Abstract
In this mini-review of our research group’s activity, the application of 57Fe Mössbauer spectroscopy in studies of electronic structure, coordination environment, and magnetic interactions in an interesting series of Fe(II/III) compounds selected is discussed. We selected two prominent phenomena that arose during investigations of selected groups of compounds carried out at different periods of time: (1) very high magnetic hyperfine fields observed at low temperatures; (2) changes in the oxidation state of the central iron atom of complexes in the solid state during interactions with gaseous O2/H2O mixtures, resulting in spin crossover (SCO).
1. Introduction
One very interesting field of modern molecular chemistry is the preparation and investigation of properties of specially designed single molecules, metal clusters, or groups of molecules that exhibit cooperative effects and extended magnetic interactions [1,2,3]. These characteristics are important for applications such as molecular spintronics, quantum technology, organometallic frameworks, and 2D materials [4,5]. The high-performance information processing devices of the future, such as high-capacity data storage equipment, require compounds that can form molecular-level clusters or thin films that can be deposited on a dielectric base.
Single-molecule magnets (SMMs) known to date [3,6,7,8,9] usually contain clusters of paramagnetic ions bound to simple ligands (oxide, alkoxide, and halide), surrounded by organic molecular ligands. In the case of SMMs, there are also organometallic compounds [6] that show superparamagnetism below a certain temperature corresponding to the molecular size range. Many SMM compounds contain iron, such as the so-called {FeIII4}, {FeIII8}, and {DyFeIII3} compounds.
Mössbauer spectroscopy is truly preferred as the most informative technique for the study of such compounds [10,11,12,13,14]. In nuclear physics today, Mössbauer spectroscopy [15,16,17,18,19] has become a widely used method for materials analysis, because it can be used to measure hyperfine interactions between electrons and nuclei with an accuracy of 13-15 orders of magnitude. Measurements of Mössbauer spectral parameters provide information on local electron density, local electric field gradients, and the local effective magnetic field (Figure 1). For iron-containing inorganic compounds, solid state phases, materials, or alloys, 57Fe Mössbauer spectroscopy has a great advantage over other methods because it allows researchers to obtain information about (1) the metal oxidation state; (2) the spin state; (3) the iron center microenvironment; and (4) the absence/presence and origin of hyperfine magnetic fields. Thus, Mössbauer spectroscopy is suitable for detecting and distinguishing magnetic coupling (ferromagnetic, antiferromagnetic, and ferrimagnetic) between metal centers. This task can be achieved by examining the exchange interactions between coupled centers using an external magnetic field [20]. In addition, spin relaxation processes in Fe-containing compounds can be monitored.
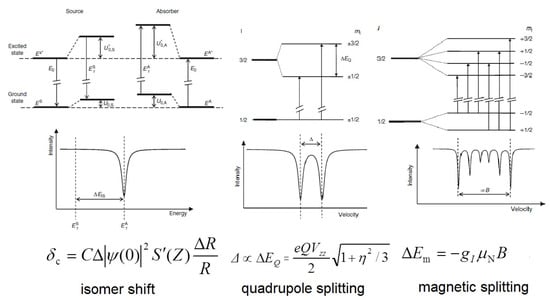
Figure 1.
Energy level diagrams for hyperfine interactions at 3/2→1/2 nuclear spin transition and a schematic representation of the corresponding Mössbauer parameters in the spectrum. In the formulas, |Ψ(0)|2 is the s electronic density, R is the radius of the nucleus, S’(Z) is the Shirley factor, Q is the nuclear quadrupole moment, Vzz is the principal component of electric field gradient, η is the asymmetry parameter, gI is the nuclear gyromagnetic factor, μN is the nuclear magneton and B is the magnetic flux density.
Spin transition may occur in transition metal complexes with an octahedral inner sphere of the central atom if the metal ion can acquire a low-spin (LS) state, as well as in high-spin (HS) states with a small enthalpy difference. The most commonly studied spin transition molecules are FeII compounds [21]. Spin transition can be initiated by changing the temperature, pressure, or visible light exposure. The schematic distribution of the electrons on the 3d shell of FeII in the low-spin and high-spin configurations and their associated enthalpy curves is shown in Figure 2, with vibrational levels indicated. At low temperatures, the low-enthalpy, low-spin state is more stable, but if the temperature exceeds a certain value, the high-spin electron structure will become the more thermodynamically stable state. Spin transition is a molecular phenomenon that depends on intermolecular interactions. The whole crystal lattice is involved in the process of the spin transition of the central iron ion. Mössbauer spectroscopy is well-positioned as a diagnostic method to determine spin transitions in iron complexes [21,22]. Since both FeII and FeIII are distinguishable by Mössbauer measurements, different isomer shift and quadrupole splitting values are characteristic for low- and high-spin compounds. Namely, the lower isomer shift is characteristic of the LS states for both FeII and FeIII, while the quadrupole splitting was found to be lower in LS FeII compounds and higher in LS FeIII compounds as compared to the relevant HS states [21]. For example, the isomer shifts typical of LS FeII compounds are δ = −0.1 to 0.2 mm/s, the quadrupole splitting is Δ = 0.2 to 1.9 mm/s, and while the typical isomer shifts of HS FeII compounds are δ = 0.9 to 1.3 mm/s, the quadrupole splitting is in the range Δ = 2.0–2.7 mm/s at room temperature [16,19]. Notably, the Mössbauer parameter ranges usually do not overlap each other.
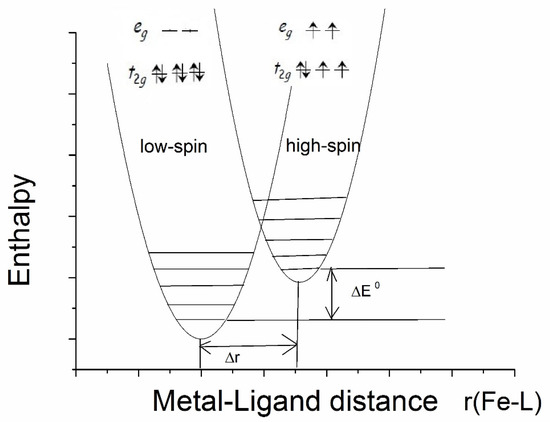
Figure 2.
