.1 | 232 mA cm−2 (0.6 V vs. RHE) | 103.0 | - | - | - |
| [53] | Pd(5%)/Vulcan carbon | 0.4 M HCOOH + 1 M KNO3 (pH 5) 5 mV s−1 | 0.1 | 0.6 mA cm−2 (0.6 V vs. RHE) | 0.5 | 0.7 | 1.4 | 3.4 |
| 0.4 M HCOOH + 1 M KNO3 (pH 13) 5 mV s−1 | 0.2 | 3.3 mA cm−2 (0.7 V vs. RHE) | 2.4 | 2.2 | 1.9 | - | ||
| [115] | Pd/C | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.1 | 108.8 mA cm−2 (0.7 V vs. RHE) | 14.6 | - | - | - |
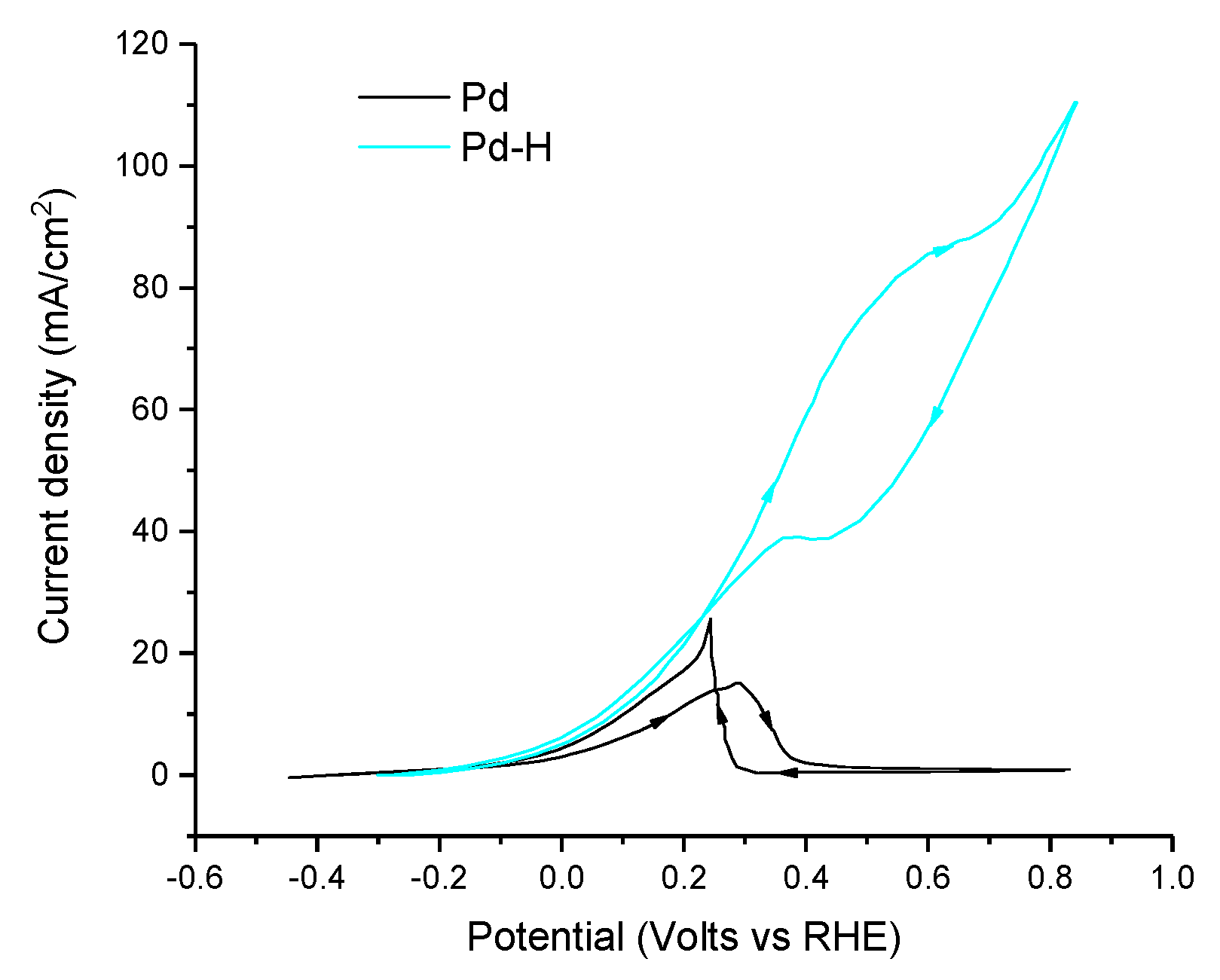
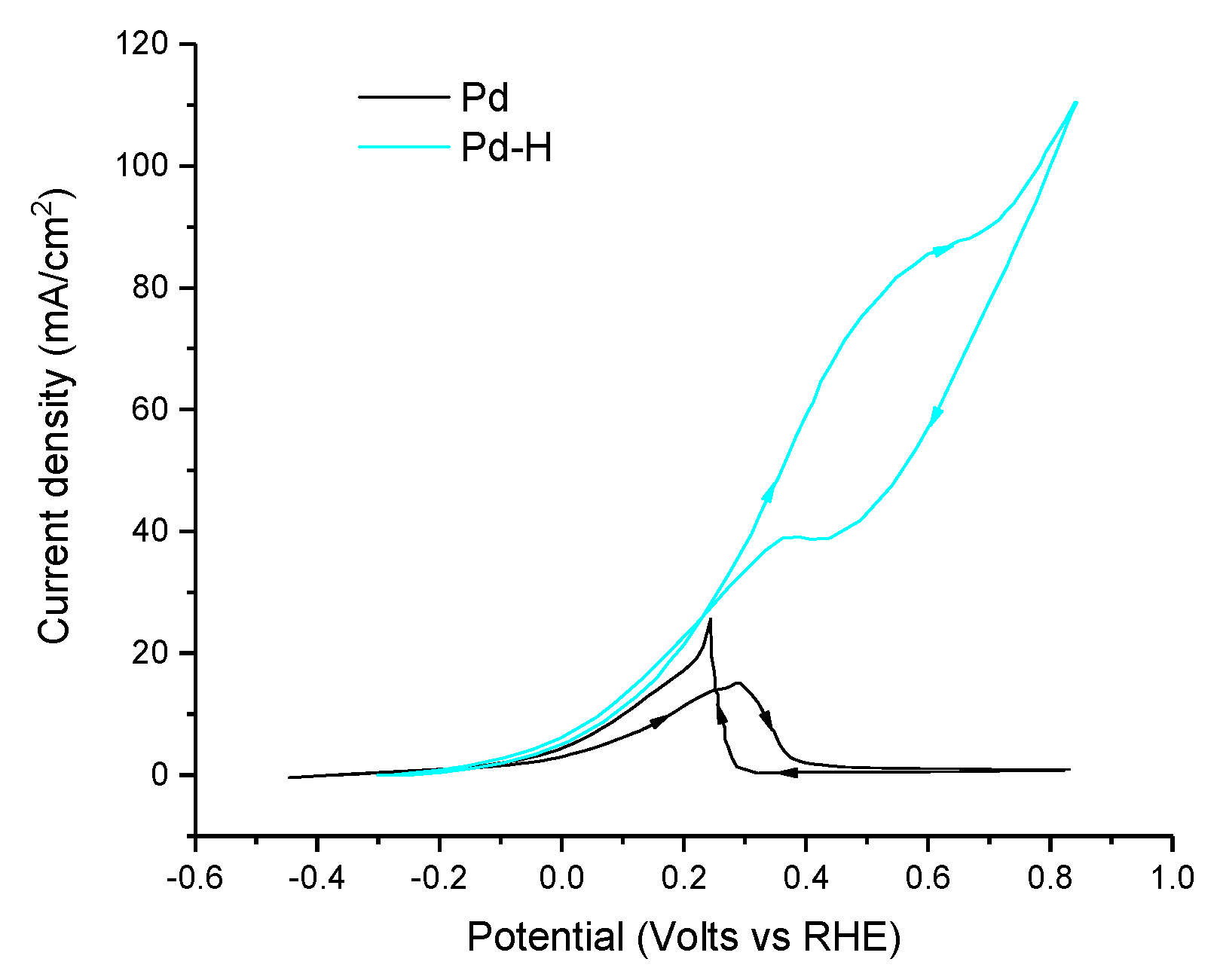
| [116] | Pd(20%)-H/Vulcan carbon | 0.5 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | 0.2 | 71.0 mA cm−2 (0.8 V vs. RHE) | <1.0 | <1.0 | - | - |
| [117] | Pd(20%)/C | 1 M HCOONa + 1 M NaOH (pH ≈ 14.0) 20 mV s−1 | 0.2 | 40.0 mA cm−2 (0.8 V vs. RHE) | 28.0 | 25.0 | 24.0 | 22.8 |
| [118] | Pd(25%)/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 4.6 mA cm−2 (0.7 V vs. RHE) | <0.5 | <0.5 | - | - |
| [119] | Pd(30%)/Vulcan carbon | 0.1 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | 0.2 | 14.7 mA cm−2 (0.5 V vs. RHE) | <1.0 | <1.0 | - | - |
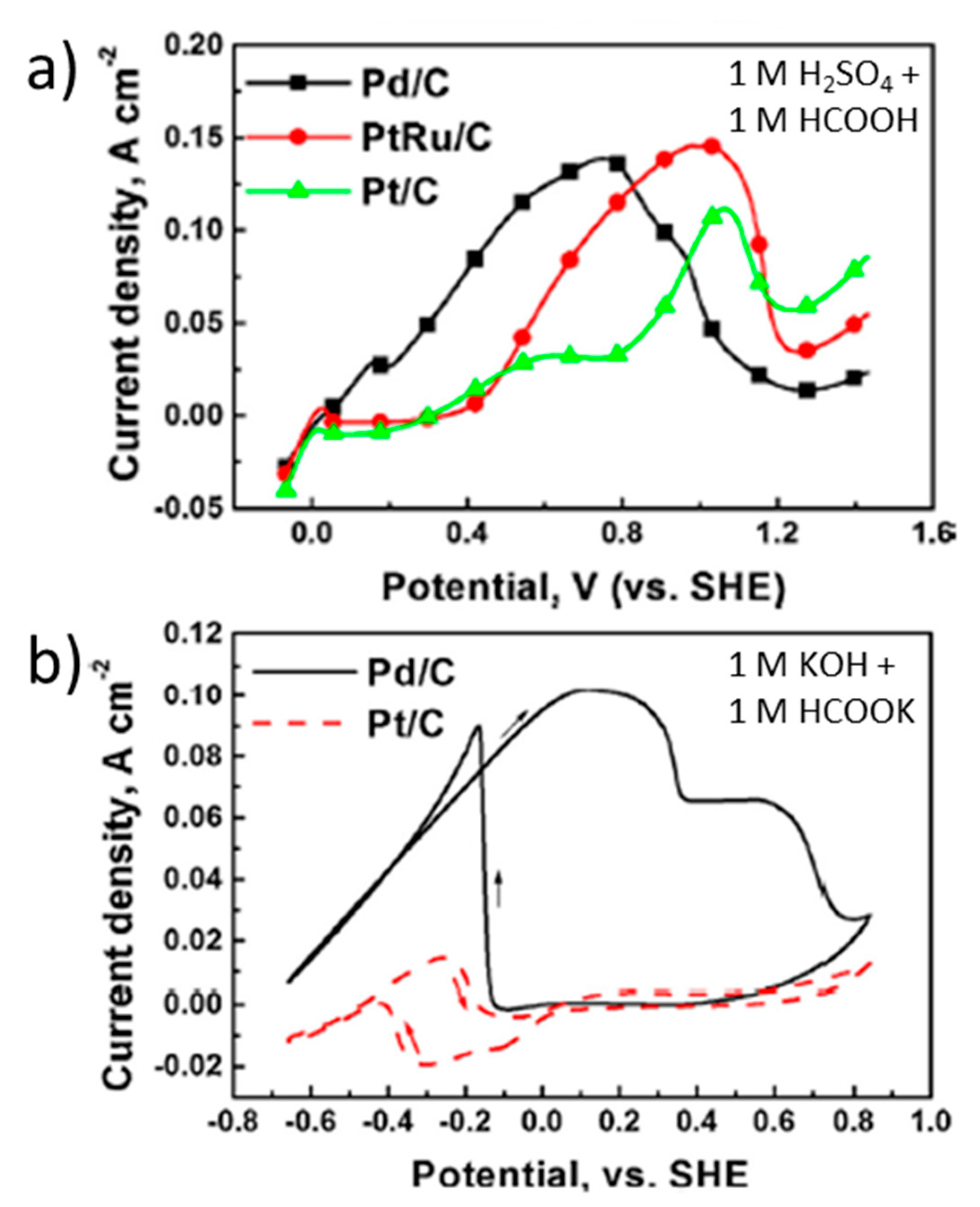
| [70] | Pd(40%)/Vulcan carbon | 1 M HCOOH + 1 M H2SO4 (pH ≈ 0) 20 mV s−1 | 0.1 | 140.0 mA cm−2 (0.8 V vs. RHE) | 60.0 | 16.0 | 20.0 | - |
| 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 20 mV s−1 | 0.2 | 102.0 mA cm−2 (1.0 V vs. RHE) | 101.1 | 65.8 | 65.5 | 27.1 | ||
| [120] | Pd/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.4 | 23.0 mA cm−2 (0.8 V vs. RHE) | <3.0 | - | - | - |
| [121] | Pd/Vulcan carbon | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 3.6 mA cm−2 (0.3 V vs. RHE) | 0.5 | 0.7 | - | - |
