
Effects of Specific Inhibitors for CaMK1D on a Primary Neuron Model for Alzheimer’s Disease


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
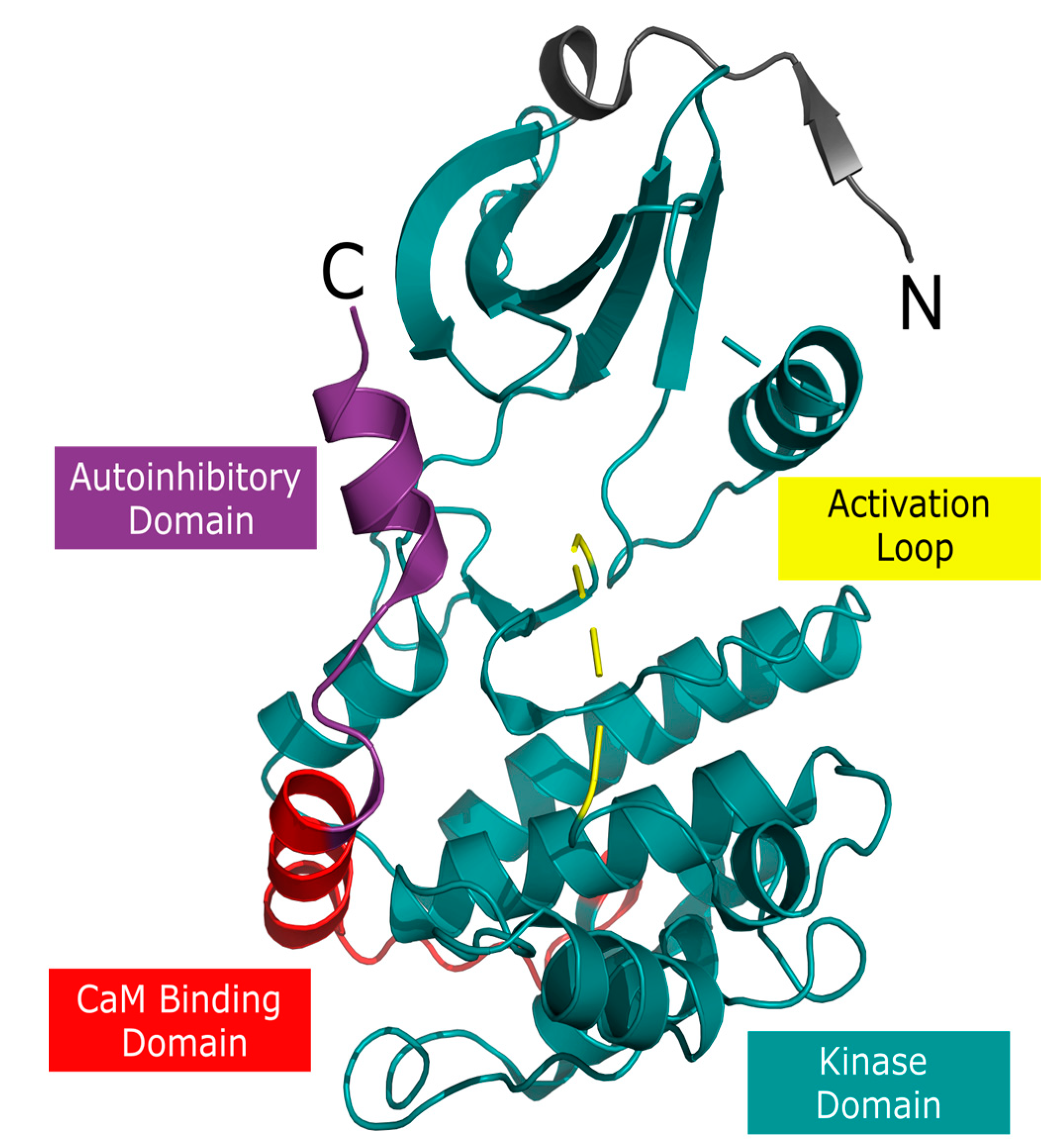
1. Introduction
2. Results
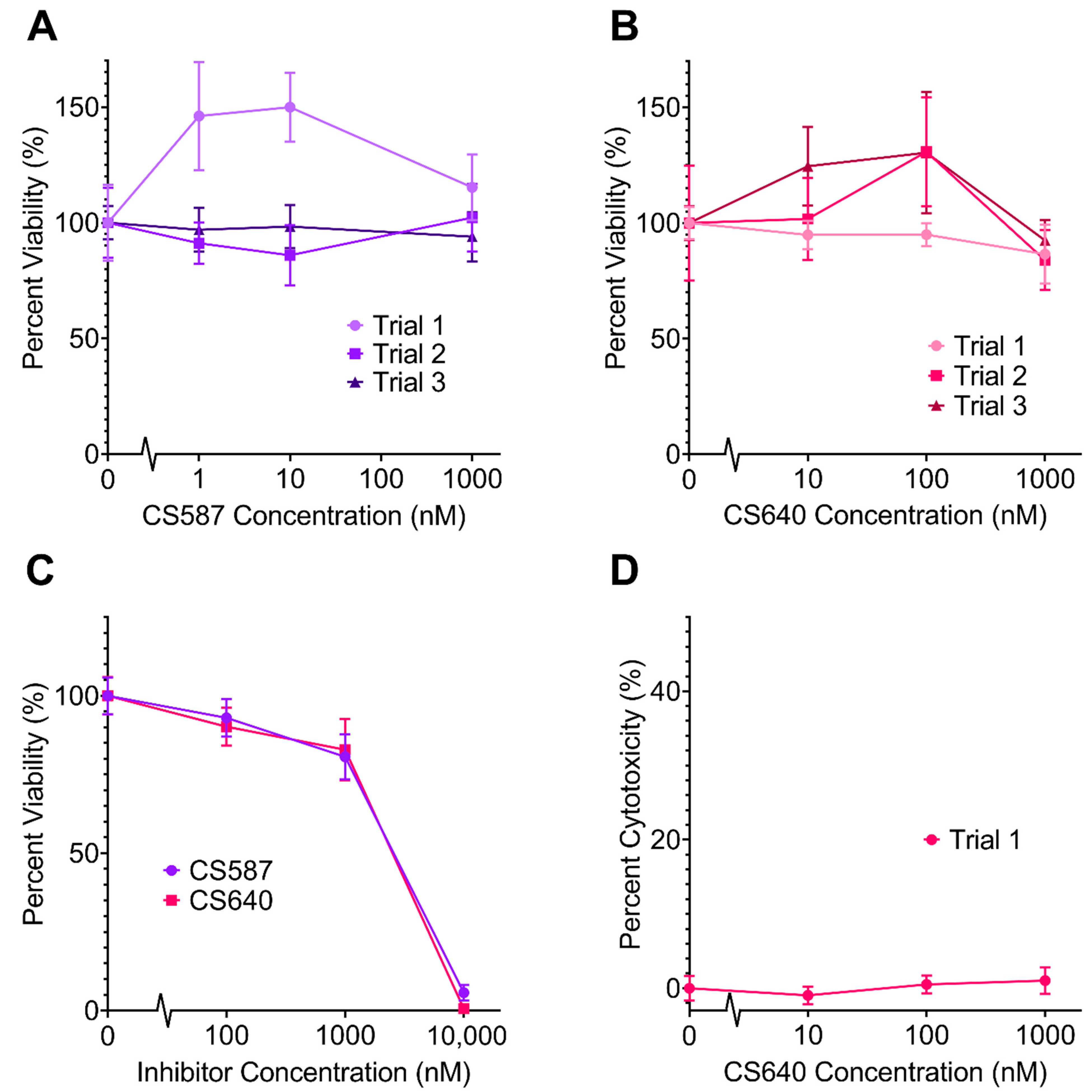
2.1. Neurotoxicity of CaMK1D Inhibitors
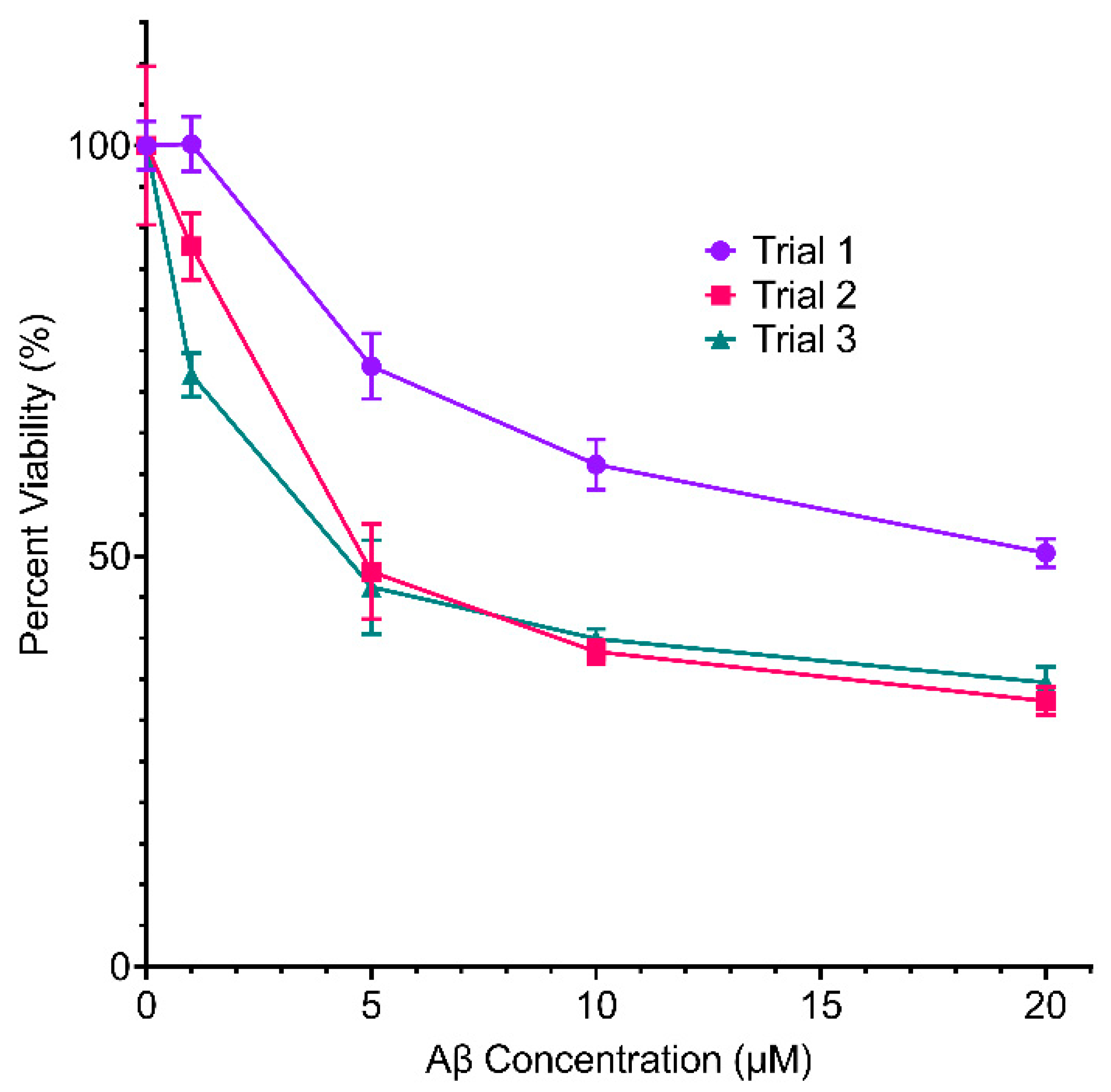
2.2. Effect of Inhibitors on Aβ Toxicity
2.3. Effect of Inhibitors on Tau Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Mouse Primary Cortical Neuron Cell Culture Preparation
4.2. Treatment of Mouse Primary Neuron Cell Cultures
4.3. MTT Assays
4.4. LDH Assays
4.5. Western Blots
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Towards a Dementia Plan: A WHO Guide; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Panza, F.; Loupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Ceyzériat, K.; Zilli, T.; Millet, P.; Frisoni, G.B.; Garibotto, V.; Tournier, B.B. Learning from the past: A review of clinical trials targeting amyloid, tau and neuroinflammation in Alzheimer’s disease. Curr. Alzheimer Res. 2020, 17, 112–125. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug. Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of neurodegeneration—Insights from familial Alzheimer’s disease. Semin Cell Dev. Biol. 2020, 105, 75–85. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, X.; Xia, W.; Zhang, Y.; Wang, C. Targeting Amyloidogenic Processing of APP in Alzheimer’s Disease. Front. Mol. Neurosci 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gómez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H. Alzheimer’s Disease: A short introduction to the calmodulin hypothesis. AIMS Neurosci. 