Distribution of electrons on a 3d shell in low-spin and high-spin configurations in FeII and their enthalpy curves as a function of iron-ligand distance.
In this mini-review, we present some of our research group’s recent results on spin transitions and magnetic coupling in oxygenated iron(II)/(III) phthalocyanines and two-coordinate linear iron(II) complexes, which were studied with extensive use of Mössbauer spectroscopy.
2. Extreme High Hyperfine Field and Ferromagnetic Interaction in Linear, Two-Fold Coordinated Single-Molecule Magnetic Iron Complexes
Molecular magnetism, which is the focus of current scientific interest with possible technological aspects, is mostly present in molecules with unusual geometry and bonding anisotropy [1,2,3,23], such as two-coordinate iron(II) complexes with linear geometry that possess interesting electrical and magnetic properties [24,25,26,27,28,29,30,31,32,33,34]. In order to create such an unusually low coordination number and stabilize low-valent metal centers, a special set of bulky, protective ligands was designed, as shown in Chart 1. These are sterically demanding methanide (L1) and amide (L2–L5) ligands, which become mono-anions upon deprotonation (Chart 1). Thus, complexes of FeL2 type were formed, and only one, FeII[C(SiMe3)3]2 (Fe(L1)2 later), represents a highly air and moisture sensitive organometallic compound [32], the structure of which is presented in Figure 3. The protective ligand is therefore trimethyl(trisilyl)methanide. Other FeL2 complexes (L = L2–L5) are formally low-coordination, number-unstable Werner-type complexes. In this structure, the iron atom forms a linear C-Fe-C array with a 180 °C-Fe-C angle. Three tri-methylated silicon atoms forming an excellent protective shield are bound to two carbon atoms connected to the metal center (Figure 3).
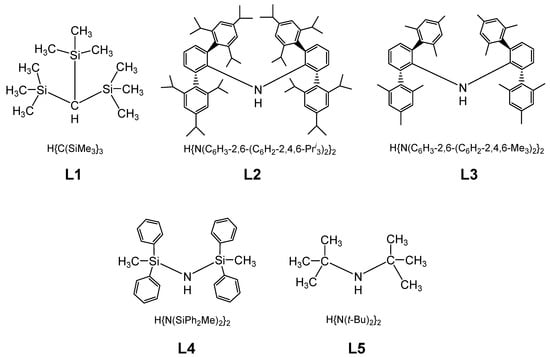
Chart 1.
Chemical structures used in the preparation of sterically protected linear and quasi-linear iron(II) amido and methanido complexes.
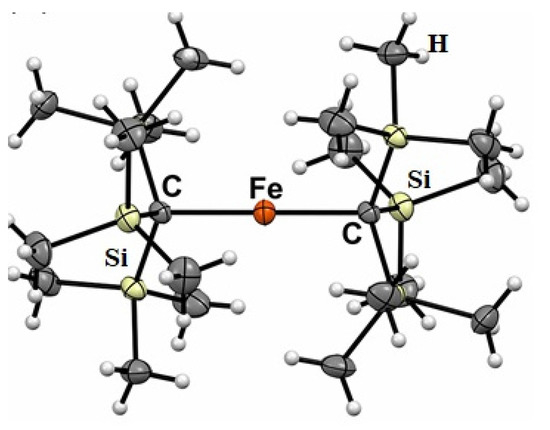
Figure 3.
Molecular structure of FeII[C(SiMe3)3]2 (Fe(L1)2).
In the FeII[C(SiMe3)3]2 (Fe(L1)2) molecule, an unusually large 57Fe hyperfine magnetic field was detected at low temperatures [26,27,28]. At first, we obtained this Fe(L1)2 compound and characterized it using 57Fe-Mössbauer spectroscopy, which is known for accurate measurement of hyperfine magnetic field strength at the iron site, and by other physical methods that helped to confirm and explain the extremely high magnetic field at the central atom [25,26,33,34]. The results of the geometry optimization and electronic structure determination performed by density functional theory (DFT) calculations were in agreement with the geometry determined by X-ray analysis. Furthermore, the calculations indicated that the HS state of iron was energetically favorable. The DFT analysis showed that there was a significant charge transfer from the iron to the two methanide L1 C(SiMe3)3− ligands. According to the calculations, the electron configuration of the Fe atom in the test compound was [Ar]3d5.83 4s0.72, where the 4s electron density was only slightly polarized and the vast majority of unpaired electrons occupied the 3d subshell [25,26]. In the 57Fe-Mössbauer spectrum of Fe(L1)2 at 20 K, a component with an extraordinary large internal magnetic field strength of 157.5 T was observed (Figure 4) [25,26].
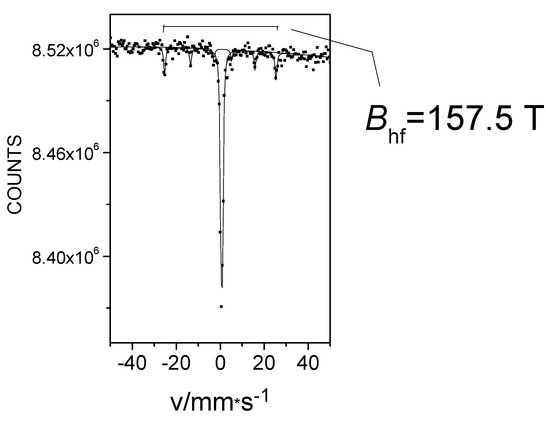
Figure 4.
The 57Fe Mössbauer spectrum of Fe(L1)2 at 20 K.