| Pd/Graphene | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 7.7 mA cm−2 (0.4 V vs. RHE) | 2.4 | 2.0 | - | - | |
| Pd/N-graphene/CNT | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 17.6 mA cm−2 (0.4 V vs. RHE) | 4.2 | 3.7 | - | - | |
| [122] | Pd(13%)/Reduced graphene oxide | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 13.2 mA cm−2 (0.4 V vs. RHE) | 1.4 | 1.5 | - | - |
| [117] | Pd(20%)/Reduced graphene oxide | 1 M HCOONa + 1 M NaOH (pH ≈ 14.0) 20 mV s−1 | 0.2 | 57.0 mA cm−2 (0.8 V vs. RHE) | 36.0 | 15.0 | 6.4 | 6.6 |
| [73] | Pd/TiO2 | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 10 mV s−1 | 0.1 | 13.0 mA cm−2 (0.4 V vs. RHE) | 2.3 | 1.2 | - | - |
| [123] | Pd-H wire | 0.24 M HCOONa + 0.24 M NaOH (pH ≈ 13.4) 50 mV s−1 | 0.1 | 15.1 mA cm−2 (0.8 V vs. RHE) | 1.2 | 1.0 | - | - |
| [74] | Pd(111) | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.1 | 2.9 mA cm−2 (0.47 V vs. RHE) | 2.0 | - | - | - |
| Pd monolayer/Ir(111) iii | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.1 | 4.3 mA cm−2 (0.4 V vs. RHE) | 1.5 | - | - | - | |
| Pd monolayer/ Au(111) iii | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.1 | 15 mA cm−2 (0.7 V vs. RHE) | 5.9 | - | - | - | |
| Pd monolayer/ Pt(111) iii | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 50 mV s−1 | 0.1 | 55 mA cm−2 (0.97 V vs. RHE) | 4.6 | - | - | - | |
| [124] | PdBi nanoparticles | 0.1 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 7.6 mA cm−2 (0.4 V vs. RHE) | 3.5 | 2.0 | - | - |
