
Preclinical Investigation in Neuroprotective Effects of the GPR55 Ligand VCE-006.1 in Experimental Models of Parkinson’s Disease and Amyotrophic Lateral Sclerosis

 , , , , , , and
, , , , , , and 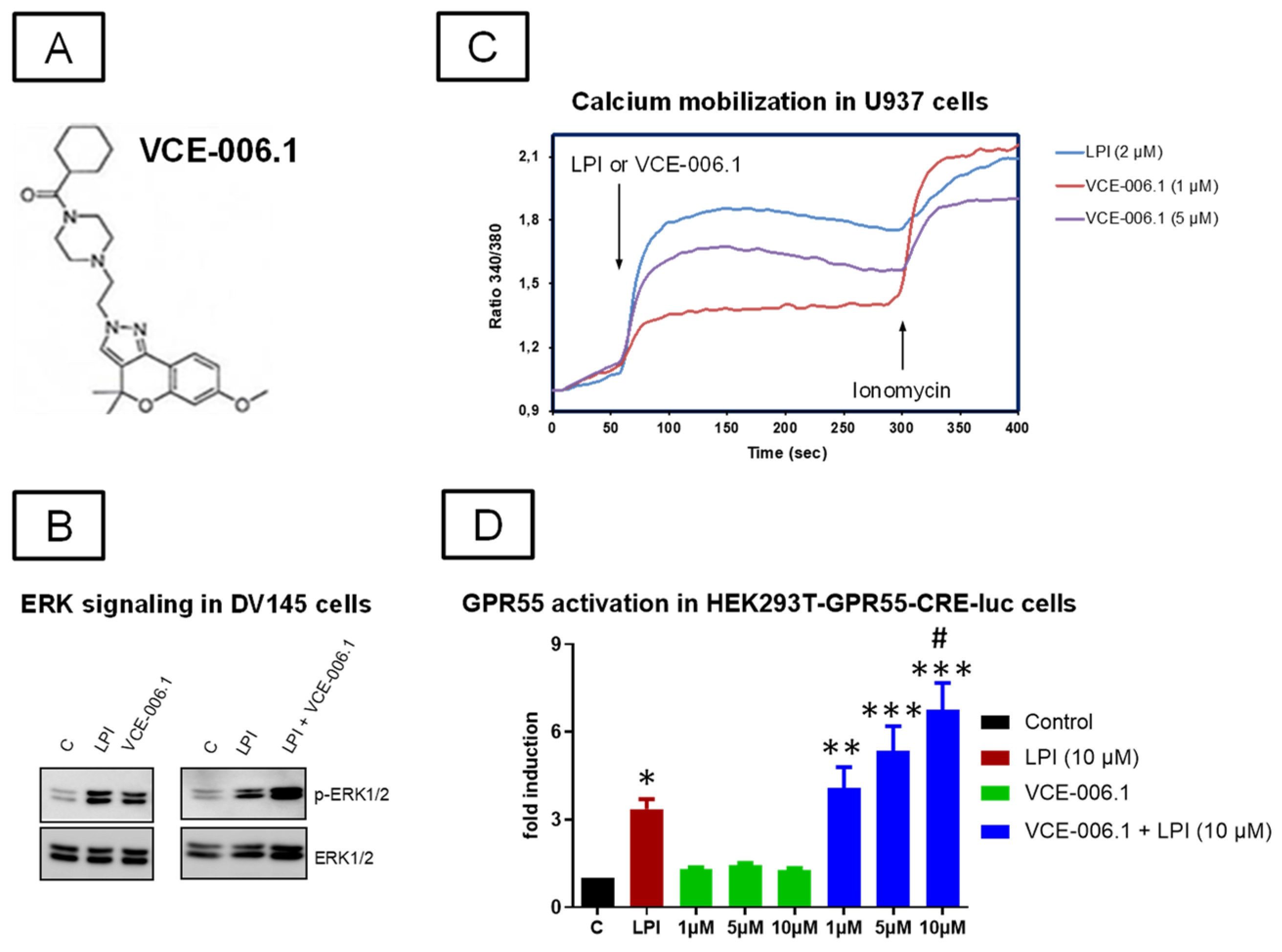{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
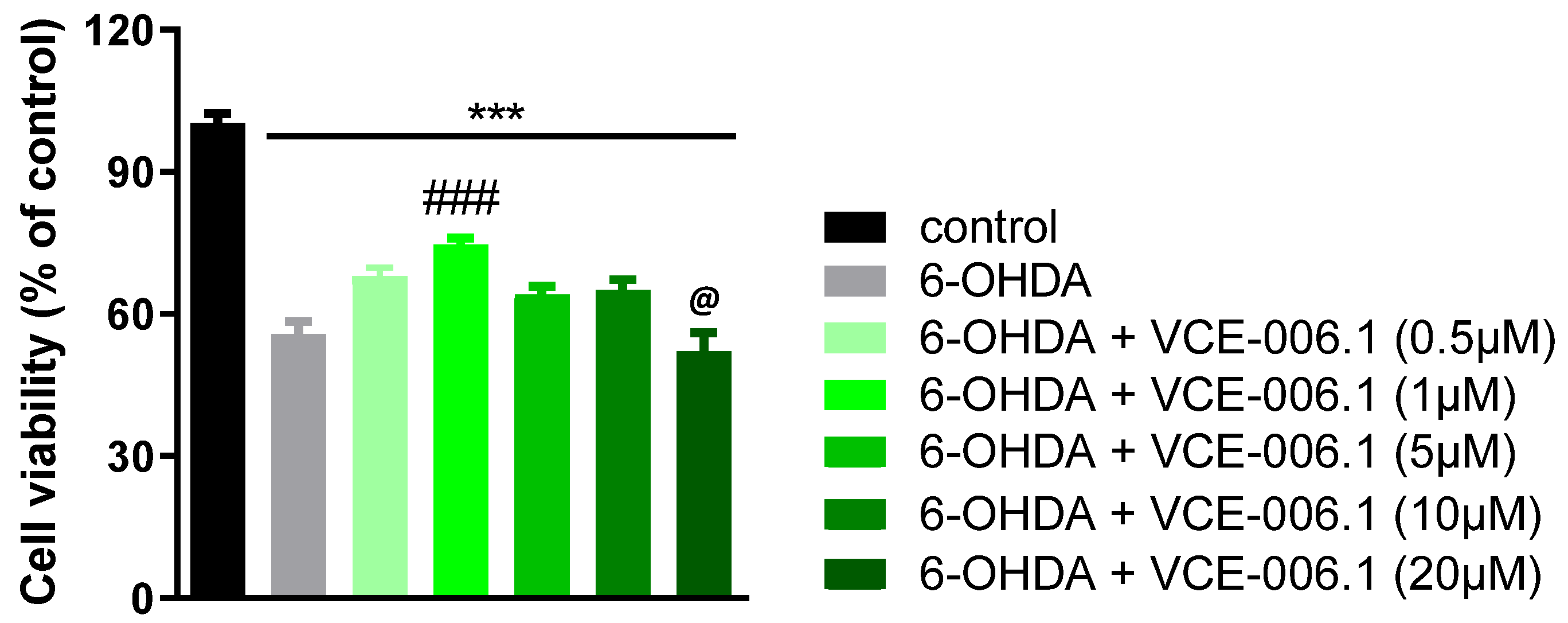
2.1. Studies on PAM Activity of VCE-006.1
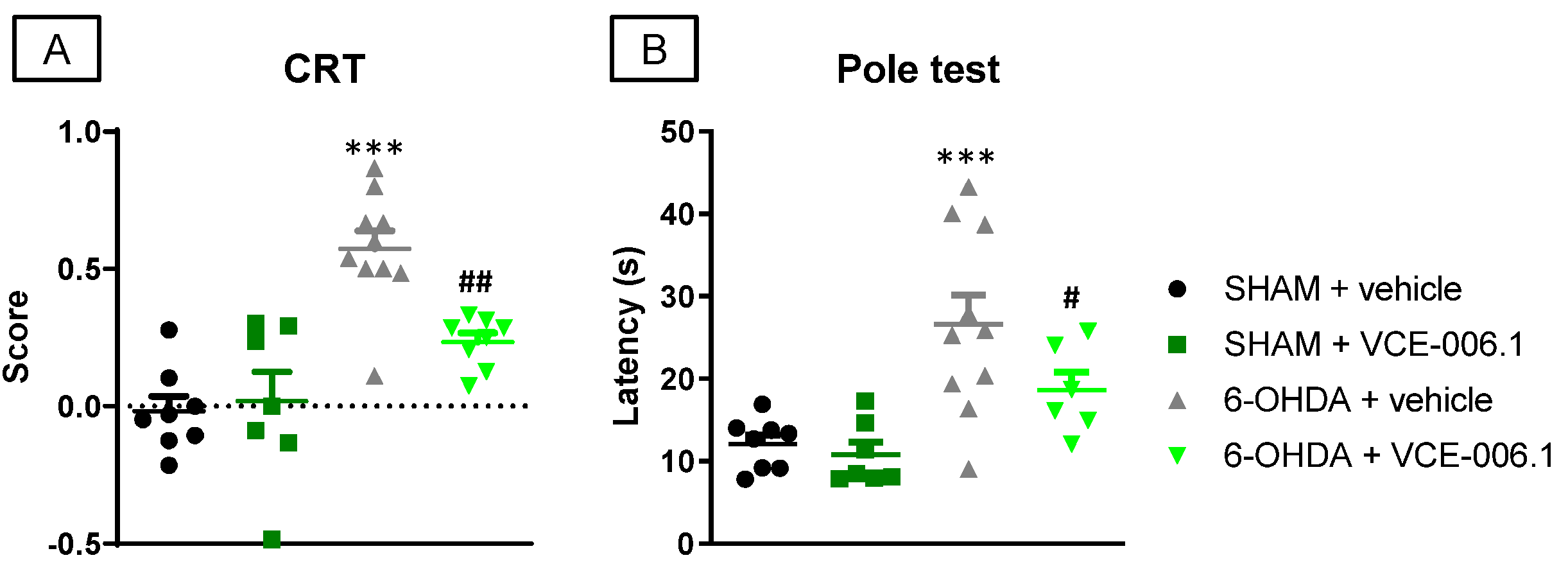
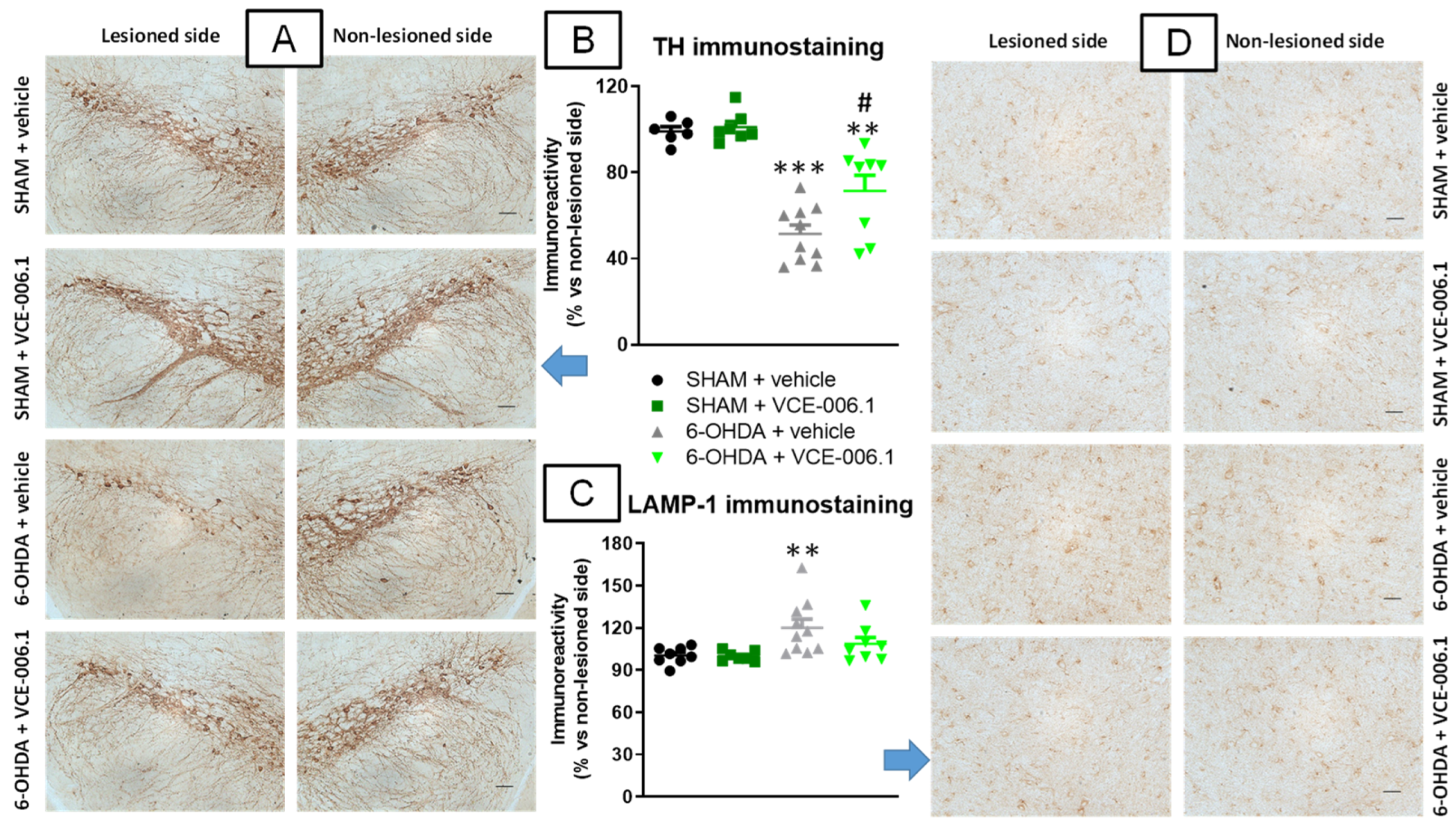
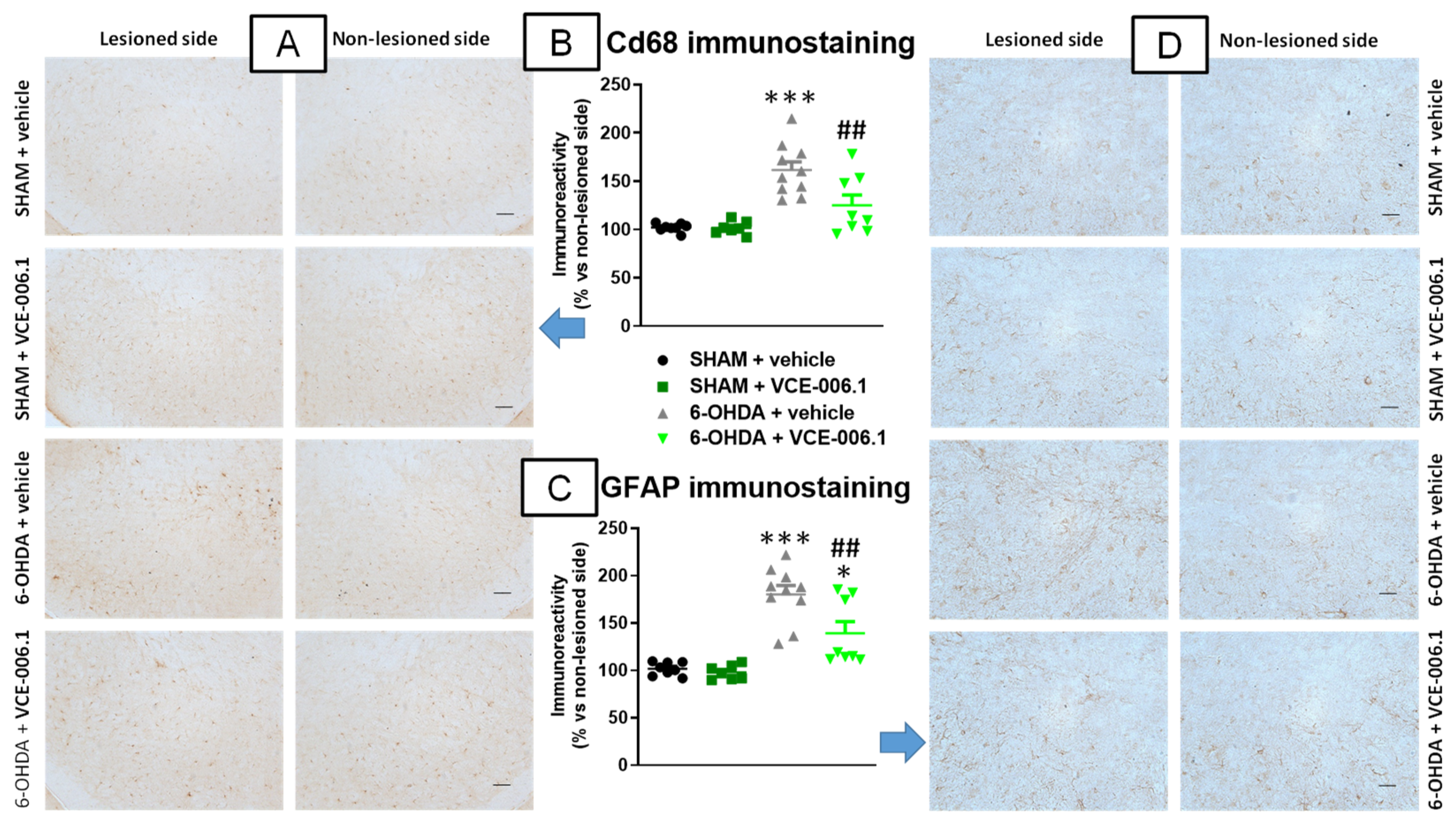
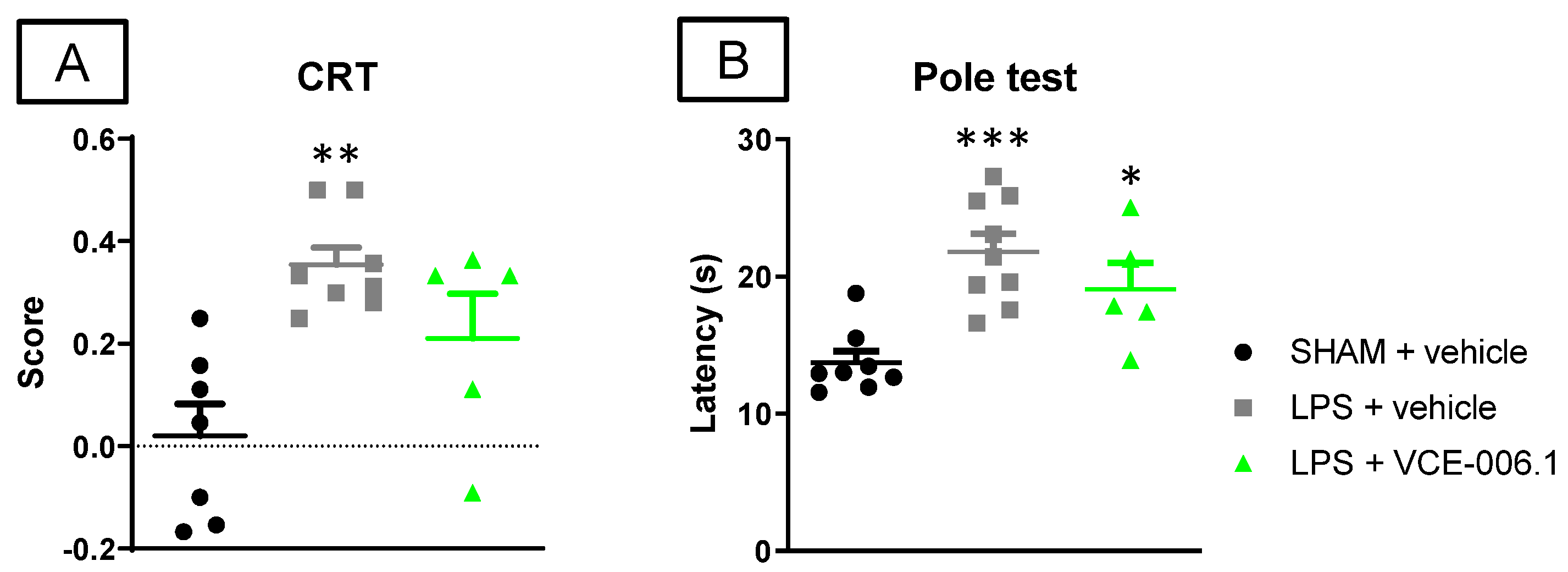
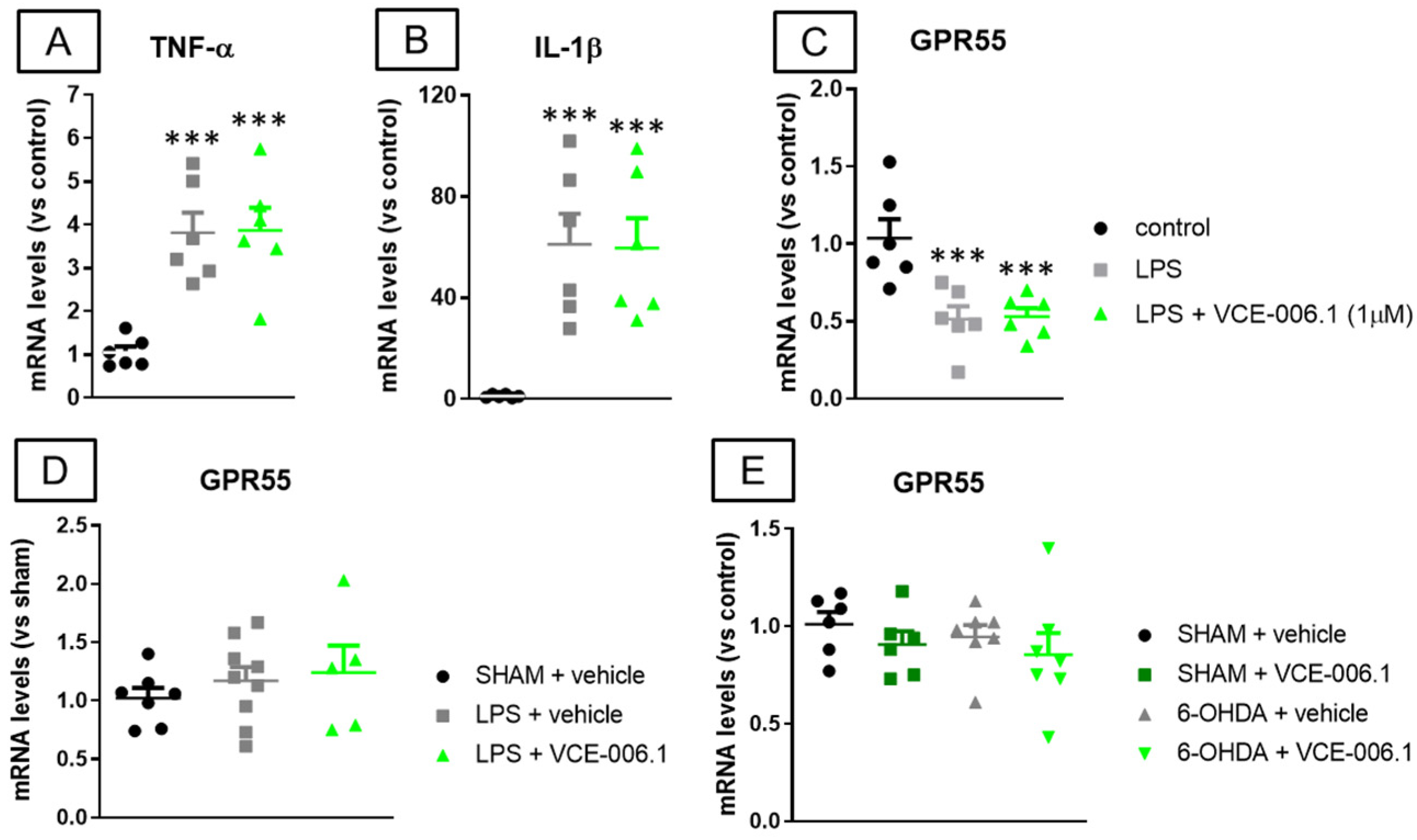
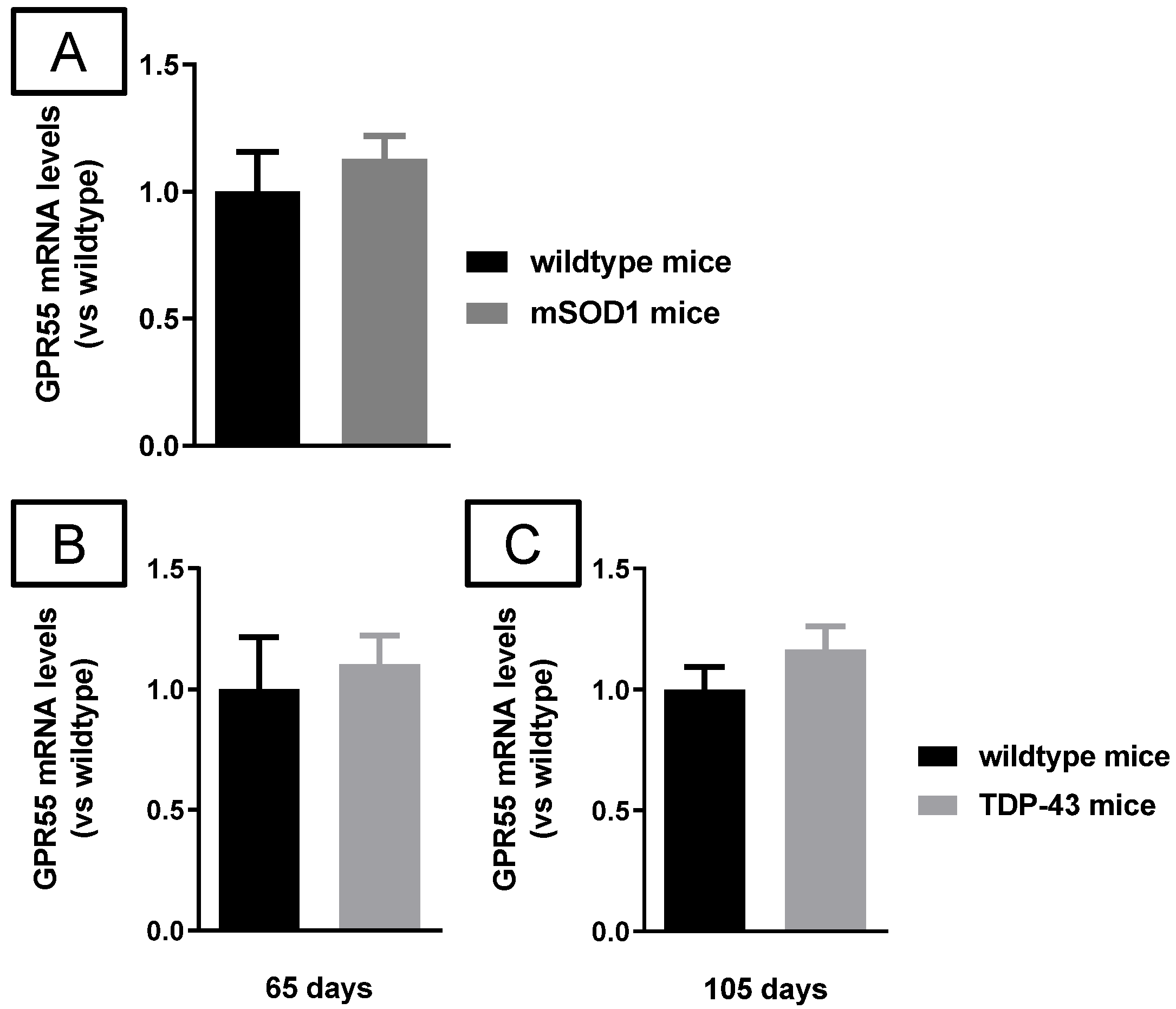
2.2. Studies in Experimental PD
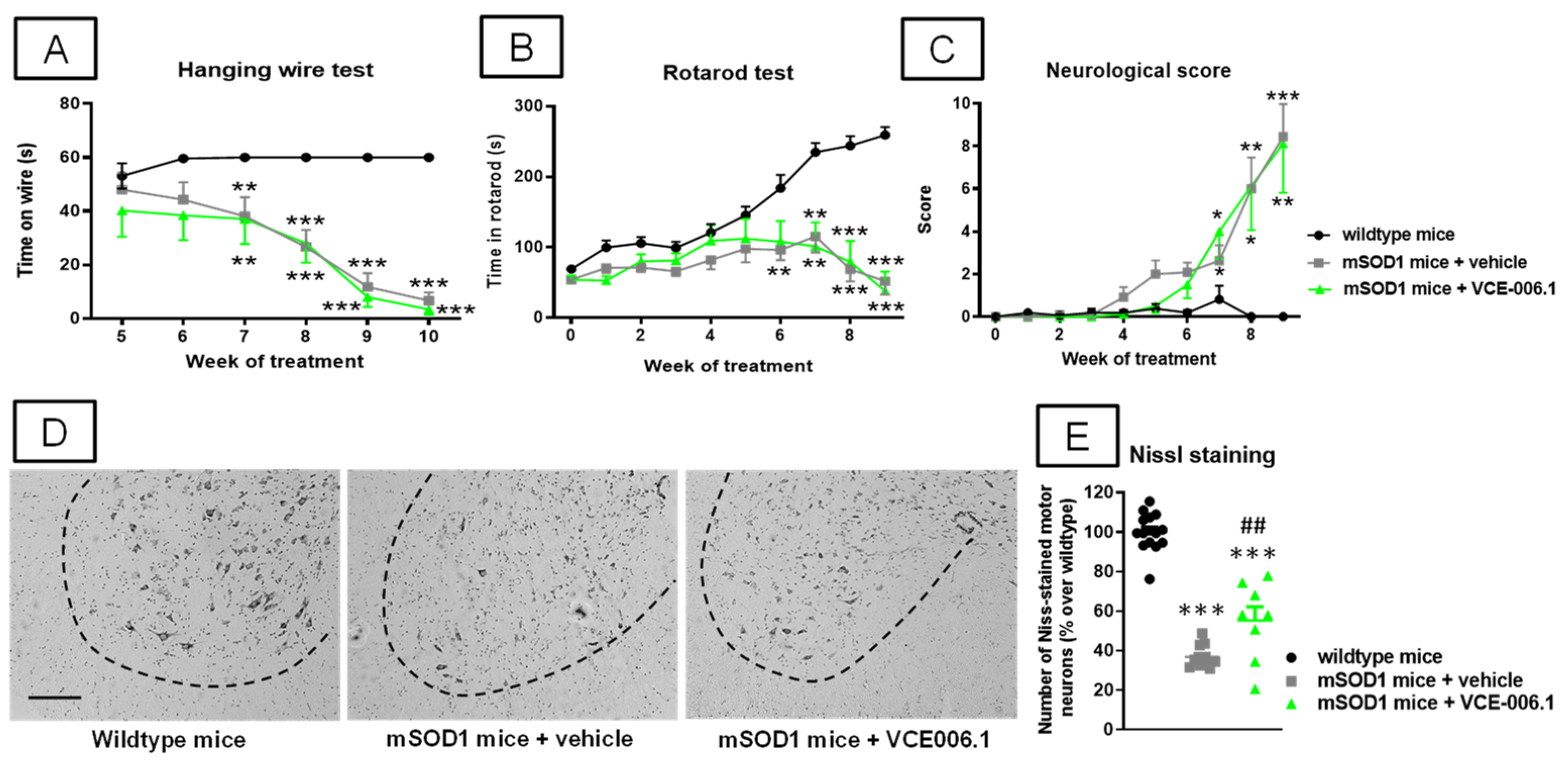
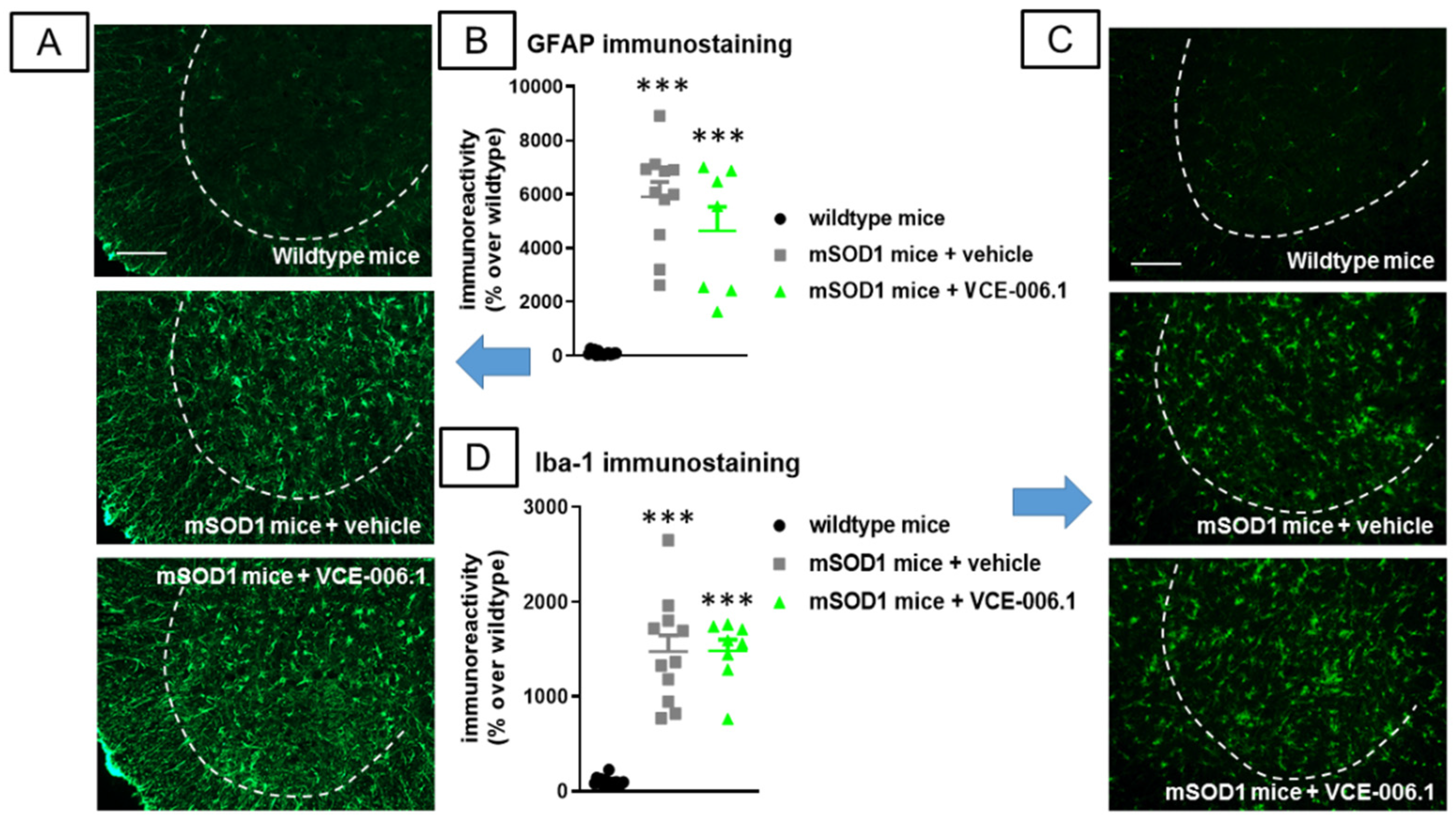
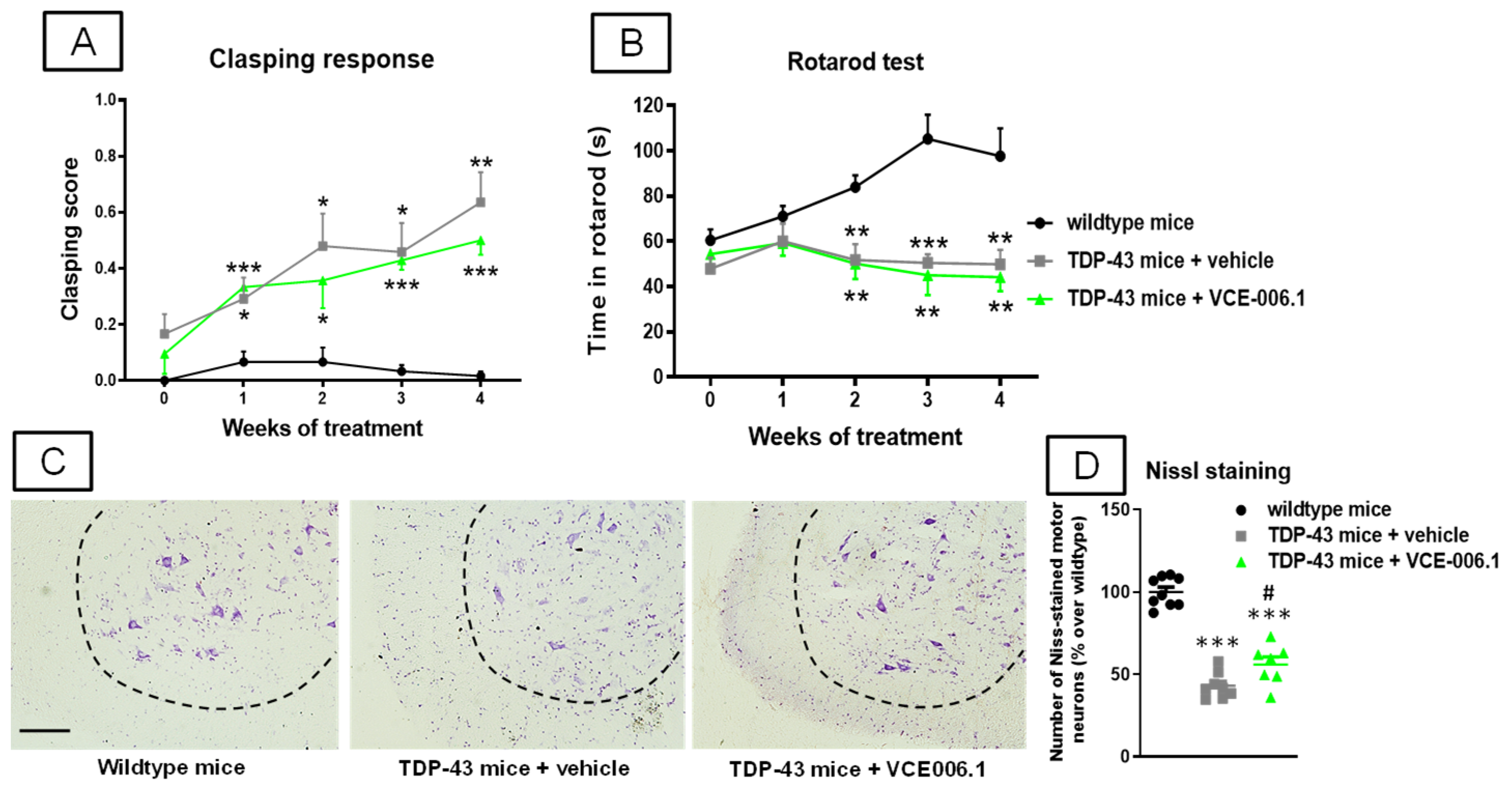
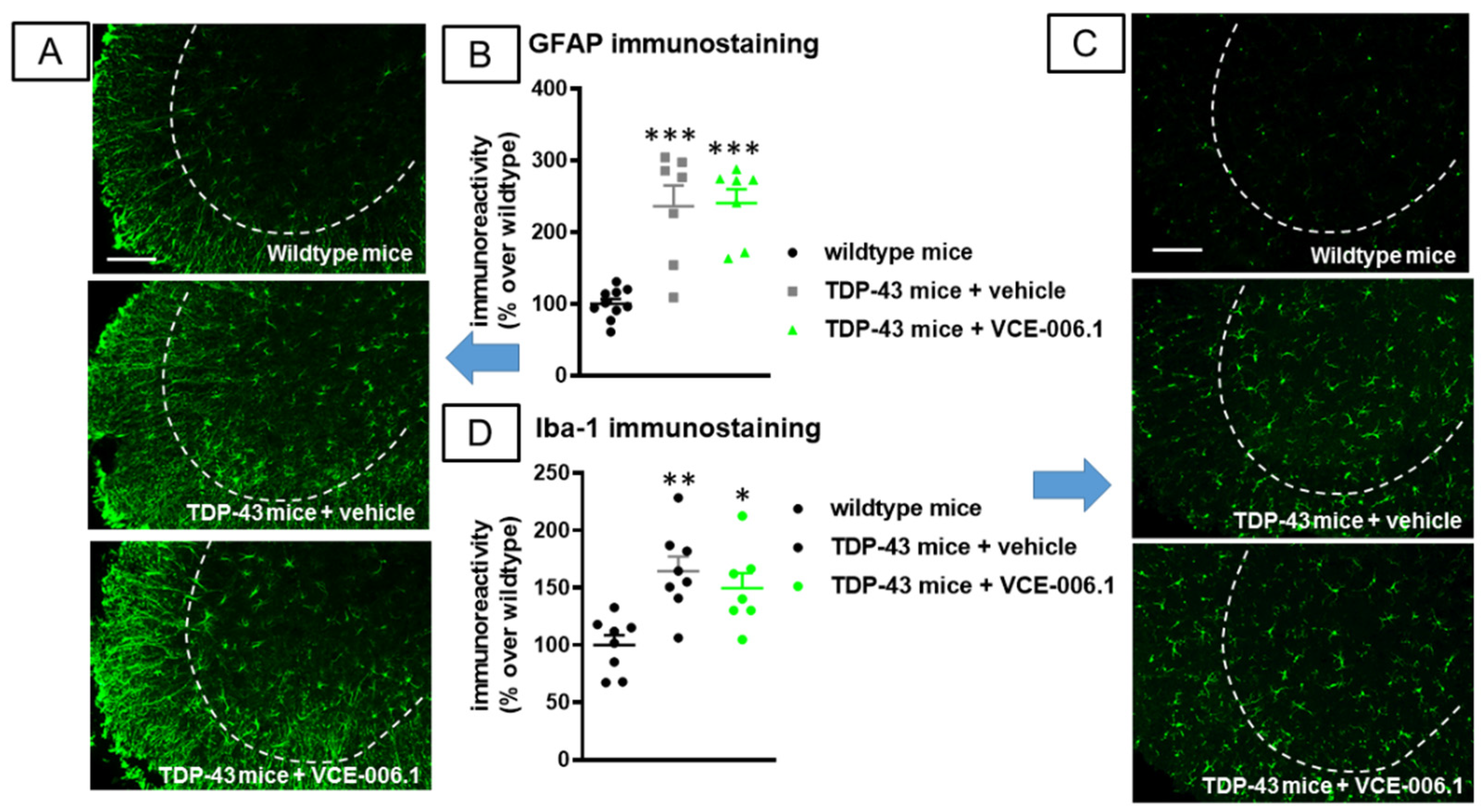
2.3. Studies in Experimental ALS
3. Discussion
4. Materials and Methods
4.1. Synthesis and Characterization as PAM of VCE-006.1 in Cell-Based Assays
4.1.1. Determination of ERK 1/2 Activation
4.1.2. Ca2+ Mobilization Assay
4.1.3. cAMP Signaling Induced by GPR55 Activation
4.2. Animals and Cell Experiments
4.2.1. PD Experiments
4.2.2. ALS Experiments
4.3. Behavioral Recording
4.3.1. Pole Test
4.3.2. Cylinder Rearing Test
4.3.3. Neurological Score
4.3.4. Rotarod Test
4.3.5. Clasping Response
4.3.6. Hanging Wire Test
4.4. Histological Procedures
4.4.1. Tissue Slicing
4.4.2. Immunohistochemistry Analysis in the PD Experiment
4.4.3. Nissl Staining
4.4.4. Immunofluorescence Analysis in the ALS Experiment
4.5. Real Time qRT-PCR Analysis
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Fernández-Ruiz, J.; Moro, M.A.; Martinez-Orgado, J. Cannabinoids in Neurodegenerative Disorders and Stroke/Brain Trauma: From Preclinical Models to Clinical Applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Ruiz, J. The biomedical challenge of neurodegenerative disorders: An opportunity for cannabinoid-based therapies to improve on the poor current therapeutic outcomes. Br. J. Pharmacol. 2018, 176, 1370–1383. [Google Scholar] [CrossRef]
- Chiarlone, A.; Bellocchio, L.; Blázquez, C.; Resel, E.; Soria-Gómez, E.; Cannich, A.; Ferrero, J.J.; Sagredo, O.; Benito, C.; Romero, J.; et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc Natl Acad Sci USA 2014, 111, 8257–8262. [Google Scholar] [CrossRef] [Green Version]
- Hiebel, C.; Behl, C. The complex modulation of lysosomal degradation pathways by cannabinoid receptors 1 and 2. Life Sci. 2015, 138, 3–7. [Google Scholar] [CrossRef]
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB1 Agonist ACEA Protects Neurons and Reduces the Cognitive Impairment of AβPP/PS1 Mice. J. Alzheimer’s Dis. 2012, 30, 439–459. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-heteromer mediated regulation of endocannabinoid signaling in activated microglia. Role of CB1 and CB2 receptors and relevance for Alzheimer’s disease and levodopa-induced dyskinesia. Brain Behav Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef]