The high internal magnetic field observed in Fe(L1)2 was surprising because the contribution of the Fermi contact field to the internal magnetic field generally remains below 55 T in conventional iron compounds [18]. It is known [20] that hyperfine magnetic fields measured at the site of the iron nucleus have three main contributors: the Fermi contact field (Bc), the dipole field, (Bd) and the orbital field (BL). Compared to the Fermi contact field, the contribution of the dipole field is usually small and often has an opposite direction. As with many transition metals, the orbital moment is “frozen” in the 3d shell of the iron in the crystal lattice due to its interaction with the crystal fields of conventional geometries—tetragonal, octahedral, etc. As a result, such “frozen” orbitals do not normally contribute to the hyperfine field. The occasionally occurring small orbital contributions in iron compounds may be related to the spin-orbit coupling in systems with less traditional geometries such as tetrahedral, trigonal, or linear. The extremely high magnetic field value detected by the Mössbauer measurements may be explained by the effect of non-frozen 3d orbital contributions in structures with unusual geometry [26]. Based on the alternating current (AC) magnetic susceptibility measurements, the compound Fe(L1)2 can be viewed as a single-molecule magnet with a U = | D | S2 = 181 cm−1 energy barrier [30]. In clusters of SMM molecules containing multiple iron atoms, the exchange interaction within the cluster is essential [6]. it is not clear whether or not an exchange interaction-based magnetism could occur in an Fe(L1)2 single-molecule magnet with only a single iron atom within a complex. To study this possibility, Mössbauer measurements in an externally applied magnetic field were taken to distinguish between different magnetic couplings.
Figure 5 shows the 57Fe Mössbauer spectra of Fe(L1)2 measured at 5 K, without an applied magnetic field and under an external magnetic field of 5T. Both spectra consist two sextets, which provide evidence for the presence of two phases in that particular sample. Sextet S1, which has an unusually high magnetic field, is the fingerprint of the iron atom linearly coordinated by the two carbon atoms in Fe(L1)2. The inner sextet, S2, belongs to the ferrihydrite (Fe2O3∙0.5H2O) present as a second phase in the sample, which was an impurity that most likely originated as a result of minor hydrolysis of the main compound by moisture. The internal magnetic field manifested by the S1 sextet was found to have increased by the value of the applied external magnetic field with which it interacted. At the same time, the spectrum of the ferrihydrite remained unchanged [35] (Figure 5). Thus, the low-temperature exchange interaction in Fe(L1)2 is confirmed to be of a ferromagnetic type, in agreement with the hysteresis clearly observed in magnetization measurements of the same system [33].
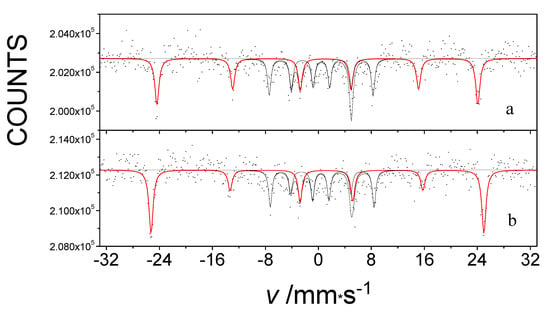
Figure 5.
The 57Fe Mössbauer spectra of an impure Fe(L1)2 system measured at 5 K without an external magnetic field (a) and in a 5 T Bext external magnetic field (b) applied parallel to the gamma rays.
Further exploration of this topic of unusually high hyperfine internal magnetic fields at low temperatures led to the preparation of other related compounds in which the C-atom in methanide L1 was replaced by a nitrogen atom in a series of similarly sterically hindered amides L2–L5 (Chart 1). New valuable information was obtained from a series of similarly unstable two-coordinate FeII complexes. The structure of one of them, FeII[N(SiPh2Me)2]2 (Fe(L4)2), is shown in Figure 6. It is interesting to note that in the complexes Fe(L2)2 and Fe(L5)2, both of which have extremely bulky protective metal center ligands (Chart 1), the N-Fe-N angle warrants their linear description, while linearity is lost for the other two amides, Fe(L3)2 and Fe(L4)2 (Table 1).
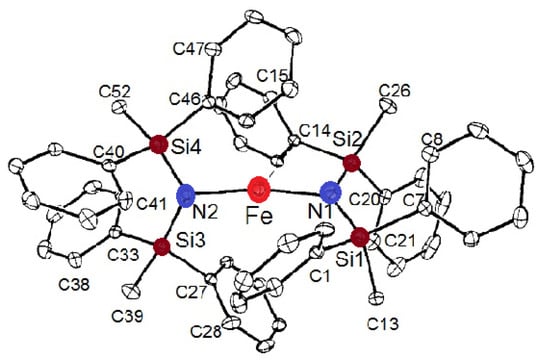
Figure 6.
The molecular structure of FeII[N(SiPh2Me)2]2 (Fe(L4)2).

Table 1.
Hyperfine field values and N-Fe-N bond angles in quasi-linear Fe(II) amido-compounds.
In the absence of an applied magnetic field, the 57Fe Mössbauer spectrum of Fe(L4)2 appears as a broadened single line (Figure 7, top). However, when an external magnetic field was applied, an unusually high internal magnetic field [34] was detected in the Mössbauer spectra, similar to the case of the compound Fe(L1)2. The Mössbauer spectra exhibited the classic magnetic 6-line splitting depicted in Figure 7. This observation also provides evidence for the considerable magnitude of the orbital momentum on the metal center in the Fe(L4)2 molecule. The hyperfine magnetic field value was greater than 90 T, much higher than what could be expected from the contribution of the Bc Fermi contact field and the Bd dipole field to the hyperfine field for the high-spin state of FeII in this complex. Consequently, the occurrence of the unusually high non-frozen orbital moment of the corresponding BL orbital contribution caused the observed high hyperfine field.