| PdCd nanoparticles | 0.1 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 25.1 mA cm−2 (0.3 V vs. RHE) | 3.7 | 3.3 | - | - | |
| [125] | Pd71In29 | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.2 | 18.4 mA cm−2 (0.5 V vs. RHE) | 1.2 | - | - | - |
| [126] | Pd54Ag46 (mixed) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 12.2 mA cm−2 (0.7 V vs. RHE) | <1.0 | - | - | - |
| Pd54Ag46 (core-shell) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 31.0 mA cm−2 (0.7 V vs. RHE) | <1.0 | - | - | - | |
| [71] | PdCu nanoparticles | 0.5 M HCOOK + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.5 | 84.6 mA cm−2 (1.2 V vs. RHE) | 65.1 | 77.1 | 38.2 | 18.6 |
| [118] | Pd67Ag33/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.1 | 6.6 mA cm−2 (0.6 V vs. RHE) | <0.5 | <0.5 | - | - |
| Pd72Ce28/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.1 | 19.4 mA cm−2 (0.6 V vs. RHE) | 0.8 | 0.7 | - | - | |
| Pd70Cu30/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 4.3 mA cm−2 (0.6 V vs. RHE) | <0.5 | <0.5 | - | - | |
| Pd63Co37/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 3.5 mA cm−2 (0.6 V vs. RHE) | <0.5 | <0.5 | - | - | |
| Pd65Ni35/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 3.4 mA cm−2 (0.6 V vs. RHE) | <0.5 | <0.5 | - | - | |
| Pd2.3Co/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.3 | 38.0 mA cm−2 (0.8 V vs. RHE) | 8.6 | - | - | - | |
| [120] | PdNi/Vulcan carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 74.0 mA cm−2 (0.8 V vs. RHE) | 11.0 | - | - | - |
| PdNi/Ketjen carbon | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 117.0 mA cm−2 (0.8 V vs. RHE) | 62.0 | - | - | - | |