- Crunfli, F.; Vrechi, T.A.; Costa, A.P.; Torrão, A.S. Cannabinoid Receptor Type 1 Agonist ACEA Improves Cognitive Deficit on STZ-Induced Neurotoxicity Through Apoptosis Pathway and NO Modulation. Neurotox. Res. 2019, 35, 516–529. [Google Scholar] [CrossRef]
- Chung, Y.C.; Bok, E.; Huh, S.H.; Park, J.Y.; Yoon, S.H.; Kim, S.R.; Kim, Y.S.; Maeng, S.; Park, S.H.; Jin, B.K. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 2011, 187, 6508–6517. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Rial, S.; García-Gutiérrez, M.S.; Molina, J.A.; Pérez-Nievas, B.G.; Ledent, C.; Leiva, C.; Leza, J.C.; Manzanares, J. Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol. Aging 2011, 32, 631–645. [Google Scholar] [CrossRef]
- Abood, M.E.; Rizvi, G.; Sallapudi, N.; McAllister, S.D. Activation of the CB1 cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neurosci. Lett. 2001, 309, 197–201. [Google Scholar] [CrossRef]
- Zhao, P.; Ignacio, S.; Beattie, E.C.; Abood, M.E. Altered presymptomatic AMPA and cannabinoid receptor trafficking in motor neurons of ALS model mice: Implications for excitotoxicity. Eur. J. Neurosci. 2008, 27, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, S.; De Chiara, V.; Musella, A.; Cozzolino, M.; Bernardi, G.; Maccarrone, M.; Mercuri, N.B.; Carrì, M.T.; Centonze, D. Abnormal sensitivity of cannabinoid CB1 receptors in the striatum of mice with experimental amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 83–90. [Google Scholar] [CrossRef]
- Blázquez, C.; Chiarlone, A.; Sagredo, O.; Aguado, T.; Pazos, M.R.; Resel, E.; Palazuelos, J.; Julien, B.; Salazar, M.; Börner, C.; et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington’s disease. Brain 2011, 134, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Maya-López, M.; Colín-González, A.L.; Aguilera, G.; De Lima, M.E.; Colpo-Ceolin, A.; Rangel-Lopez, E.; Villeda-Hernández, J.; Rembao-Bojórquez, D.; Túnez, I.; Luna-López, A.; et al. Neuroprotective effect of WIN55,212-2 against 3-nitropropionic acid-induced toxicity in the rat brain: Involvement of CB1 and NMDA receptors. Am. J. Transl. Res. 2017, 9, 261–274. [Google Scholar]
- Ruiz-Calvo, A.; Maroto, I.B.; Bajo-Grañeras, R.; Chiarlone, A.; Gaudioso, Á.; Ferrero, J.J.; Resel, E.; Sánchez-Prieto, J.; Rodríguez-Navarro, J.A.; Marsicano, G.; et al. Pathway-specific control of striatal neuron vulnerability by corticostriatal cannabinoid CB1 receptors. Cereb. Cortex 2018, 28, 307–322. [Google Scholar] [CrossRef]
- Rossi, S.; Furlan, R.; De Chiara, V.; Muzio, L.; Musella, A.; Motta, C.; Studer, V.; Cavasinni, F.; Bernardi, G.; Martino, G.; et al. Cannabinoid CB1 receptors regulate neuronal TNF-α effects in experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2011, 25, 1242–1248. [Google Scholar] [CrossRef]