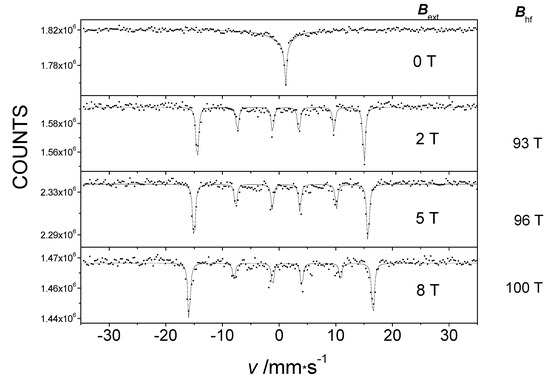
Figure 7.
The 5 K Mössbauer spectra of pure Fe(L4)2 without an external magnetic field and in an external magnetic field Bext applied parallel to the direction of the gamma rays, showing the Bhf hyperfine field.
Figure 8 shows a correlation between the induced hyperfine magnetic field and the N-Fe-N bond angle in Fe(L2)2……Fe(L5)2 molecules [28,30,34]. The value of the hyperfine field increases with an increasing N-Fe-N angle [34]. This can be explained by the fact that the value of the non-frozen orbital moment decreases with the bending of the N-Fe-N structural motif, since the iron atom interacts more with other atoms than it does with nitrogen. Therefore, Fe(L4)2 is another example of a low-coordination iron complex where quasi-free-ion magnetism occurs through a non-frozen orbital moment.
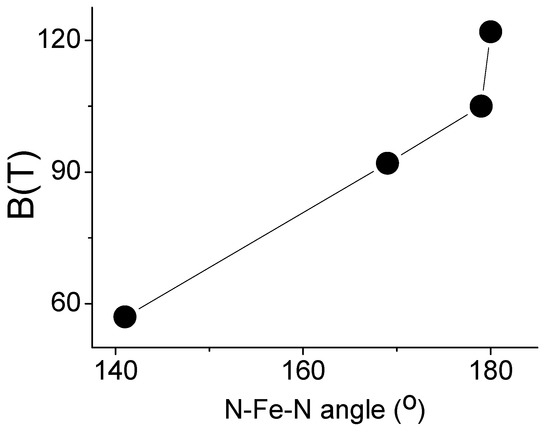
Figure 8.
Graph depicting the relationship between the N-Fe-N angle and the magnitude of the internal magnetic field in a series of FeL2 amide complexes (Table 1).
The effect of the applied magnetic field on the Mössbauer spectra of Fe(L4)2 indicates a ferromagnetic exchange interaction [34] similar to that shown above for Fe(L1)2 [33]. The magnetization curves support the notion that magnetism is based on the exchange interaction and correlate well with the presented Mössbauer spectroscopy data [34].
In conclusion, we observed an unusually high internal hyperfine magnetic field on the central atom centers in a series of the linear and quasi-linear crystalline two-coordinated iron (II) methanido and amido complexes. This field can be observed either by simple cooling of the compound to low temperatures (as in Fe(L1)2) or by applying an external magnetic field with an order of magnitude weaker than that of the generated internal hyperfine field (as in Fe(L2-L5)2 molecules). Several factors were found to affect the observed magnetic coupling: (1) ligand field symmetry/geometry, (2) ligand field strength, (3) composition of the compound (type of ligand used), (4) overall molecular geometry with respect to the linearity of the two-coordinate moiety, and (5) spin–orbit coupling and orbital contribution stemming from the iron center.
Further investigations of similar systems are necessary to better understand the effect of these factors on the very large Bhf observed in such compounds.
3. Temperature-Dependent Spin Transitions and Antiferromagnetic Interaction in Iron Phthalocyanines Loaded with Oxygen in the Solid State
As a result of our collaboration with scientists from Drexel University (Philadelphia, PA, USA), we found a new method for the encapsulation of ferromagnetic iron carbides into carbon nanotubes by pyrolysis of the iron(II) phthalocyanines, FePc, in both the α-and β-forms [36]. This complex has a layered structure, with an FeII center in a square-planar environment with very short Fe-N(ring) bonds of 1.926 Å (Figure 9). The FePc units form a slipped π-stacked arrangement in the crystal where Fe is in very long Fe-N distance (3.24 A) from the N1 atom of an adjacent molecule [37].
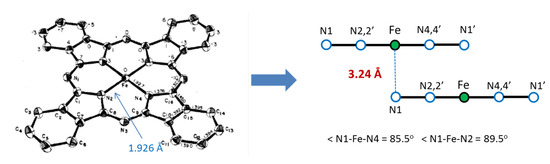
Figure 9.
Molecular structure of β-FePc (left panel) and its crystal packing, showing the layered structure and some details of its geometry (from ref. [37]).
As new materials, iron carbides obtained this way have received considerable attention in recent years for potential technological applications in the development of magnetic data storage materials, as toners in xerography, and as a component of ferrofluids [38]. We carried out investigations to better understand the formation mechanism of iron carbides encapsulated in carbon nanoparticles, with the most intriguing results presented below.
In several experiments, solid iron(II) phthalocyanine was reacted with oxygen at room temperature and slightly above room temperature under dry and wet conditions. The oxidation of the metal centers suggested the incorporation of oxygen into the crystal lattice of the initial compound, which was examined by X-ray photoelectron spectroscopy (XPS), X-ray diffraction. and Mössbauer spectroscopy.
As mentioned above, the initial compound in β-form has a metal center FeII with intermediate spin (S = 1, from [37]) in what is basically a square-planar environment with very long axial contacts with Pc molecules from adjacent layers. This creates a significant electric field gradient, EFG, leading to a large amount of quadrupole splitting Δ in the spectrum. In the first round of experiments, the room temperature Mössbauer spectra of the initial, pure, and oxygenated β-iron(II) phthalocyanines (displayed in Figure 10a) clearly show that the progressive oxidation of the starting compound over time led to the appearance of new chemical environments (Figure 10b–d). The new species are associated with different oxygenation products with much smaller quadrupole splitting values as compared to the quadrupole splitting, Δ = 2.54 ± 0.02 mm/s, of the initial pure β-iron(II) phthalocyanine (Figure 10a). The number and occurrence of the components depend on the oxygen uptake and increase with the amount of oxygen incorporated. After treatment with O2, the new spectral components had a significantly smaller quadrupole splitting Δ (Figure 10). Based on these observations, we tentatively ruled out the chemical oxidation process and suggest that oxygen can be incorporated between layers of the solid β-iron(II) phthalocyanine instead. Interestingly, this process does not occur in solution, which clearly indicates the role of a layered solid matrix. Regarding the location of oxygen, four different microenvironments were detected in the oxygenated β-form of iron (II) phthalocyanine [39] in our experiments. Among the components formed by oxygen uptake, the μ-oxo- and μ-peroxo-bridged species were identified.