| [122] | Pd3(15%)Cu1(3%)/Reduced graphene oxide | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 22.9 mA cm−2 (0.4 V vs. RHE) | 2.3 | 1.8 | - | - |
| [73] | PdPt/TiO2 | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 10 mV s−1 | 0.1 | 12.8 mA cm−2 (0.3 V vs. RHE) | 8.3 | 3.7 | - | - |
| [126] | Pd72Ag19Ni9 (mixed) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 33.7 mA cm−2 (0.8 V vs. RHE) | <2.0 | - | - | - |
| [126] | Pd60Ag20Ni20 (alloyed) | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.2 | 99.6 mA cm−2 (0.8 V vs. RHE) | 18.8 | - | - | - |
| [127] | PdAg nanotubes | 0.5 M HCOOH + 0.1 M HClO4 (pH ≈ 1.0) 100 mV s−1 | 0.1 | 3.8 mA cm−2 (0.6 V vs. RHE) | 0.7 | 0.5 | 0.6 | - |
| [115] | Pd2Ag1 aerogel | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.2 | 27.5 mA cm−2 (0.8 V vs. RHE) | 2.0 | 1.6 | - | - |
| [128] | Pd50Ag50 aerogel | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.1 | 17.9 mA cm−2 (0.7 V vs. RHE) | 0.8 | - | - | - |
| [128] | Pd50Cu50 aerogel | 1 M HCOOK + 1 M KOH (pH ≈ 14.0) 50 mV s−1 | 0.3 | 17.8 mA cm−2 (0.7 V vs. RHE) | <1.0 | - | - | - |
| [129] | PdCu aerogel | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 174.0 mA cm−2 (0.5 V vs. RHE) | <10.0 | <10.0 | - | - |
| [115] | Pd2Ag1Pt025 aerogel | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.1 | 60.0 mA cm−2 (0.7 V vs. RHE) | <2.0 | 3.4 | - | - |
| [129] | B-PdCuAu nanospine assembly | 0.5 M HCOOH + 0.5 M H2SO4 (pH ≈ 0.3) 50 mV s−1 | 0.1 | 23.2 mA cm−2 (0.6 V vs. RHE) | <1.0 | <1.0 | - | - |
