
New Methods for the Synthesis of Spirocyclic Cephalosporin Analogues


Abstract
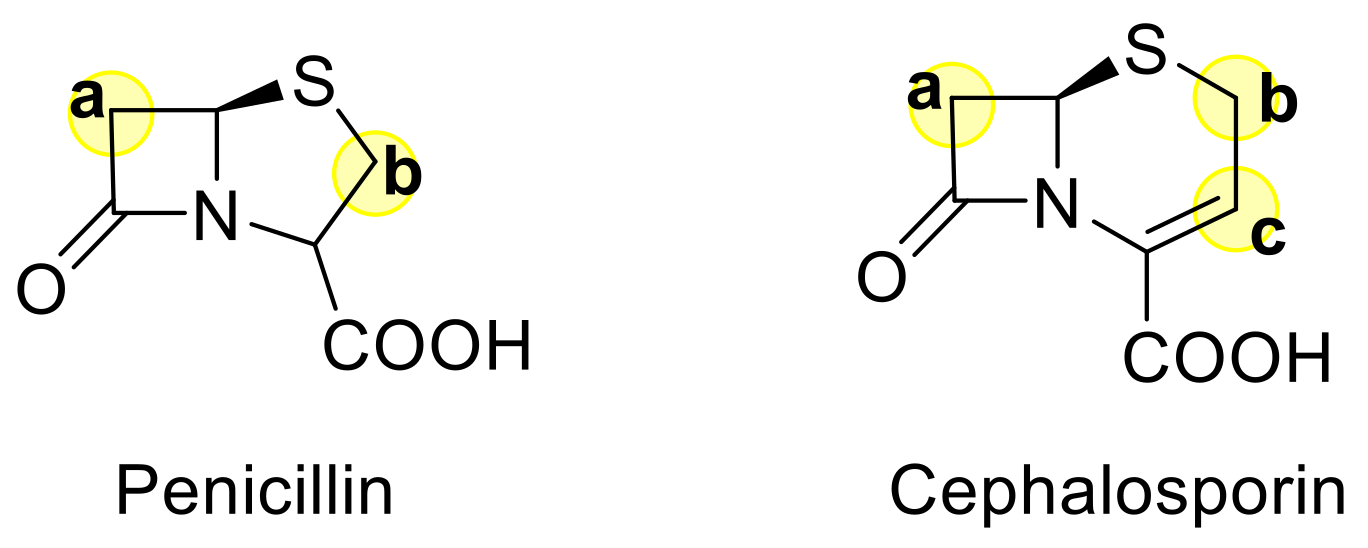
:1. Introduction
2. Results and Discussion
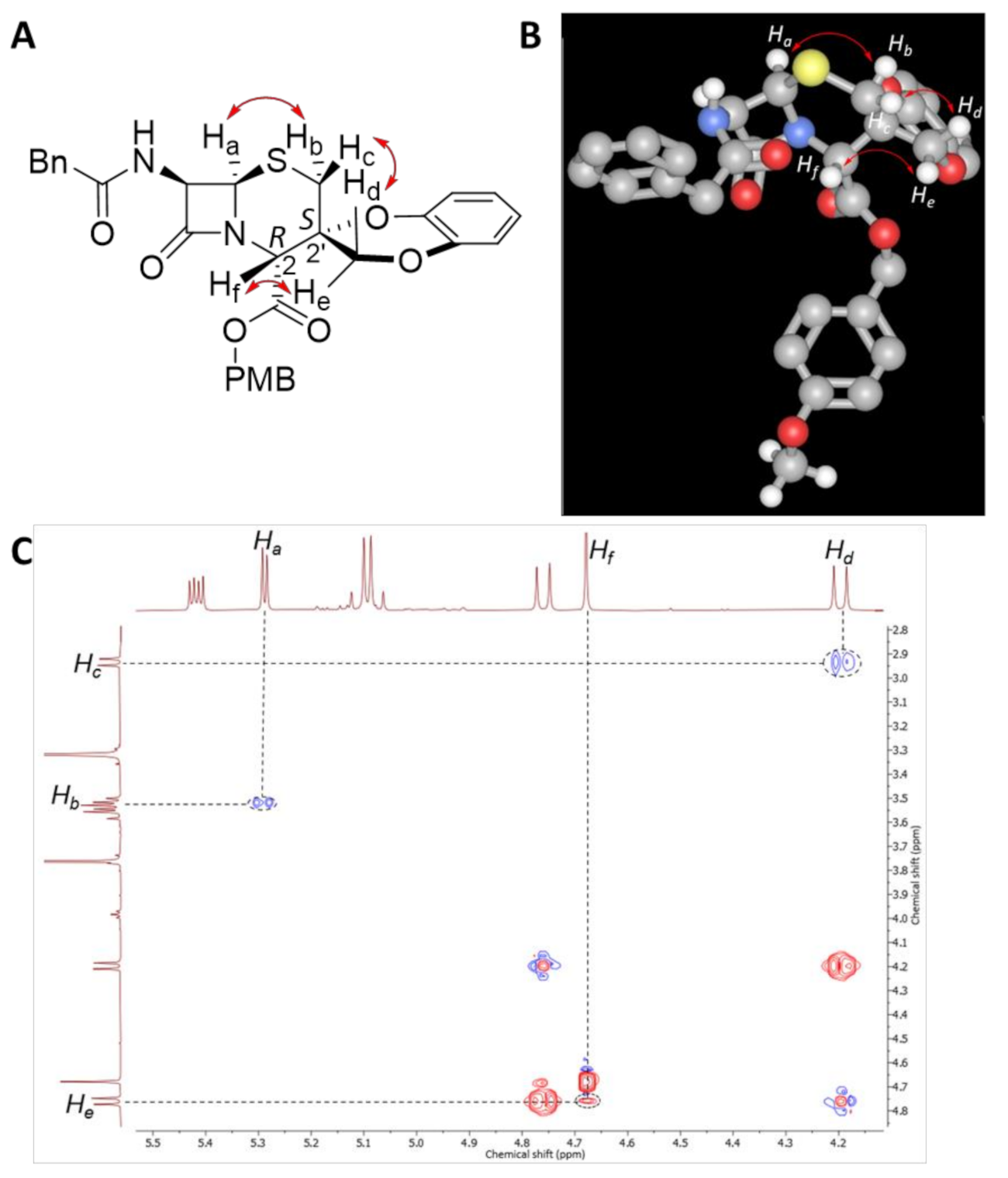
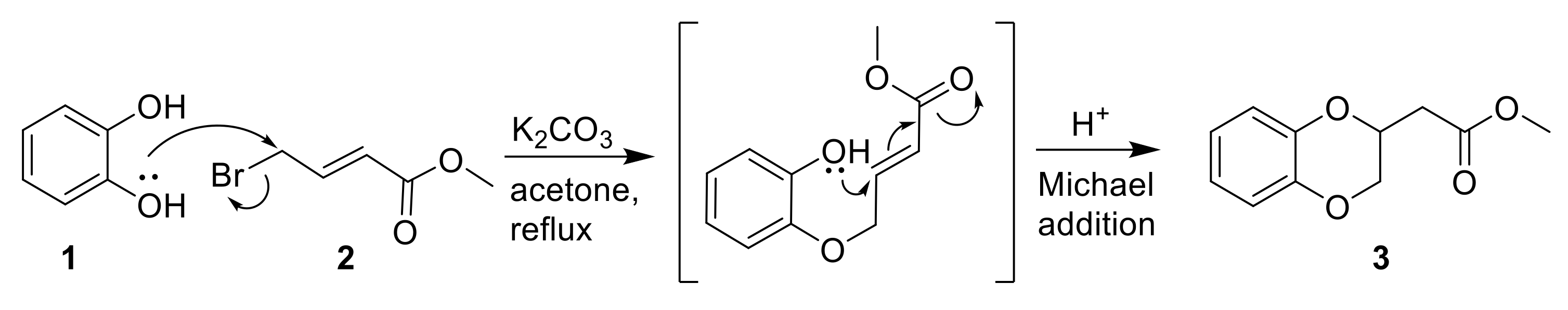
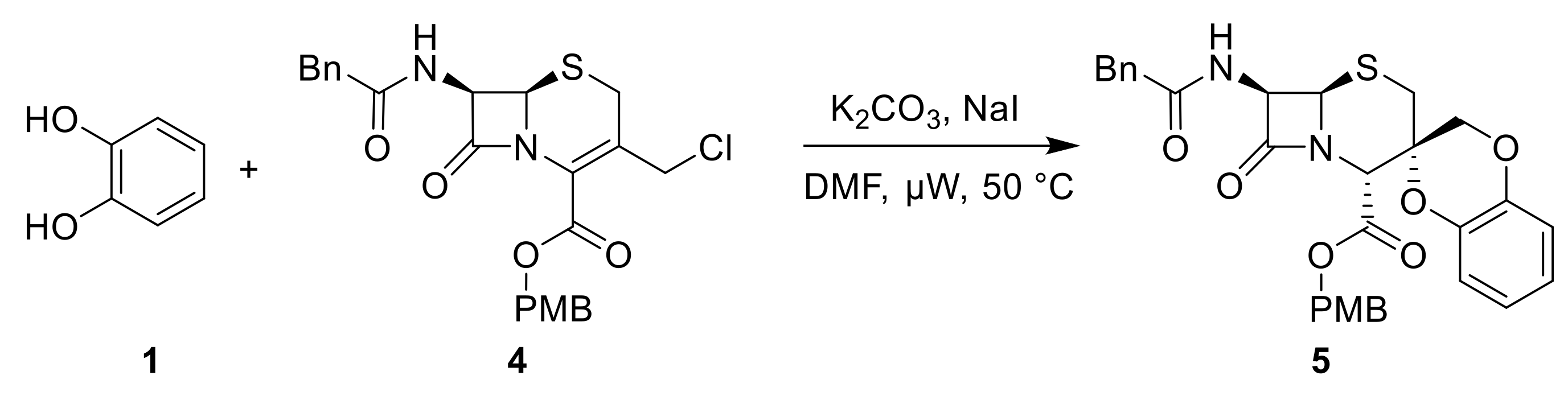
2.1. Reaction Scope for Generating Spiro-Cephalosporins
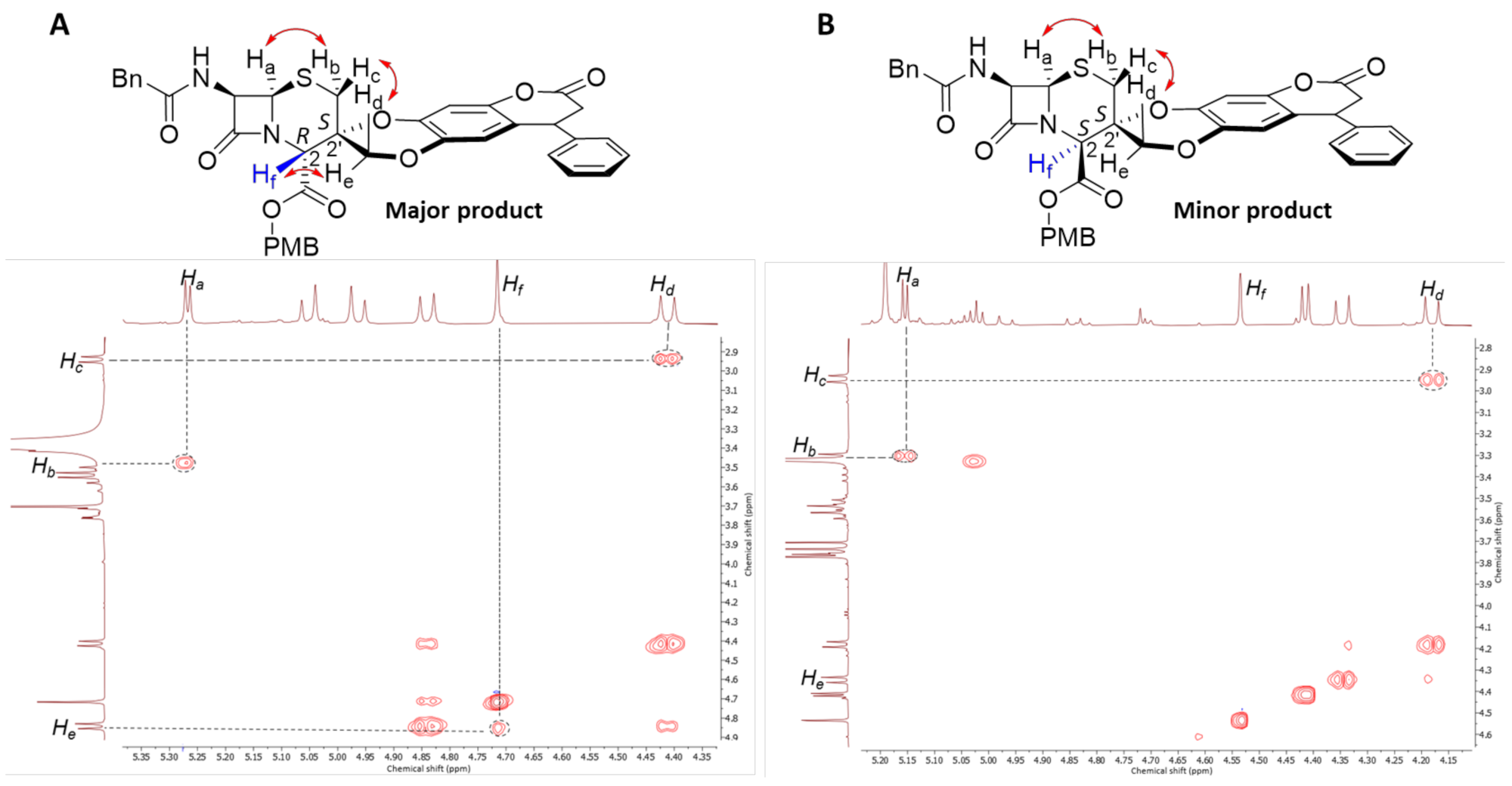
2.2. Functionalisation of Spiro-Cephalosporins
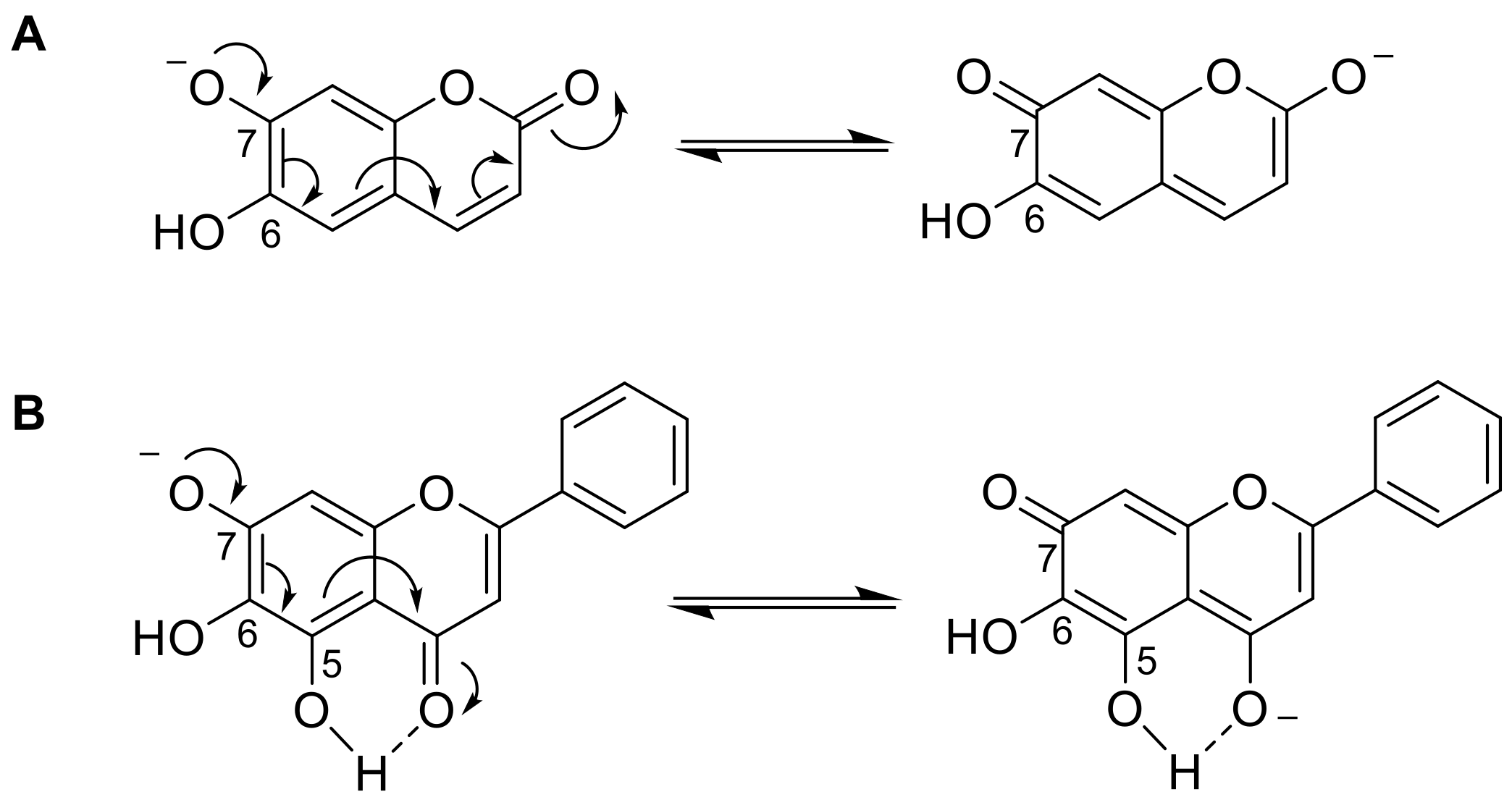
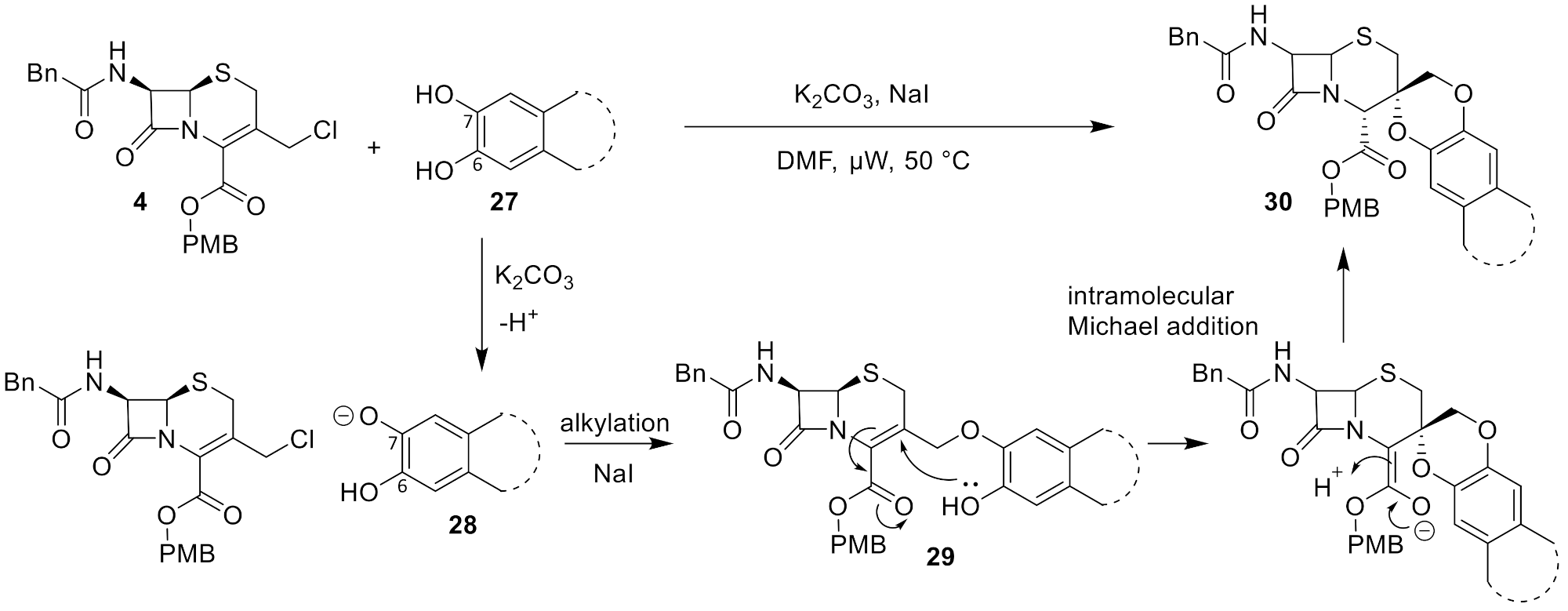
2.3. Proposed Mechanism
3. Materials and Methods
3.1. General Information
3.2. Typical Procedure for the Spiro-Cyclisation Reaction
3.3. Characterisation of Spiro-Cephalosporin Products
3.4. Typical Procedure for PMB Deprotection
3.5. Characterisation of PMB Deprotected Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Baeyer, A. Systematik und Nomenclatur Bicyclischer Kohlenwasserstoffe. Ber. Dtsch. Chem. Ges. 1900, 33, 3771–3775. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.B.; Rønnest, M.H.; Larsen, T.O.; Clausen, M.H. The Chemistry of Griseofulvin. Chem. Rev. 2014, 114, 12088–12107. [Google Scholar] [CrossRef] [Green Version]
- Dugar, S.; Clader, J.W.; Chan, T.-M.; Davis, H. Substituted 2-azaspiro [5.3] nonan-1-ones as potent cholesterol absorption inhibitors: Defining a binding conformation for SCH 48461. J. Med. Chem. 1995, 38, 4875–4877. [Google Scholar] [CrossRef]
- Kambara, T.; Tomioka, K. Formal Asymmetric Synthesis of a Cholesterol Absorption Inhibitor Bearing a 2-Azaspiro [3.5] nonan-1-one Moiety. J. Org. Chem. 1999, 64, 9282–9285. [Google Scholar] [CrossRef]