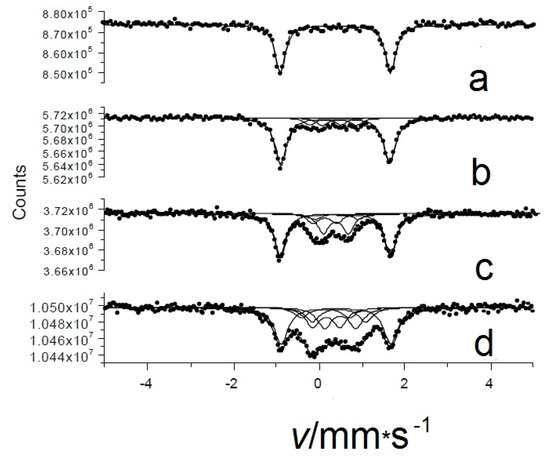
Figure 10.
Room temperature Mössbauer spectra of pristine oxygen-free (a) and oxygen-loaded β-iron(II) phthalocyanines: oxygenated at room temperature in an O2/wet atmosphere for 14 days (b) at 50 °C in an O2/wet atmosphere for 14 days (c) and at 50 °C in an O2/dry atmosphere for 92 days (d).
In our next study of the oxygenated β-iron(II) phthalocyanine, we observed the formation of an oxidized FeIII species, and we also found the temperature-dependent spin transition (SCO) for one of the FeIII components [40,41]. This process is illustrated in a series of overlaid spectra shown in Figure 11. When the temperature was raised from 77 to 292 K, the relative area of the low-spin component c decreased, and the area of the high-spin component d increased.
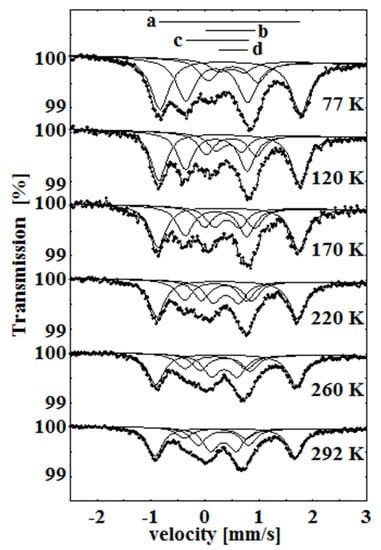
Figure 11.
Temperature dependence of the Mössbauer spectra of β-iron phthalocyanines treated with oxygen in wet atmosphere at 50 °C for over 6 weeks. As the temperature increases, a transition is observed between the low-spin component c and the high-spin component d.
Continuing our research, we were able to see only the temperature-dependent spin transition occurring among oxygenated/oxidized species incorporated between the layers of the initial compound [42]. This is seen in the Mössbauer spectra displayed in Figure 12, showing the gradual increase in component b at the expense of component c with increasing temperature. The Mössbauer measurements indicated that the treatment of β-iron phthalocyanine in oxygen resulted in the formation of new species, corresponding to FeIIIPc oxygen adducts that are in a dynamic-temperature-dependent spin equilibrium (SCO). Thus, variable temperature Mössbauer spectra showed that as the temperature increased, the low-spin species transformed into the high-spin species (Figure 14) [42].
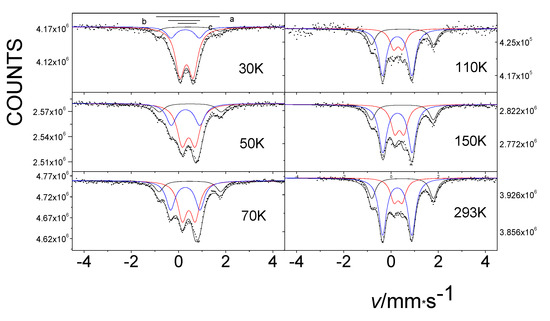
Figure 12.
Temperature dependence of the Mössbauer spectra of β-iron phthalocyanines loaded with oxygen in an O2/H2O stream at 50 °C for 75 days. As the temperature increases, a transition is observed between the low-spin component b and the high-spin component c.
In light of our experience with β-iron phthalocyanines, we attempted to introduce oxygen into the α-iron-phthalocyanine polymorph under similar conditions. There is no chemical difference in bonding in either form, but there is a difference in crystal packing, which is reflected in a different angle of orientation of slipped π-stacks. In our earlier studies, it was impossible to insert any significant amount of oxygen between the layers of this solid compound. However, in our later work, we were able to achieve this. A comparison of the Mössbauer spectra of pure oxygen-free and oxygen-treated α-iron phthalocyanines at 80 K is shown in Figure 13. The D1 doublet, characteristic of the oxygen-free original sample (Figure 13a), represents the spectroscopic signature of iron(II) centers, and treatment with oxygen led to their dramatic decrease (Figure 13b,c). The corresponding FeII component was not detectable at room temperature after treatment. For all species in this series, the value of the isomer shift δ is remarkably similar, indicating that the oxidation state of metal did not change. We have for the first time succeeded in effectively oxygenating α-iron(II) phthalocyanine by an appropriate treatment with oxygen, such that all iron atoms have oxygen neighbors (Figure 13c) [43].