| [130] | Pd (interstitial B) | 0.5 M HCOOK + 1 M KOH (pH ≈ 14.0) 100 mv s−1 | 0.2 | 90 mA cm−2 (0.8 V vs. RHE) | 5 | 5 | - | - |
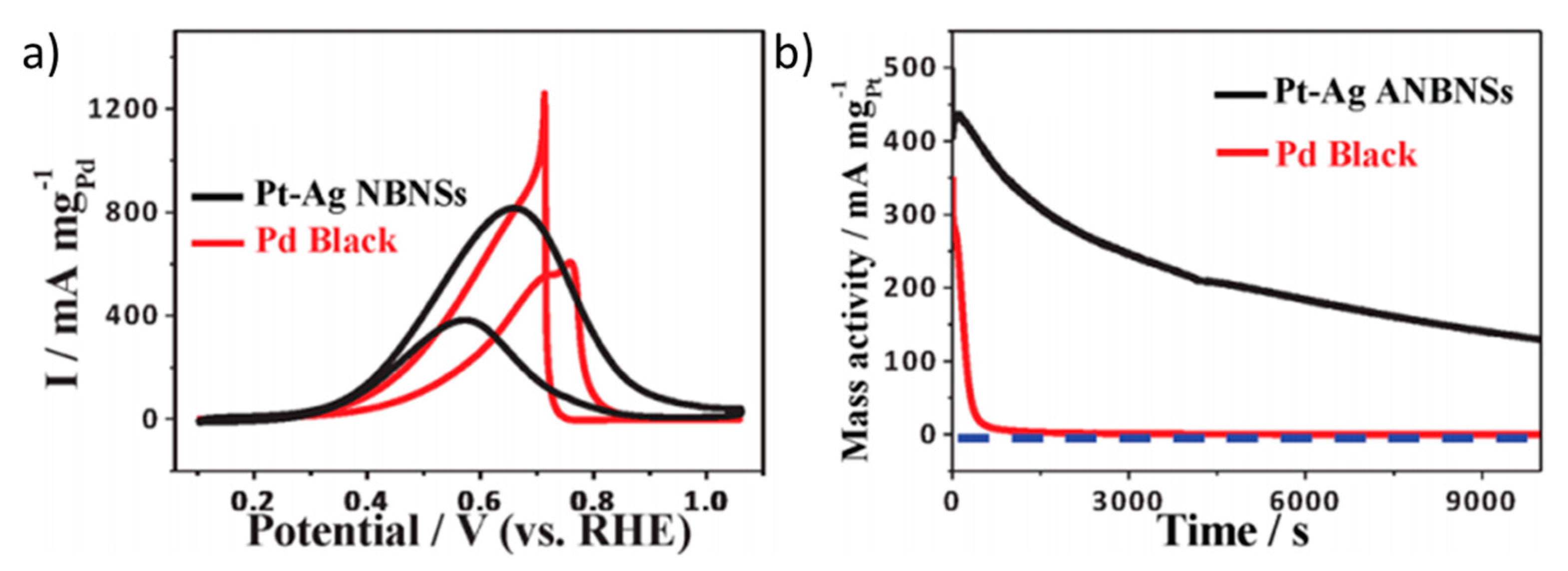
| [71] | PdCuPt (hierarchical zigzag-branched urchin-like superstructure) | 0.5 M HCOOH + 0.5 M KOH (pH ≈ 13.7) 50 mV s−1 | 0.5 | 102.4 mA cm−2 (1.2 V vs. RHE) | 75.4 | 101.9 | 75.7 | 22.1 |
| Ref. | Catalyst | Experimental Conditions i | Eonset (V vs. RHE) ii | jmax (mA cm−2) at Epeak (V vs. RHE) ii | j (mA cm−2) ii at | |||
|---|---|---|---|---|---|---|---|---|
| 1.00 V vs. RHE | 1.20 V vs. RHE | 1.40 V vs. RHE | 1.60 V vs. RHE | |||||
| [60] | Au(111) | 0.05 M HCOOH + 0.2 M NaPi (pH 3.1) 10 mV s−1 | 0.5 | - | 0.1 | 0.2 | 0.2 | - |
| [58] | Au (polycrystalline bead) | 0.1 M HCOOH + 0.5 M Na2SO4 (pH 3.6) 50 mV s−1 | 0.3 | 0.3 mA cm−2 (1.5 V vs. RHE) | 0.3 | 0.3 | 0.3 | <0.1 |
| [54] | Au disc | 0.1 M HCOOK + 0.2 M K2SO4 (pH ≈ 3.8) 50 mV s−1 | 0.7 | - | 0.9 | 1.5 | - | - |
| [136] | Au (1 1 1) | 0.1 M HCOOH + 0.1 M Py (pH = 3.4) 10 mV s−1 | 0.4 | 3.2 mA cm−2 (0.7 V vs. RHE) | 0.4 | 0.2 | - | - |
| [137] | Rh | 0.1 M HCOOH + 1.0 M NaOH (pH ≈ 14) 5 mV s−1 | 0.2 | 12.5 mA cm−2 (0.6 V vs. RHE) | 2 | 2 | 2 | - |
| [138] | Rh | 0.5 M HCOOH + 0.5M H2SO4 (pH ≈ 0) 50 mV s−1 | 0.6 | 0.5 mA cm−2 (0.8 V vs. RHE) | - | - | - | - |
| [139] | Ir (1 1 1) | 1 M HCOOH + 0.5 M H2SO4 (pH ≈ 0) solution under He flow | 0.3 | - | - | - | - | - |
| [140] | Os/GC | 1 M HCOOH 0.5 M NaClO4 1 mV s−1 | 0.6 | 0.2 mA cm−2 (0.8 V vs. RHE) | - | - | - | - |
| [141] | IrO2 | 0.75 M HCOOH + 1 M H2SO4 (pH ≈ 0) 100 mV s−1 | 1.2 | - | - | - | 0.5 | - |