- Howell, K.; Devita, R.J.; Garcia-Calvo, M.; Meurer, R.D.; Lisnock, J.; Bull, H.G.; McMasters, D.R.; McCann, M.E.; Mills, S.G. Spiroimidazolidinone NPC1L1 inhibitors. Part 2: Structure-activity studies and in vivo efficacy. Bioorganic Med. Chem. Lett. 2010, 20, 6929–6932. [Google Scholar] [CrossRef]
- Khalil, E.M.; Ojala, W.H.; Pradhan, A.; Nair, V.D.; Gleason, W.B.; Mishra, R.K.; Johnson, R.L. Design, Synthesis, and Dopamine Receptor Modulating Activity of Spiro Bicyclic Peptidomimetics ofl-Prolyl-l-leucyl-glycinamide. J. Med. Chem. 1999, 42, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Macias, A.; Ramallal, A.M.; Alonso, E.; del Pozo, C.; González, J. Synthesis of Enantiopure Pyrrolidine-Derived Peptidomimetics and Oligo-β-peptides via Nucleophilic Ring-Opening of β-Lactams†. J. Org. Chem. 2006, 71, 7721–7730. [Google Scholar] [CrossRef]
- Bhagwanth, S.; Mishra, S.; Daya, R.; Mah, J.; Mishra, R.K.; Johnson, R.L. Transformation of Pro-Leu-Gly-NH2 Peptidomimetic Positive Allosteric Modulators of the Dopamine D2 Receptor into Negative Modulators. ACS Chem. Neurosci. 2012, 3, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Venkatraman, S.; Zheng, Y.; McKeever, B.M.; Dillard, L.W.; Singh, S.B. Structure-Based Design of β-Site APP Cleaving Enzyme 1 (BACE1) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2013, 56, 4156–4180. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Hunt, K.W.; Newhouse, B.; Watts, R.J.; Liu, X.; Vigers, G.; Smith, D.; Rhodes, S.P.; Brown, K.D.; Otten, J.N.; et al. 8-Tetrahydropyran-2-yl Chromans: Highly Selective Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitors. J. Med. Chem. 2014, 57, 10112–10129. [Google Scholar] [CrossRef]