- Moreno-Martet, M.; Feliú, A.; Espejo-Porras, F.; Mecha, M.; Carrillo-Salinas, F.J.; Fernández-Ruiz, J.; Guaza, C.; de Lago, E. The disease-modifying effects of a Sativex-like combination of phytocannabinoids in mice with experimental autoimmune encephalomyelitis are preferentially due to Δ9-tetrahydrocannabinol acting through CB1 receptors. Mult. Scler. Relat. Disord. 2015, 4, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Ruiz, J.; Romero, J.; Velasco, G.; Tolón, R.M.; Ramos, J.A.; Guzmán, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. CB2 Cannabinoid Receptor As Potential Target against Alzheimer’s Disease. Front. Neurosci. 2016, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- López, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vázquez, C.; Amores, M.; Ruiz-Pérez, G.; García-García, E.; et al. Cannabinoid CB2 receptors in the mouse brain: Relevance for Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Magham, S.V.; Krishnamurthy, P.T.; Shaji, N.; Mani, L.; Balasubramanian, S. Cannabinoid receptor 2 selective agonists and Alzheimer’s disease: An insight into the therapeutic potentials. J. Neurosci. Res. 2021, 99, 2888–2905. [Google Scholar] [CrossRef] [PubMed]
- Galán-Ganga, M.; Rodríguez-Cueto, C.; Merchán-Rubira, J.; Hernández, F.; Ávila, J.; Posada-Ayala, M.; Lanciego, J.L.; Luengo, E.; Lopez, M.G.; Rábano, A.; et al. Cannabinoid receptor CB2 ablation protects against TAU induced neurodegeneration. Acta Neuropathol. Commun. 2021, 9, 90. [Google Scholar] [CrossRef]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.; Pertwee, R.; Fernández-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol. 2011, 163, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB2 receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Cai, Q.; Zhang, J.; He, X.; Liu, Y.; Zhu, R.; Jin, L. AM1241 alleviates MPTP-induced Parkinson’s disease and promotes the regeneration of DA neurons in PD mice. Oncotarget 2017, 8, 67837–67850. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Moore, D.H.; Makriyannis, A.; Abood, M.E. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur. J. Pharmacol. 2006, 542, 100–105. [Google Scholar] [CrossRef]
- Shoemaker, J.L.; Seely, K.A.; Reed, R.L.; Crow, J.P.; Prather, P.L. The CB2 cannabinoid agonist AM-1241 prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis when initiated at symptom onset. J. Neurochem. 2006, 101, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espejo-Porras, F.; García-Toscano, L.; Rodríguez-Cueto, C.; Santos-García, I.; de Lago, E.; Fernandez-Ruiz, J. Targeting glial cannabinoid CB2 receptors to delay the progression of the pathological phenotype in TDP-43 (A315T) transgenic mice, a model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2019, 176, 1585–1600. [Google Scholar] [CrossRef]