| [63] | La0.8Sr0.2CoO3 | 2.1 M HCOOH + 0.5 M KNO3 (pH ≈ 1.7) 40 mV s−1 | 1.1 | 2.2 mA cm−2 (1.4 V vs. RHE) | <0 | <0.1 | 2.2 | - |
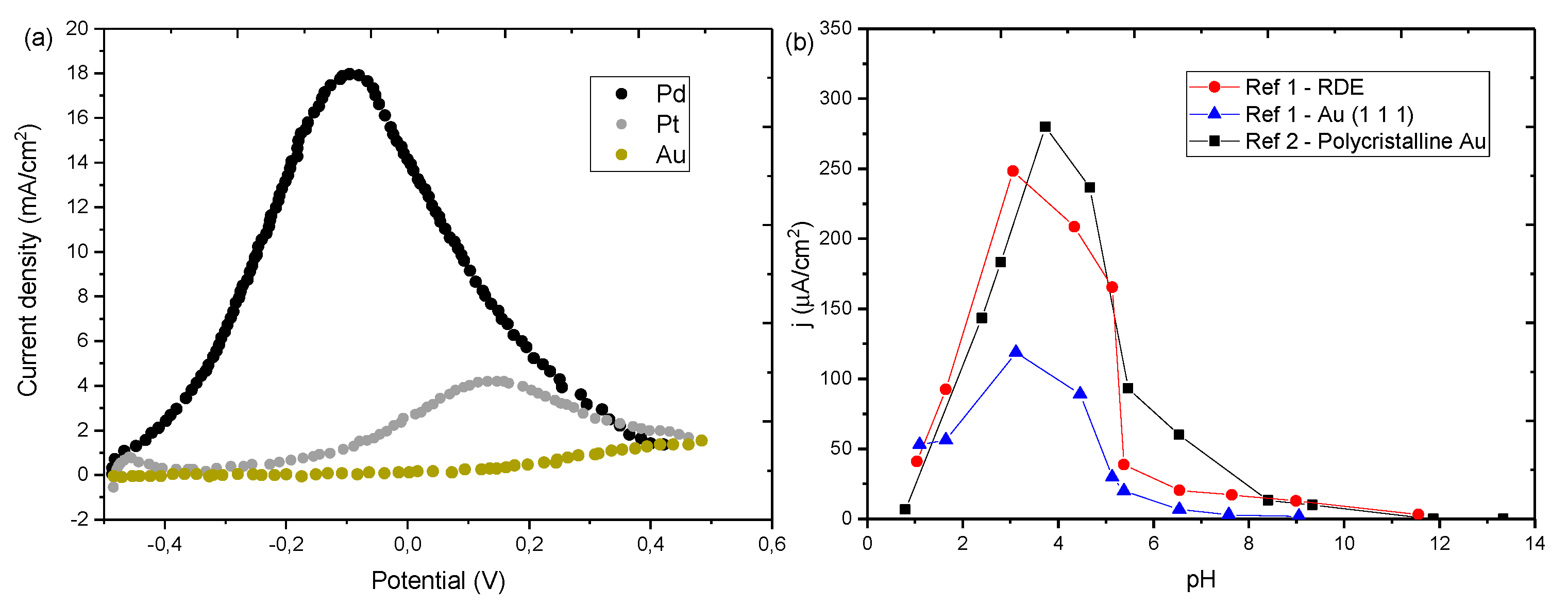
| [51] | NiO/rGO | 0.3 M HCOOH solution (pH 3.5) 100 mV s−1 | 0.2 | 8.9 mA cm−2 (0.6 V vs. RHE) | 3.2 | - | - | - |
| [142] | WC | 6 M HCOOH + 3 M H2SO4 (pH ≈ 0) iii | - | 60.0 mA cm−2 (0.3 V vs. RHE) iv | - | - | - | - |
| [143] | WC | 3 M HCOOH + 1 M H2SO4 (pH ≈ 0) iii | - | 0.3 μA cm−2 (0.3 V vs. RHE) v | - | - | - | - |
| [144] | WC | 1 M HCOOH + 0.1 M KCl (pH 5.0), 2 mV s−1 | 0.4 | - | - | - | - | - |
| [145] | WS2, MoS2 | low currents, not specified | ||||||
| [53] | CoFe Prussian Blue/SnO2:F | 0.4 M HCOOH + 1 M KNO3 (pH 5) 5 mV s−1 | 1.2 | - | - | 0.2 | 10.1 | 39.4 |
| 1 M HCOOH + 1 M KNO3 (pH 5) 5 mV s−1 | 1.2 | - | - | 1.0 | 27.7 | 97.3 | ||
| 0.4 M HCOOH + 1 M KNO3 (pH 13) 5 mV s−1 | 1.2 | - | - | 0.3 | 6.0 | 25.4 | ||
| 1 M HCOOH + 1 M KNO3 (pH 13) 5 mV s−1 | 1.2 | - | - | 1.0 | 13.3 | 34 | ||
| [146] | Ir/GNP | 0.5M H2SO4 + 1.0M HCOOH, (pH ≈ 0) 50mV s−1 | 0.2 | 4.2 mA cm−2 (0.7 V vs. RHE) | - | - | - | - |
| Ir50Zn50/GNP | 0.5M H2SO4 +1.0M HCOOH, (pH ≈ 0) 50mV s−1 | 0.2 | 4.0 mA cm−2 (0.5 V vs. RHE) | - | - | - | - | |
| [138] | Rh nano-chains | 0.5M HCOOH +0.5M H2SO4 (pH ≈ 0) 50 mV s−1 | 0.4 | 1.9 mA cm−2 (0.7 V vs. RHE) | 0.2 | 0.4 | 0.3 | - |
| [147] | Au + Pin | 1.0 M HCOOH + 0.5 M H2SO4 (pH ≈ 0)100 mV s−1 | 0.3 | 0.1 mA cm−2 (0.7 V vs. RHE) | 0.04 | - | - | - |
| [148] | PANI-MnO2 | 0.5M HCOOH + 0.5M H2SO4 (pH ≈ 0) 10mV s−1 | - | - | - | - | - | - |
| [149] | SnO2 + Pin | 1.0 M HCOOH + 0.5 M H2SO4 (pH ≈ 0) | 0. 4 | 0.2 mA cm−2 (0.6 V vs. RHE) | - | - | - | - |