- Dineen, T.A.; Chen, K.; Cheng, A.C.; Derakhchan, K.; Epstein, O.; Esmay, J.; Hickman, D.; Kreiman, C.; Marx, I.E.; Wahl, R.C.; et al. Inhibitors of β-site amyloid precursor protein cleaving enzyme (BACE1): Identification of (S)-7-(2-fluoropyridin-3-yl)-3-((3-methyloxetan-3-yl)ethynyl)-5′H-spiro[chromeno[2,3-b]pyridine-5,4′-oxazol]-2′-amine (AMG-8718). J. Med. Chem. 2014, 57, 9811–9831. [Google Scholar] [CrossRef]
- Neelamkavil, S.F.; Agrawal, S.; Bara, T.; Bennett, C.; Bhat, S.; Biswas, D.; Brockunier, L.; Buist, N.; Burnette, D.; Cartwright, M.; et al. Discovery of MK-8831, a novel spiro-proline macrocycle as a pan-genotypic HCV-NS3/4a protease inhibitor. ACS Med. Chem. Lett. 2016, 7, 111–116. [Google Scholar] [CrossRef]
- Link, J.O.; Taylor, J.G.; Xu, L.; Mitchell, M.; Guo, H.; Liu, H.; Kato, D.; Kirschberg, T.; Sun, J.; Squires, N.; et al. Discovery of Ledipasvir (GS-5885): A Potent, Once-Daily Oral NS5A Inhibitor for the Treatment of Hepatitis C Virus Infection. J. Med. Chem. 2014, 57, 2033–2046. [Google Scholar] [CrossRef]
- Ye, N.; Chen, H.; Wold, E.A.; Shi, P.-Y.; Zhou, J. Therapeutic Potential of Spirooxindoles as Antiviral Agents. ACS Infect. Dis. 2016, 2, 382–392. [Google Scholar] [CrossRef]
- Das, P.; Boone, S.; Mitra, D.; Turner, L.; Tandon, R.; Raucher, D.; Hamme, A.T. Synthesis and biological evaluation of fluoro-substituted spiro-isoxazolines as potential anti-viral and anti-cancer agents. RSC Adv. 2020, 10, 30223–30237. [Google Scholar] [CrossRef]
- Guardia, A.; Baiget, J.; Cacho, M.; Pérez, A.; Ortega-Guerra, M.; Nxumalo, W.; Khanye, S.D.; Rullas, J.; Ortega, F.; Jiménez, E.; et al. Easy-To-Synthesize Spirocyclic Compounds Possess Remarkable in vivo Activity against Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 11327–11340. [Google Scholar] [CrossRef]