2019, 6, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Slifer, M.; Martin, E.R.; Schnetz-Boutaud, N.; Bartlett, J.; Anderson, B.; Züchner, S.; Gwirtsman, H.; Gilbert, J.R.; Pericak-Vance, M.A.; et al. Genomic convergence to identify candidate genes for Alzheimer disease on chromosome 10. Hum. Mutat. 2009, 30, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Floudas, C.S.; Um, N.; Kamboh, M.I.; Barmada, M.M.; Visweswaran, S. Identifying genetic interactions associated with late-onset Alzheimer’s disease. BioData Min. 2014, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhu, Y.; Yang, J.; Li, L.; Wu, H.; De Jager, P.L.; Jin, P.; Bennett, D.A. A genome-wide profiling of brain DNA hydroxymethylation in Alzheimer’s disease. Alzheimers Dement. 2017, 13, 674–688. [Google Scholar] [CrossRef]
- Bernstein, A.I.; Lin, Y.; Street, R.C.; Lin, L.; Dai, Q.; Yu, L.; Bao, H.; Gearing, M.; Lah, J.J.; Nelson, P.T.; et al. 5-Hydroxymethylation-associated epigenetic modifiers of Alzheimer’s disease modulate Tau-induced neurotoxicity. Hum. Mol. Genet. 2016, 25, 2437–2450. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Chen, L.; Shen, Q.; Yu, H.; Pei, S.; Yangting, Z.; He, X.; Wang, Q.; Li, D. 5-Hydroxymethylcytosine Signatures in Circulating Cell-Free DNA as Diagnostic Biomarkers for Late-Onset Alzheimer’s Disease. Preprints Lancet 2021, 1–32. [Google Scholar] [CrossRef]
- Riascos, D.; Nicholas, A.; Samaeekia, R.; Yukhananov, R.; Mesulam, M.M.; Bigio, E.H.; Weintraub, S.; Guo, L.; Geula, C. Alterations of Ca2+ -responsive proteins within cholinergic neurons in aging and Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokumitsu, H.; Enslen, H.; Soderling, T.R. Characterization of a Ca2+/Calmodulin-dependent Protein Kinase Cascade: Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 1995, 270, 19320–19324. [Google Scholar] [CrossRef] [Green Version]