| [150] | Ir1-NC | 0.5 M H2SO4 +0.5 M HCOOH (pH ≈ 0) 50 mV s−1 | 0.4 | 12.9 A mg−1 (0.7 V vs. RHE) vi | 3.5 | 2 | - | - |
| [151] | Rh1-NC | 0.5 M H2SO4 + 0.5 M HCOOH (pH ≈ 0) 10 mV s−1 | 0.2 | 16.1 A mg−1 (0.7 V vs. RHE) vi | 8 | - | - | - |
| Catalyst Properties | Solution Conditions | Expected Electrochemical Performance Parameters ii | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Catalyst Loading (mg cm−2) | pH i | Buffer/Electrolyte | Formate Concentration | Scan Rate (mV s−1) | Onset Potential (V) | Maximum Current Density (mA cm−2) | ECSA (cm2 g−1) (Pt or Pd) | ECSA-Normalized Maximum Current Density (mA cm−2) (Pt or Pd) | Mass-Normalized Maximum Current Density (A mg−1) (Pt or Pd) | Ref. |
| Pt disk | N/A | 0–4.5 | 1 M HClO4–0.1 M H2SO4–0.2 M KPi | 0.1 HCOOH–1 M HCOONa | 20–50 | 0.2–0.5 | 1.0–2.9 | - | - | N/A | [40,51,52,55] |
| Pt(20%)/C | - | 0.3–1 | 0.5 M H2SO4–0.1 M HClO4 | 0.5 M HCOOH | 50 | 0.2–0.3 | 13.1–13.5 | - | - | - | [67,68] |
| Pt(20%)Bi/Vulcan carbon | 0.026 | 1 | 0.1 M HClO4 | 0.25 M HCOOH | 50 | 0.1 | 46.1 | - | - | 9.06 | [69] |
| Pt1Ru1(40%)/Vulcan carbon | 1.6 | ~0 | 1 M H2SO4 | 1 M HCOOH | 20 | 0.3 | 145 | - | - | 0.34 | [70] |
| Pt(2.6 at.%)AuCu | - | 1 | 0.1 M HClO4 | 2 M HCOOH | 50 | 0.2 | - | 249 | 162.1 (at 1.3 V) | 40.3 (at 1.3 V) | [80] |
| Pd disk | N/A | 0.3 | 0.5 M H2SO4 | 1 M HCOOH | 50–70 | ~0.2 | ~3 | - | - | N/A | [30,110] |
| Pd(40%)/Vulcan carbon | 1.6 | ~0 | 1 M H2SO4 | 1 M HCOOH | 20 | 0.1 | 140 | - | - | 0.22 | [70] |
| 14 | 1 M KOH | 1 M HCOOK | 20 | 0.2 | 102 | - | - | 0.16 | |||