- Blondiaux, N.; Moune, M.; Desroses, M.; Frita, R.; Flipo, M.; Mathys, V.; Soetaert, K.; Kiass, M.; Delorme, V.; Djaout, K.; et al. Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science 2017, 355, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Urzúa, A.; Echeverría, J.; Rezende, M.C.; Wilkens, M. Antibacterial properties of 3 H-spiro[1-benzofuran-2,1′-cyclohexane] derivatives from Heliotropium filifolium. Molecules 2008, 13, 2385–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandanayaka, V.P.; Prashad, A.S.; Yang, Y.; Williamson, R.T.; Lin, Y.I.; Mansour, T.S. Spirocyclopropyl β-lactams as mechanism-based inhibitors of serine β-lactamases. Synthesis by rhodium-catalyzed cyclopropanation of 6-diazopenicillanate sulfone. J. Med. Chem. 2003, 46, 2569–2571. [Google Scholar] [CrossRef]
- Mani, K.S.; Kaminsky, W.; Rajendran, S.P. A facile atom economic one pot multicomponent synthesis of bioactive spiro-indenoquinoxaline pyrrolizines as potent antioxidants and anti-cancer agents. New J. Chem. 2018, 42, 301–310. [Google Scholar] [CrossRef]
- Islam, M.S.; Al-Majid, A.M.; El-Senduny, F.F.; Badria, F.A.; Rahman, A.F.M.M.; Barakat, A.; Elshaier, Y.A.M.M. Synthesis, Anticancer Activity, and Molecular Modeling of New Halogenated Spiro [pyrrolidine-thiazolo-oxindoles] Derivatives. Appl. Sci. 2020, 10, 2170. [Google Scholar] [CrossRef] [Green Version]
- Hati, S.; Tripathy, S.; Dutta, P.K.; Agarwal, R.; Srinivasan, R.; Singh, A.; Singh, S.; Sen, S. Spiro[pyrrolidine-3,3-oxindole] as potent anti-breast cancer compounds: Their design, synthesis, biological evaluation and cellular target identification. Sci. Rep. 2016, 6, 32213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najim, N.; Bathich, Y.; Zain, M.M.; Hamzah, A.S.; Shaameri, Z. Evaluation of the Bioactivity of Novel Spiroisoxazoline Type Compounds against Normal and Cancer Cell Lines. Molecules 2010, 15, 9340–9353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.-J.; Tice, C.M. The utilization of spirocyclic scaffolds in novel drug discovery. Expert Opin. Drug Discov. 2016, 11, 831–834. [Google Scholar] [CrossRef] [Green Version]