- Rodríguez-Cueto, C.; Gómez-Almería, M.; García Toscano, L.; Romero, J.; Hillard, C.J.; de Lago, E.; Fernández-Ruiz, J. Inactivation of the CB2 receptor accelerated the neuropathological deterioration in TDP-43 transgenic mice, a model of amyotrophic lateral sclerosis. Brain. Pathol. 2021, 31, e12972. [Google Scholar] [CrossRef]
- Rodríguez-Cueto, C.; García-Toscano, L.; Santos-García, I.; Gómez-Almería, M.; Gonzalo-Consuegra, C.; Espejo-Porras, F.; Fernández-Ruiz, J.; de Lago, E. Targeting the CB2 receptor and other endocannabinoid elements to delay disease progression in amyotrophic lateral sclerosis. Br. J. Pharmacol. 2021, 178, 1373–1387. [Google Scholar] [CrossRef]
- Sagredo, O.; González, S.; Aroyo, I.; Pazos, M.R.; Benito, C.; Lastres-Becker, I.; Romero, J.P.; Tolón, R.M.; Mechoulam, R.; Brouillet, E.; et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: Relevance for Huntington’s disease. Glia 2009, 57, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain 2009, 132, 3152–3164. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, J.; Truong, J.; Bouchard, K.; Dunkelberger, D.; Desrayaud, S.; Moussaoui, S.; Tabrizi, S.J.; Stella, N.; Muchowski, P.J. Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J. Neurosci. 2012, 32, 18259–18268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.; Gómez-Cañas, M.; Navarro, G.; Hurst, D.P.; Carrillo-Salinas, F.J.; Lagartera, L.; Pazos, R.; Goya, P.; Reggio, P.H.; Guaza, C.; et al. Chromenopyrazole, a versatile cannabinoid scaffold with in vivo activity in a model of multiple sclerosis. J. Med. Chem. 2016, 59, 6753–6771. [Google Scholar] [CrossRef] [Green Version]
- Alberti, T.B.; Barbosa, W.L.; Vieira, J.L.; Raposo, N.R.; Dutra, R.C. (-)-β-Caryophyllene, a CB2 receptor-selective phytocannabinoid, suppresses motor paralysis and neuroinflammation in a murine model of multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 691. [Google Scholar] [CrossRef]
- Mecha, M.; Carrillo-Salinas, F.J.; Feliú, A.; Mestre, L.; Guaza, C. Perspectives on Cannabis-Based Therapy of Multiple Sclerosis: A Mini-Review. Front. Cell. Neurosci. 2020, 14, 34. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Iannotti, F.; Vitale, R. The Endocannabinoid System and PPARs: Focus on Their Signalling Crosstalk, Action and Transcriptional Regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef]
- García, C.; Gómez-Cañas, M.; Burgaz, S.; Palomares, B.; Gómez-Gálvez, Y.; Palomo-Garo, C.; Campo, S.; Ferrer-Hernández, J.; Pavicic, C.; Navarrete, C.; et al. Benefits of VCE-003.2, a cannabigerol quinone derivative, against inflammation-driven neuronal deterioration in experimental Parkinson’s disease: Possible involvement of different binding sites at the PPARγ receptor. J. Neuroinflamm. 2018, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junior, N.C.F.; dos-Santos-Pereira, M.; Guimarães, F.S.; Del Bel, E. Cannabidiol and Cannabinoid Compounds as Potential Strategies for Treating Parkinson’s Disease and l-DOPA-Induced Dyskinesia. Neurotox. Res. 2020, 37, 12–29. [Google Scholar] [CrossRef]