- Soderling, T.R. The Ca2+–calmodulin-dependent protein kinase cascade. Trends Biochem. Sci. 1999, 24, 232–236. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Tokumitsu, H.; Inuzuka, H.; Murata-Hori, M.; Hosoya, H.; Kobayashi, R. Identification and characterization of novel components of a Ca2+/calmodulin-dependent protein kinase cascade in HeLa cells. FEBS Lett. 2003, 550, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, H.; Kamata, A.; Nishimura, H.; Kasahara, J.; Owada, Y.; Takeuchi, Y.; Watanabe, M.; Fukunaga, K.; Kondo, H. Prominent expression and activity-dependent nuclear translocation of Ca2+/calmodulin-dependent protein kinase Iδ in hippocampal neurons. Eur. J. Neurosci. 2005, 22, 2697–2707. [Google Scholar] [CrossRef]
- Fromont, C.; Atzori, A.; Kaur, D.; Hashmi, L.; Greco, G.; Cabanillas, A.; Nguyen, H.V.; Jones, D.H.; Garzón, M.; Varela, A.; et al. Discovery of Highly Selective Inhibitors of Calmodulin-Dependent Kinases That Restore Insulin Sensitivity in the Diet-Induced Obesity in Vivo Mouse Model. J. Med. Chem. 2020, 63, 6784–6801. [Google Scholar] [CrossRef] [PubMed]
- Kamata, A.; Sakagami, H.; Tokumitsu, H.; Owada, Y.; Fukunaga, K.; Kondo, H. Spatiotemporal expression of four isoforms of Ca2+/calmodulin-dependent protein kinase I in brain and its possible roles in hippocampal dendritic growth. Neurosci. Res. 2007, 57, 86–97. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack Jr, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Calvet, M.; Karikari, T.K.; Ashton, N.J.; Lantero Rodríguez, J.; Milà-Alomà, M.; Gispert, J.D.; Salvadó, G.; Minguillon, C.; Fauria, K.; Shekari, M.; et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol. Med. 2020, 12, e12921. [Google Scholar] [CrossRef]