- Shennan, B.D.A.; Smith, P.W.; Ogura, Y.; Dixon, D.J. A modular and divergent approach to spirocyclic pyrrolidines. Chem. Sci. 2020, 11, 10354–10360. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The use of spirocyclic scaffolds in drug discovery. Bioorganic Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [Green Version]
- Rios, R. Enantioselective methodologies for the synthesis of spiro compounds. Chem. Soc. Rev. 2012, 41, 1060–1074. [Google Scholar] [CrossRef]
- Ding, A.; Meazza, M.; Guo, H.; Yang, J.W.; Rios, R. New development in the enantioselective synthesis of spiro compounds. Chem. Soc. Rev. 2018, 47, 5946–5996. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.K.; Hanhan, N.V.; Ball-Jones, N.R. Asymmetric Catalysis for the Synthesis of Spirocyclic Compounds. ACS Catal. 2013, 3, 540–553. [Google Scholar] [CrossRef]
- Pawlowski, R.; Skorka, P.; Stodulski, M. Radical-Mediated Non-Dearomative Strategies in Construction of Spiro Compounds. Adv. Synth. Catal. 2020, 362, 4462–4486. [Google Scholar] [CrossRef]
- Bycroft, B.W.; Shute, R.E.; Begley, M.J. Novel β-lactamase inhibitory and antibacterial 6-spiro- epoxypenicillins. J. Chem. Soc. Chem. Commun. 1988, 274–276. [Google Scholar] [CrossRef]
- Turos, E.; Long, T.E.; Heldreth, B.; Leslie, J.M.; Reddy, G.S.K.; Wang, Y.; Coates, C.; Konaklieva, M.; Dickey, S.; Lim, D.V.; et al. N-Thiolated β-lactams: A new family of anti-Bacillus agents. Bioorganic Med. Chem. Lett. 2006, 16, 2084–2090. [Google Scholar] [CrossRef]