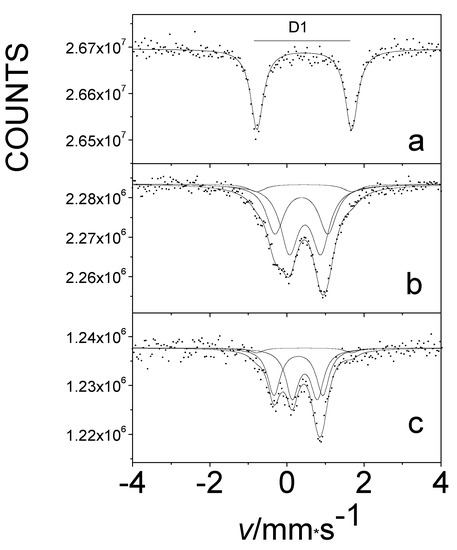
Figure 13.
The Mössbauer spectra at 80 K of pristine oxygen-free (a) and oxygen-loaded α-iron phthalocyanines, treated at 50 °C in O2 for 75 days (b) and at 50 °C in O2/H2O for 75 days (c).
Furthermore, we found that one of the components in the Mössbauer spectrum of the oxygen-treated α-iron(II)-phthalocyanine showed a typical six-line magnetic splitting pattern, as shown in Figure 14 [42,43]. To investigate the origin of the spectroscopic sextet that arose simply by cooling of this sample, the effect of an applied external magnetic field on the sample’s internal magnetic field was measured. These observations are well demonstrated by the Mössbauer effect measurements conducted at 5 K (Figure 15).
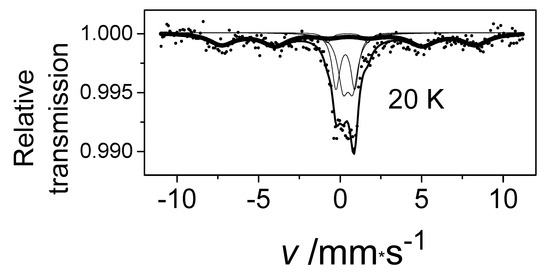
Figure 14.
The Mössbauer spectrum of α-iron phthalocyanine loaded with oxygen in O2/H2O stream at 50 °C for 75 days measured at 20 K.
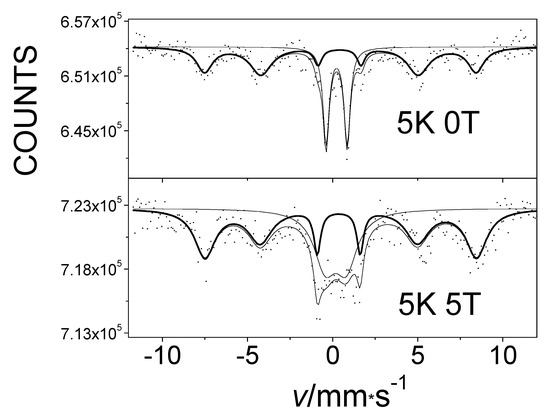
Figure 15.
The Mössbauer spectra at 5 K of α-iron-phthalocyanine treated with oxygen in an O2/H2O stream at 50 °C for 75 days, without an external magnetic field (top) and at 5 T in an external magnetic field applied in parallel with gamma radiation direction (bottom).
An analysis of spectra showed that the magnitude of the internal magnetic field remained unchanged when an external magnetic field was applied. This confirms the presence of an antiferromagnetic interaction in the studied system [43]. The antiferromagnetism of this sample was also verified by measurements of its magnetic susceptibility [43].
In summary, the antiferromagnetic coupling of magnetism observed at low temperatures in the highly oxygen-loaded α-iron phthalocyanines was explained by a super-exchange interaction between the long μ-peroxo – Fe –O–O– Fe– zigzag chains connecting layers of complexes (Figure 16) [43]. We realize that this model opposes data on the existing and well-known μ-oxo Fe–O–Fe dimers and polymers observed in the chemistry of porphyrins [44], phthalocyanines, and texaphyrins [45]. Additional studies involving oxygen isotope labeling and Raman spectroscopy are necessary to elucidate the structures displayed in Figure 16 in more detail.
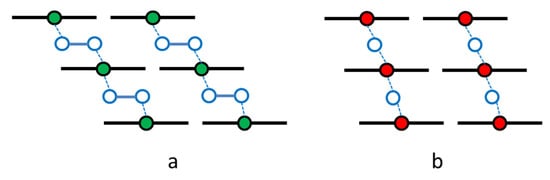
Figure 16.
Models of the oxygen binding that bridges iron centers, m-peroxo FeIIPc (a) and m-oxo FeIIIPc (b), in oxygen-loaded solid iron phthalocyanines.
4. Conclusions
This review presents the results of some of our studies of totally different iron-containing compounds and systems using 57Fe Mössbauer spectroscopy, which is shown to be a powerful tool for structural and magnetic investigations.
Author Contributions
Conceptualization and investigation, E.K.; methodology and investigation, Z.H.; formal analysis, Z.K.; investigation, R.S. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for the research carried out and presented in this mini-review was received from the NKFIH (OTKA project No K43687, K68135, K111692, K129210, K115913, K115784) and (No TÉT_16-1-2016-0002) for which the authors are very grateful.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or cited references.