| PdNi/Ketjen carbon | 0.08 | 14 | 1 M KOH | 1 M HCOOK | 50 | 0.2 | 117 | 54 | 14 | 7.8 | [120] |
| Nanoporous Pd (AlPd dealloyed) | 0.88 | 0.3 | 0.5 M H2SO4 | 0.5 M HCOOH | 10 | 0.1 | 232 | 230,000 | 1.1 | 0.262 | [114] |
| Au disk | N/A | 3.8 | 0.2 M K2SO4 | 0.1 M HCOOK | 50 | 0.7 | 1.5 (at 1.2 V) | - | - | N/A | [54] |
| CoFe Prussian Blue/SnO2:F | 0.3 mg CoFe-PB cm−2 | 5 | 1 M KNO3 | 1M HCOOH | 5 | 1.2 | 97.3 (at 1.6 V) | -- | -- | -- | [53] |
| 13 | 1 M KNO3 | 1M HCOOH | 5 | 1.2 | 34.0 (at 1.6 V) | - | - | - | |||
| Rh1-NC | 4 | ~0 | 0.5 M H2SO4 | 0.5 M HCOOH | 10 | 0.2 | - | - | - | 16.1 | [151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Folkman, S.J.; González-Cobos, J.; Giancola, S.; Sánchez-Molina, I.; Galán-Mascarós, J.R. Benchmarking Catalysts for Formic Acid/Formate Electrooxidation. Molecules 2021, 26, 4756. https://doi.org/10.3390/molecules26164756
Folkman SJ, González-Cobos J, Giancola S, Sánchez-Molina I, Galán-Mascarós JR. Benchmarking Catalysts for Formic Acid/Formate Electrooxidation. Molecules. 2021; 26(16):4756. https://doi.org/10.3390/molecules26164756
Chicago/Turabian StyleFolkman, Scott J., Jesús González-Cobos, Stefano Giancola, Irene Sánchez-Molina, and José Ramón Galán-Mascarós. 2021. "Benchmarking Catalysts for Formic Acid/Formate Electrooxidation" Molecules 26, no. 16: 4756. https://doi.org/10.3390/molecules26164756
APA StyleFolkman, S. J., González-Cobos, J., Giancola, S., Sánchez-Molina, I., & Galán-Mascarós, J. R. (2021). Benchmarking Catalysts for Formic Acid/Formate Electrooxidation. Molecules, 26(16), 4756. https://doi.org/10.3390/molecules26164756