- Sheehan, J.C.; Chacko, E.; Lo, Y.S.; Ponzi, D.R.; Sato, E. Some new spiro penicillins. J. Org. Chem. 1978, 43, 4856–4859. [Google Scholar] [CrossRef]
- Benfatti, F.; Cardillo, G.; Gentilucci, L.; Tolomelli, A. Synthesis and biological evaluation of unprecedented classes of spiro-β-lactams and azido-β-lactams as acyl-CoA:cholesterol acyltransferase inhibitors. Bioorganic Med. Chem. Lett. 2007, 17, 1946–1950. [Google Scholar] [CrossRef] [PubMed]
- Narender Reddy, A.V.; Fiakpui, C.Y.; Czajkowski, D.P.; Spevak, P.; Kaleta, J.; Micetich, R.G.; Maiti, S.N. Synthesis and human leukocyte elastase inhibitory activity of novel 2-spiro (2’,2’-diphenylcyclopropane) cephalosporin sulfones. Chem. Heterocycl. Compd. 1998, 34, 1289–1295. [Google Scholar] [CrossRef]
- Skiles, J.W.; McNeil, D. Spiro indolinone beta-lactams, inhibitors of pliovirus and rhinovirus 3C-proteinases. Tetrahedron Lett. 1990, 31, 7277–7280. [Google Scholar] [CrossRef]
- Santos, B.S.; Nunes, S.C.C.; Pais, A.A.C.C.; Pinho e Melo, T.M.V.D. Chiral spiro-β-lactams from 6-diazopenicillanates. Tetrahedron 2012, 68, 3729–3737. [Google Scholar] [CrossRef]
- Hirai, K.; Iwano, Y.; Saito, T.; Hiraoka, T.; Kishida, Y. Functionalisation of C6(7) of penicillins and cephalosporins via 1,3-dipolar intermediate. Tetrahedron Lett. 1976, 17, 1303–1306. [Google Scholar] [CrossRef]
- Santos, B.S.; Pino e Melo, T.M.V.D. Synthesis of chiral spirocyclopentenyl-β-lactams through phosphane-catalyzed [3+2] annulation of allenoates with 6- alkylidenepenicillanates. Eur. J. Org. Chem. 2013, 3901–3909. [Google Scholar] [CrossRef]
- Pitlik, J. Cycloaddition and related reactions of cephalosporin antibiotics. Bioorganic Med. Chem. 1995, 3, 1157–1181. [Google Scholar] [CrossRef]