- Burgaz, S.; García, C.; Gómez-Cañas, M.; Rolland, A.; Muñoz, E.; Fernández-Ruiz, J. Neuroprotection with the Cannabidiol Quinone Derivative VCE-004.8 (EHP-101) against 6-Hydroxydopamine in Cell and Murine Models of Parkinson’s Disease. Molecules 2021, 26, 3245. [Google Scholar] [CrossRef]
- Burgaz, S.; García, C.; Gómez-Cañas, M.; Navarrete, C.; García-Martín, A.; Rolland, A.; Del Río, C.; Casarejos, M.J.; Muñoz, E.; Gonzalo-Consuegra, C.; et al. Neuroprotection with the cannabigerol quinone derivative VCE-003.2 and its analogs CBGA-Q and CBGA-Q-Salt in Parkinson’s disease using 6-hydroxydopamine-lesioned mice. Mol. Cell Neurosci. 2021, 110, 103583. [Google Scholar] [CrossRef]
- Cueto, C.R.; Santos-García, I.; García-Toscano, L.; Espejo-Porras, F.; Bellido, M.; Fernández-Ruiz, J.; Munoz, E.; de Lago, E. Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1G93A transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem. Pharmacol. 2018, 157, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 2012, 63, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dong, Z.; Liu, S. β-Caryophyllene Ameliorates the Alzheimer-Like Phenotype in APP/PS1 Mice through CB2 Receptor Activation and the PPARγ Pathway. Pharmacology 2014, 94, 1–12. [Google Scholar] [CrossRef]
- Kallendrusch, S.; Kremzow, S.; Nowicki, M.; Grabiec, U.; Winkelmann, R.; Benz, A.; Kraft, R.; Bechmann, I.; Dehghani, F.; Koch, M. The G protein-coupled receptor 55 ligand l-α-lysophosphatidylinositol exerts microglia-dependent neuroprotection after excitotoxic lesion. Glia 2013, 61, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.D.; Zuluaga-Ramirez, V.; Gajghate, S.; Winfield, M.; Sriram, U.; Rom, S.; Persidsky, Y. Activation of GPR55 induces neuroprotection of hippocampal neurogenesis and immune responses of neural stem cells following chronic, systemic inflammation. Brain Behav. Immun. 2019, 76, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Minamihata, T.; Takano, K.; Moriyama, M.; Nakamura, Y. Lysophosphatidylinositol, an Endogenous Ligand for G Protein-Coupled Receptor 55, Has Anti-inflammatory Effects in Cultured Microglia. Inflammation 2020, 43, 1971–1987. [Google Scholar] [CrossRef]
- Celorrio, M.; Rojo-Bustamante, E.; Fernández-Suárez, D.; Sáez, E.; Estella-Hermoso de Mendoza, A.; Müller, C.E.; Ramírez, M.J.; Oyarzábal, J.; Franco, R.; Aymerich, M.S. GPR55: A therapeutic target for Parkinson’s disease? Neuropharmacology 2017, 125, 319–332. [Google Scholar] [CrossRef]