Acknowledgments
The authors are grateful to Libor Machala and Jiri Pechousek (Palacky University, Olomouc) for the low-temperature external magnetic field recording of Mössbauer spectra and for the magnetochemical measurements, to Nikolay Gerasimchuk for the helpful comments and discussion, as well as to Amar Nath (University of North Carolina, Asheville) for the preparation of the oxygenated iron phthalocyanine samples and for participation in the discussion of the results.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kahn, O. Molecular Magnetism; VCH Publishers: New York, NY, USA, 1993. [Google Scholar]
- Itoh, K.; Kinoshita, M. Molecular Magnetism; Kodansha and Gordon & Breach: Tokyo, Japan; Amsterdam, The Netherlands, 2001. [Google Scholar]
- Sessoli, R.; Gatteschi, D.; Caneschi, A.; Novak, M.A. Magnetic bistability in a metal-ion cluster. Nature 1993, 365, 141–143. [Google Scholar] [CrossRef]
- Coronado, E. Molecular magnetism: From chemical design to spin control in molecules, materials and devices. Nat. Rev. Mater. 2019. [Google Scholar] [CrossRef]
- Bogani, L.; Wersdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Sessoli, R.; Tsai, H.L.; Shake, A.R.; Wang, S.; Vincent, J.B.; Folting, K.; Gatteschi, D.; Christou, G.; Hendrickson, D.N. High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 1993, 115, 1804–1816. [Google Scholar] [CrossRef]
- del Barco, E.; Kent, A.D.; Hill, S.; North, J.M.; Dalal, N.S.; Rumberger, E.M.; Hendrickson, D.N.; Chakov, N.; Christou, G. Magnetic Quantum Tunneling in the Single-Molecule Magnet Mn12-Acetate. J. Low Temp Phys. 2005, 140, 119. [Google Scholar] [CrossRef]
- Luenberger, M.N.; Loss, D. Quantum computing in molecular magnets. Nature 2001, 401, 789. [Google Scholar] [CrossRef]
- Cianchi, L.; Del Giallo, F.; Lantieri, M.; Moretti, P.; Spina, G.; Caneschi, A. Spin dynamics of the magnetic dimer [Fe(OMe)(dpm)2]2 studied using Mössbauer spectroscopy. Phys. Rev. 2004, B69, 014418. [Google Scholar] [CrossRef]
- Cianchi, L.; Del Giallo, F.; Lantieri, M.; Moretti, P.; Spina, G. Study of the [Fe(OMe)(dpm)2]2 dimer in the presence of a magnetic field by using Mössbauer spectroscopy. Hyperfine Interact. 2006, 168, 1115. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron(II) complex. Nat. Chem. 2011, 5, 2013. [Google Scholar]
- Abbas, G.; Lan, Y.; Mereacre, V.; Wernsdorfer, W.; Clerac, R.; Buth, G.; Sougrati, M.T.; Grandjean, F.; Long, G.J.; Anson, C.E.; et al. Magnetic and 57Fe Mössbauer Study of the Single Molecule Magnet Behavior of a Dy3Fe7 Coordination Cluster. Inorg. Chem. 2009, 48, 9345–9355. [Google Scholar] [CrossRef]
- Molins, E.; Gich, M.; Tejada, J.; Grenèche J., M.; Macià, F. Zero-field quantum tunneling relaxation of the molecular spin in Fe8 observed by 57Fe Mössbauer spectrometry. Europhys. Lett. 2014, 108, 47004. [Google Scholar] [CrossRef]
- Kuzmann, E.; Homonnay, Z.; Nagy, S.; Nomura, K. Mössbauer Spectroscopy. In Handbook of Nuclear Chemistry; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R., Rösch, F., Eds.; Springer: New York, NY, USA; Heidelberg, Germany, 2011. [Google Scholar]
- Gütlich, P.; Eckhard, B.; Trautwein, A.X. Mössbauer Spectroscopy and Transition Metal Chemistry; Springer: New York, NY, USA, 2011. [Google Scholar]
- Vértes, A.; Burger, K.; Korecz, L. Mössbauer Spectroscopy; Akadémiai Kiadó-Elsevier: Budapest, Hungary; Amsterdam, The Netherlands, 1979. [Google Scholar]
- Greenwood, N.N.; Gibb, T.C. Mössbauer Spectroscopy; Chapman & Hall: London, UK, 1971. [Google Scholar]
- Stevens, J.G.; Stevens, V.E. Mössbauer Effect Data Index (MERDI); Adam Hilger: London, UK, 1966–1979. [Google Scholar]
- Gonser, U. Mössbauer Spectroscopy. Springer: New York, NY, USA, 1975. [Google Scholar]
- Gütlich, P. Spin crossover in iron(II)-complexes. In Metal Complexes. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1981. [Google Scholar]
- Dézsi, I.; Molnár, B.; Tarnóczi, T.; Tompa, K. On the magnetic behaviour of iron(II)-bis-(1,10 phenantroline)-thiocyanate between −190° and 30° C. J. Inorg. Nucl. Chem. 1967, 29, 2486. [Google Scholar] [CrossRef]
- Holynska, M. Single Molecule Magnets Single-Molecule Magnets: Molecular Architectures and Building Blocks for Spintronics; Wiley-VCH Verlag GmbH & Co.: Marburg, Germany, 2019. [Google Scholar]
- Reiff, W.M.; LaPointe, A.M.; Witten, E.H. Virtual Free Ion Magnetism and the Absence of Jahn−Teller Distortion in a Linear Two-Coordinate Complex of High-Spin Iron(II). J. Am. Chem. Soc. 2004, 126, 10206. [Google Scholar] [CrossRef]
- Kuzmann, E.; Szalay, R.; Vértes, A.; Homonnay, Z.; Pápai, I.; de Chatel, P.; Szepes, L. 157 T internal magnetic field in Fe[C(SiMe3)3]2 compound at 20 K. Hyperfine Interact. 2008, 185, 185–189. [Google Scholar] [CrossRef]