- Yang, D.-P.; Ji, H.-F.; Tang, G.-Y.; Ren, W.; Zhang, H.-Y. How Many Drugs Are Catecholics. Molecules 2007, 12, 878–884. [Google Scholar] [CrossRef]
- Ivanova, G.; Bratovanova, E.; Petkov, D. Catechol as a nucleophilic catalyst of peptide bond formation. J. Pept. Sci. 2002, 8, 8–12. [Google Scholar] [CrossRef]
- Cabiddu, S.; Floris, C.; Melis, S.; Sotgiu, F.; Cerioni, G. Heterocyclic compound studies. II. Synthesis of 1,4-benzodioxin, 1,4-benzoxathiin, 1,4-benzoxazine and 1,4-benzothiazine derivatives. J. Heterocycl. Chem. 1986, 23, 1815–1820. [Google Scholar] [CrossRef]
- van Berkel, S.S.; Brem, J.; Rydzik, A.; Salimraj, R.; Cain, R.; Verma, A.; Owens, R.J.; Fishwick, C.W.G.; Spencer, J.; Schofield, C.J. Assay Platform for Clinically Relevant Metallo-β-lactamases. J. Med. Chem. 2013, 56, 6945–6953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
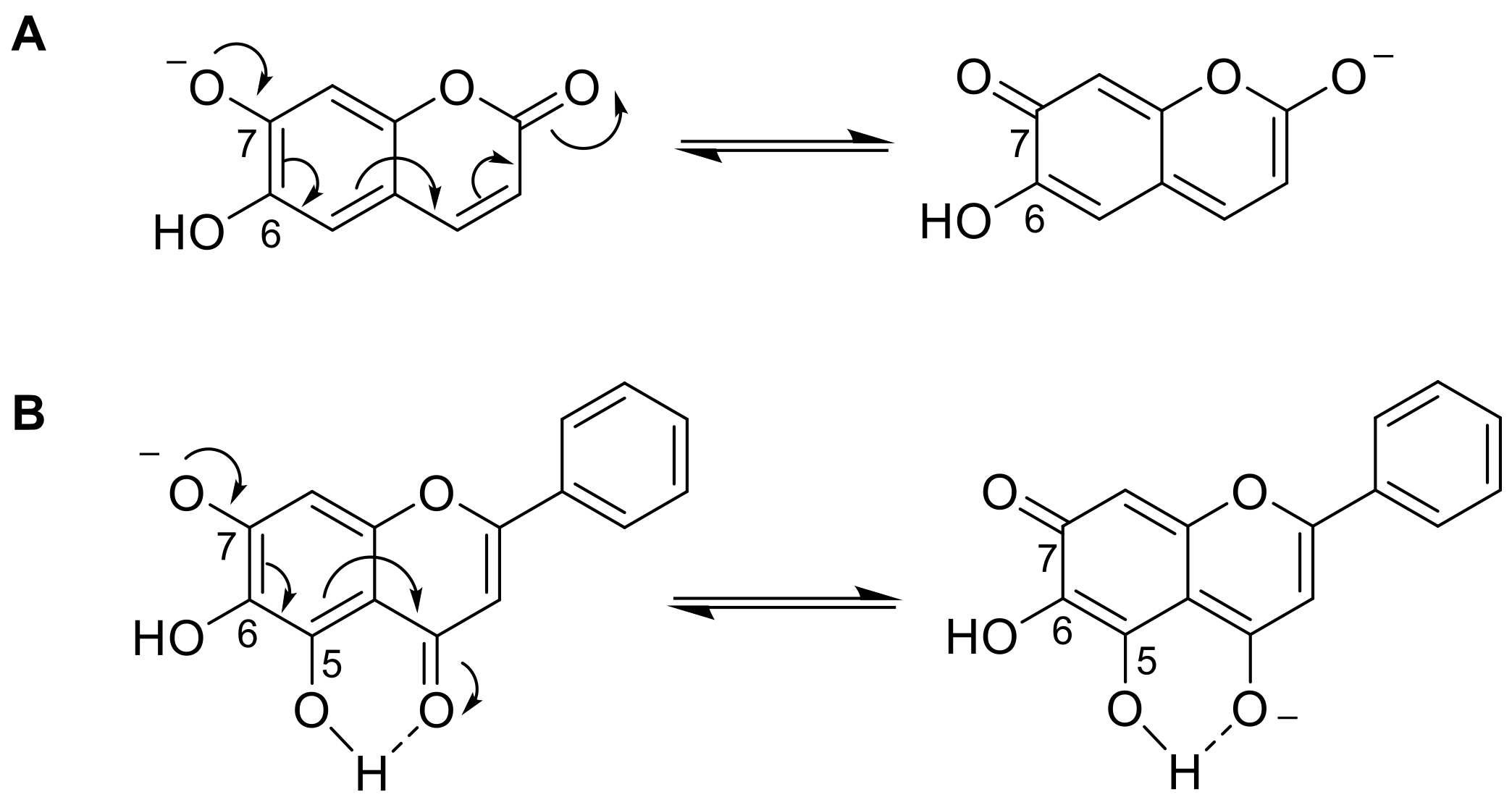
- Nowak, P.M.; Sagan, F.; Mitoraj, M.P. Origin of Remarkably Different Acidity of Hydroxycoumarins—Joint Experimental and Theoretical Studies. J. Phys. Chem. B 2017, 121, 4554–4561. [Google Scholar] [CrossRef] [PubMed]
- Musialik, M.; Kuzmicz, R.; Pawłowski, T.S.; Litwinienko, G. Acidity of Hydroxyl Groups: An Overlooked Influence on Antiradical Properties of Flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- McNab, H.; Ferreira, E.S.B.; Hulme, A.N.; Quye, A. Negative ion ESI–MS analysis of natural yellow dye flavonoids—An isotopic labelling study. Int. J. Mass Spectrom. 2009, 284, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Schweigert, N.; Zehnder, A.J.B.; Eggen, R.I.L. Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ. Microbiol. 2001, 3, 81–91. [Google Scholar] [CrossRef]
- Dhumrongvaraporn, S. Synthesis of mono- and dialkyl catechol ethers. Master’ Thesis, Rochester Institute of Technology, Rochester, NY, USA; p. 1983.