- Martínez-Pinilla, E.; Aguinaga, D.; Navarro, G.; Rico, A.J.; Oyarzábal, J.; Sánchez-Arias, J.A.; Lanciego, J.L.; Franco, R. Targeting CB1 and GPR55 Endocannabinoid Receptors as a Potential Neuroprotective Approach for Parkinson’s Disease. Mol. Neurobiol. 2019, 56, 5900–5910. [Google Scholar] [CrossRef]
- Wu, C.S.; Chen, H.; Sun, H.; Zhu, J.; Jew, C.P.; Wager-Miller, J.; Straiker, A.; Spencer, C.; Bradshaw, H.; Mackie, K.; et al. GPR55, a G-Protein Coupled Receptor for Lysophosphatidylinositol, Plays a Role in Motor Coordination. PLoS ONE 2013, 8, e60314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55-a putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Lauckner, J.E.; Jensen, J.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.; Reggio, P.H. An update on non-CB1, non-CB2 cannabinoid related G-protein-coupled receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.Z.; He, L. Neuro-psychopharmacological perspective of Orphan receptors of Rhodopsin (class A) family of G protein-coupled receptors. Psychopharmacology 2017, 234, 1181–1207. [Google Scholar] [CrossRef] [PubMed]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, & Psi;GPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198 . [Google Scholar] [CrossRef]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Lysophosphatidylinositols, from Cell Membrane Constituents to GPR55 Ligands. Trends Pharmacol. Sci. 2018, 39, 586–604. [Google Scholar] [CrossRef]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Jagerovic, N. Advances towards the Discovery of GPR55 Ligands. Curr. Med. Chem. 2016, 23, 2087–2100. [Google Scholar] [CrossRef]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Mendez-Díaz, M.; Prospero-García, O. Advances in the physiology of GPR55 in the Central Nervous System. Curr. Neuropharmacol. 2017, 15, 771–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henstridge, C.M.; Balenga, N.A.; Kargl, J.; Andradas, C.; Brown, A.J.; Irving, A.; Sanchez, C.; Waldhoer, M. Minireview: Recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol. Endocrinol. 2011, 25, 1835–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.; Whyte, L.S.; Chicharro, R.; Gómez-Cañas, M.; Pazos, M.R.; Goya, P.; Irving, A.J.; Fernández-Ruiz, J.; Ross, R.A.; Jagerovic, N. Identification of novel GPR55 modulators using cell-impedance-based label-free technology. J. Med. Chem 2016, 59, 1840–1853. [Google Scholar] [CrossRef]
- Jagerovic, N.; Morales, P.; Ross, R.; Whyte, L. Selective Modulators of the Activity of the gpr55 Receptor: Chromenopyrazole Derivatives. Patent WO2016177922A1, 27 April 2016. [Google Scholar]
- Szliszka, E.; Czuba, Z.P.; Domino, M.; Mazur, B.; Zydowicz, G.; Krol, W. Ethanolic Extract of Propolis (EEP) Enhances the Apoptosis- Inducing Potential of TRAIL in Cancer Cells. Molecules 2009, 14, 738–754. [Google Scholar] [CrossRef]
- Pietr, M.; Kozela, E.; Levy, R.; Rimmerman, N.; Lin, Y.H.; Stella, N.; Vogel, Z.; Juknat, A. Differential changes in GPR55 during microglial cell activation. FEBS Lett. 2009, 583, 2071–2076. [Google Scholar] [CrossRef] [Green Version]