- Song, M.; Rauw, G.; Baker, G.; Kar, S. Memantine protects rat cortical cultured neurons against β-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Amritraj, A.; Wang, Y.; Revett, T.J.; Vergote, D.; Westaway, D.; Kar, S. Role of Cathepsin D in U18666A-induced Neuronal Cell Death: Potential implication in niemann-pick type c disease pathogenesis. J. Bio Chem. 2013, 288, 3136–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fa, M.; Orozco, I.J.; Francis, Y.I.; Saeed, F.; Gong, Y.; Arancio, O. Preparation of Oligomeric beta-amyloid 1-42 and Induction of Synaptic Plasticity Impairment on Hippocampal Slices. J. Vis. Exp. 2010, 41, e1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grant, P.; Kumar, J.; Kar, S.; Overduin, M. Effects of Specific Inhibitors for CaMK1D on a Primary Neuron Model for Alzheimer’s Disease. Molecules 2021, 26, 7669. https://doi.org/10.3390/molecules26247669
Grant P, Kumar J, Kar S, Overduin M. Effects of Specific Inhibitors for CaMK1D on a Primary Neuron Model for Alzheimer’s Disease. Molecules. 2021; 26(24):7669. https://doi.org/10.3390/molecules26247669
Chicago/Turabian StyleGrant, Paige, Jitendra Kumar, Satyabrata Kar, and Michael Overduin. 2021. "Effects of Specific Inhibitors for CaMK1D on a Primary Neuron Model for Alzheimer’s Disease" Molecules 26, no. 24: 7669. https://doi.org/10.3390/molecules26247669
APA StyleGrant, P., Kumar, J., Kar, S., & Overduin, M. (2021). Effects of Specific Inhibitors for CaMK1D on a Primary Neuron Model for Alzheimer’s Disease. Molecules, 26(24), 7669. https://doi.org/10.3390/molecules26247669