- Zagorevskii, V.A.; Kirsanova, Z.D.; Persianova, I.V.; Zykov, D.A. Alkylation and some physicochemical characteristics of 6,7- and 7,8-dihydroxycoumarins. Chem. Heterocycl. Compd. 1974, 10, 1023–1028. [Google Scholar] [CrossRef]
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catechol | Major Product | Isolated Yield (%) | dr b |
|---|---|---|---|---|
| 1 |  |  | 59 | –c |
| 2 |  |  | 62 | 14:1 |
| 3 |  |  | 54 | 9:1 |
| 4 |  | 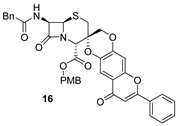 | 65 | 12:1 |
| 5 |  |  | 51 | – c |
| 6 |  |  | – d | – d |
| 7 |  |  | 28 | 8:1 |
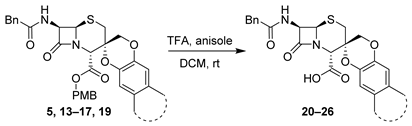
| Entry | Starting Material | Product | Isolated Yield (%) |
|---|---|---|---|
| 1 | 5 |  | 71 |
| 2 | 13 | 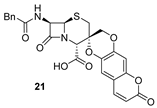 | 93 |
| 3 | 14 |  | 74 |
| 4 | 15 |  | 74 |
| 5 | 16 |  | 97 |
| 6 | 17 |  | 63 |
| 7 | 19 | 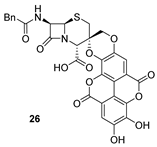 | 77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, A.X.; Horsfall, L.E.; Hulme, A.N. New Methods for the Synthesis of Spirocyclic Cephalosporin Analogues. Molecules 2021, 26, 6035. https://doi.org/10.3390/molecules26196035
Zhao AX, Horsfall LE, Hulme AN. New Methods for the Synthesis of Spirocyclic Cephalosporin Analogues. Molecules. 2021; 26(19):6035. https://doi.org/10.3390/molecules26196035
Chicago/Turabian StyleZhao, Alan X., Louise E. Horsfall, and Alison N. Hulme. 2021. "New Methods for the Synthesis of Spirocyclic Cephalosporin Analogues" Molecules 26, no. 19: 6035. https://doi.org/10.3390/molecules26196035
APA StyleZhao, A. X., Horsfall, L. E., & Hulme, A. N. (2021). New Methods for the Synthesis of Spirocyclic Cephalosporin Analogues. Molecules, 26(19), 6035. https://doi.org/10.3390/molecules26196035