- Medina-Vera, D.; Rosell-Valle, C.; López-Gambero, A.; Navarro, J.; Zambrana-Infantes, E.; Rivera, P.; Santín, L.; Suarez, J.; De Fonseca, F.R. Imbalance of Endocannabinoid/Lysophosphatidylinositol Receptors Marks the Severity of Alzheimer’s Disease in a Preclinical Model: A Therapeutic Opportunity. Biology 2020, 9, 377. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, X.; Jin, S.; Hu, J.; Wu, Y.; Li, Y.; Wu, X. Activation of GPR55 attenuates cognitive impairment and neurotoxicity in a mouse model of Alzheimer’s disease induced by Aβ1–42 through inhibiting RhoA/ROCK2 pathway. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 112, 110423. [Google Scholar] [CrossRef]
- Martínez-Pinilla, E.; Rico, A.J.; Rivas-Santisteban, R.; Lillo, J.; Roda, E.; Navarro, G.; Lanciego, J.L.; Franco, R. Expression of GPR55 and either cannabinoid CB1 or CB2 heteroreceptor complexes in the caudate, putamen, and accumbens nuclei of control, parkinsonian, and dyskinetic non-human primates. Brain Struct. Funct. 2020, 225, 2153–2164. [Google Scholar] [CrossRef]
- Fatemi, I.; Abdollahi, A.; Shamsizadeh, A.; Allahtavakoli, M.; Roohbakhsh, A. The effect of intra-striatal administration of GPR55 agonist (LPI) and antagonist (ML193) on sensorimotor and motor functions in a Parkinson’s disease rat model. Acta Neuropsychiatr. 2020, 33, 15–21. [Google Scholar] [CrossRef]
- Malek, N.; Popiolek-Barczyk, K.; Mika, J.; Przewlocka, B.; Starowicz, K. Anandamide, Acting viaCB2Receptors, Alleviates LPS-Induced Neuroinflammation in Rat Primary Microglial Cultures. Neural Plast. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Martet, M.; Espejo-Porras, F.; Fernández-Ruiz, J.; de Lago, E. Changes in endocannabinoid receptors and enzymes in the spinal cord of SOD1(G93A) transgenic mice and evaluation of a Sativex®-like combination of phytocannabinoids: Interest for future therapies in amyotrophic lateral sclerosis. CNS Neurosci. 2014, 20, 809–815. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Höglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the striatal 6-OHDA model of Parkinson’s disease in wild type and α-synuclein-deleted mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef]
- Palkovits, M.; Browstein, J. Maps and Guide to Microdissection of the Rat Brain; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Hunter, R.L.; Cheng, B.; Choi, D.Y.; Liu, M.; Liu, S.; Cass, W.A.; Bing, G. Intrastriatal lipopolysaccharide injection induces parkinsonism in C57/B6 mice. J. Neurosci Res. 2009, 87, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.-H.; Kim, S.-K.; Kwon, S.-H.; Seo, J.-Y.; Lee, B.-R.; Kim, Y.-J.; Hur, K.-H.; Kim, S.Y.; Lee, S.-Y.; Jang, C.-G. 7,8,4′-Trihydroxyisoflavone, a Metabolized Product of Daidzein, Attenuates 6-Hydroxydopamine-Induced Neurotoxicity in SH-SY5Y Cells. Biomol. Ther. 2019, 27, 363–372. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H. A high-fat jelly diet restores bioenergetic balance and extends lifespan in the presence of motor dysfunction and lumbar spinal cord motor neuron loss in TDP-43A315T mutant C57BL6/J mice. Dis. Model Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Fleming, S.M.; Ekhator, O.R.; Ghisays, V. Assessment of Sensorimotor Function in Mouse Models of Parkinson’s Disease. J. Vis. Exp. 2013, 76, e50303. [Google Scholar] [CrossRef] [Green Version]
- Guyenet, S.J.; Furrer, S.A.; Damian, V.M.; Baughan, T.D.; La Spada, A.R.; Garden, G.A. A Simple Composite Phenotype Scoring System for Evaluating Mouse Models of Cerebellar Ataxia. J. Vis. Exp. 2010, 21, e1787. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, F.J.; Lafuente, H.; Rey-Santano, M.C.; Mielgo, V.E.; Gastiasoro, E.; Rueda, M.; Pertwee, R.G.; Castillo, A.I.; Romero, J.; Martínez-Orgado, J. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr. Res. 2008, 64, 653–658. [Google Scholar] [CrossRef] [Green Version]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgaz, S.; García, C.; Gonzalo-Consuegra, C.; Gómez-Almería, M.; Ruiz-Pino, F.; Unciti, J.D.; Gómez-Cañas, M.; Alcalde, J.; Morales, P.; Jagerovic, N.; et al. Preclinical Investigation in Neuroprotective Effects of the GPR55 Ligand VCE-006.1 in Experimental Models of Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Molecules 2021, 26, 7643. https://doi.org/10.3390/molecules26247643
Burgaz S, García C, Gonzalo-Consuegra C, Gómez-Almería M, Ruiz-Pino F, Unciti JD, Gómez-Cañas M, Alcalde J, Morales P, Jagerovic N, et al. Preclinical Investigation in Neuroprotective Effects of the GPR55 Ligand VCE-006.1 in Experimental Models of Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Molecules. 2021; 26(24):7643. https://doi.org/10.3390/molecules26247643
Chicago/Turabian StyleBurgaz, Sonia, Concepción García, Claudia Gonzalo-Consuegra, Marta Gómez-Almería, Francisco Ruiz-Pino, Juan Diego Unciti, María Gómez-Cañas, Juan Alcalde, Paula Morales, Nadine Jagerovic, and et al. 2021. "Preclinical Investigation in Neuroprotective Effects of the GPR55 Ligand VCE-006.1 in Experimental Models of Parkinson’s Disease and Amyotrophic Lateral Sclerosis" Molecules 26, no. 24: 7643. https://doi.org/10.3390/molecules26247643
APA StyleBurgaz, S., García, C., Gonzalo-Consuegra, C., Gómez-Almería, M., Ruiz-Pino, F., Unciti, J. D., Gómez-Cañas, M., Alcalde, J., Morales, P., Jagerovic, N., Rodríguez-Cueto, C., de Lago, E., Muñoz, E., & Fernández-Ruiz, J. (2021). Preclinical Investigation in Neuroprotective Effects of the GPR55 Ligand VCE-006.1 in Experimental Models of Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Molecules, 26(24), 7643. https://doi.org/10.3390/molecules26247643