- Kuzmann, E.; Szalay, R.; Vértes, A.; Homonnay, Z.; Pápai, I.; de Chatel, P.; Klencsár, Z.; Szepes, L. Observation and interpretation of 157.5 T internal magnetic field in Fe[C(SiMe3)3]2 coordination compound. Struct. Chem. 2009, 20, 453–460. [Google Scholar] [CrossRef]
- Reiff, W.M.; Schulz, C.E.; Whangbo, M.-H.; Seo, J.I.; Lee, Y.S.; Potratz, G.R.; Spicer, C.W.; Girolami, G.S. Consequences of a Linear Two-Coordinate Geometry for the Orbital Magnetism and Jahn−Teller Distortion Behavior of the High Spin Iron(II) Complex Fe[N(t-Bu)2]2. J. Am. Chem. Soc. 2009, 131, 404. [Google Scholar] [CrossRef]
- Merrill, W.A.; Stich, T.A.; Brynda, M.; Yeagle, G.J.; Fettinger, J.C.; De Hont, R.; Reiff, W.M.; Schulz C., E.; Britt, R.D.; Power, P.P. Direct Spectroscopic Observation of Large Quenching of First-Order Orbital Angular Momentum with Bending in Monomeric, Two-Coordinate Fe(II) Primary Amido Complexes and the Profound Magnetic Effects of the Absence of Jahn− and Renner−Teller Distortions in Rigorously Linear Coordination. J. Am. Chem. Soc. 2009, 131, 12693–12702. [Google Scholar]
- Atanasov, M.; Zadrozny J., M.; Long, J.R.; Neese, F. A theoretical analysis of chemical bonding, vibronic coupling, and magnetic anisotropy in linear iron(II) complexes with single-molecule magnet behavior. Chem. Sci. 2013, 4, 139. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Atanasov, M.; Bryan, A.M.; Lin, C.-Y.; Rekken, B.D.; Power, P.P.; Neese, F.; Long, J.R. Slow magnetization dynamics in a series of two-coordinate iron(II) complexes. Chem. Sci. 2013, 4, 125. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron(I) complex. Nat. Chem. 2013, 5, 577–581. [Google Scholar] [CrossRef]
- La Pointe, A.M. Fe[C(SiMe3)3]2: Synthesis and reactivity of a monomeric homoleptic iron(II) alkyl complex. Inorg. Chim. Acta 2003, 345, 359–362. [Google Scholar] [CrossRef]
- Kuzmann, E.; Zoppellaro, G.; Pechousek, J.; Cuda, J.; Klencsár, Z.; Homonnay, Z.; Tucek, J.; Szalay, R.; Pápai, M.; Machala, L.; et al. Ferromagnetic Coupling in an Fe[C(SiMe3)3]2/Ferrihydrite Hetero-Mixture Molecular Magnet. Eur. J. Inorg. Chem. 2014, 2014, 3178–3183. [Google Scholar] [CrossRef]
- Kuzmann, E.; Zoppellaro, G.; Pechousek, J.; Klencsár, Z.; Machala, L.; Tucek, J.; Homonnay, Z.; Cuda, J.; Szalay, R.; Pápai, M. Magnetic coupling and relaxation in Fe[N(SiPh2Me)2]2 molecular magnet. Struct. Chem. 2016, 28, 975–983. [Google Scholar] [CrossRef]
- Guyodo, Y.; Banerjee, S.K.; Penn, R.L.; Burleson, D.; Berquo, T.S.; Seda, T.; Solheid, P. Magnetic properties of synthetic six-line ferrihydrite nanoparticles. Phys. Earth Plan. Interiors 2006, 154, 222–233. [Google Scholar] [CrossRef]
- Kopelev, N.; Chechersky, V.; Nath, A.; Wang, Y.L.; Kuzmann, E.; Zhang, B.; Via, G.H. Encapsulation of Iron Carbide in Carbon Nanocapsules. Chem. Mater. 1995, 95, 19. [Google Scholar] [CrossRef]
- Kirner, J.F.; Dow, W.; Scheidt, R.W. Molecular Stereochemistry of Two Intermediate-Spin Complexes Iron(II) Phthalocyanine and Manganese(II) Phthalocyanine. Inorg. Chem. 1976, 15, 1685–1690. [Google Scholar] [CrossRef]
- Annese, E.; DiSanto, G.; Choueikani, F.; Otero, E.; Ohresser, P. Iron Phthalocyanine and Ferromagnetic Thin Films: Magnetic Behavior of Single and Double Interfaces. ACS Omega 2019, 4, 5076–5082. [Google Scholar] [CrossRef]
- Kuzmann, E.; Homonnay, Z.; Vértes, A.; Li, S.; Yin, H.; Kubuki, S.; Wei, Y.; Nath, A.; Chen, X.; Li, J. Mössbauer studies of the interaction of oxygen with solid β-Fe(II)-phthalocyanine. J. Solid State Chem. 2003, 170, 118–123. [Google Scholar] [CrossRef]
- Kuzmann, E.; Nath, A.; Chechersky, V.; Li, S.; Wei, Y.; Chen, X.; Li, J.; Homonnay, Z.; Gál, M.; Garg, V.K.; et al. Mössbauer Study of Oxygenated Iron-Phthalocyanines, a Precursor of Magnetic Storage Material. Hyperfine Interact. 2002, 139, 631–639. [Google Scholar] [CrossRef]
- Yin, H.; Kubuki, S.; Homonnay, Z.; Kuzmann, E.; Vértes, A.; Wei, Y.; Nath, A. A Mössbauer Study of the Low Spin-High Spin Transition of an Oxygen Adduct Formed in Solid β-Fe(II)Phthalocyanine. Open Inorg. Chem. J. 2008, 2, 69–72. [Google Scholar] [CrossRef]
- Kuzmann, E.; Homonnay, Z.; Mylonakis, A.; Yin, H.; Wei, Y.; Kovács, K.; Kubuki, S.; Klencsár, Z.; Vértes, A.; Nath, A. Mössbauer study of oxygen adducts in solid Fe(II) phthalocyanines. J. Phys. Conf. Ser. 2010, 217, 12029. [Google Scholar] [CrossRef]
- Kuzmann, E.; Pechousek, J.; Cuda, J.; Yin, H.; Wei, Y.; Homonnay, Z.; Klencsár, Z.; Horváth, A.; Machala, L.; Kubuki, S.; et al. Magnetic interaction in oxygenated alpha iron phthalocyanines. AIP Conf. Proc. 2014, 1622, 97–103. [Google Scholar]
- Scheidt, W.R.; Kadish, K.M. The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 3, pp. 49–112. [Google Scholar]
- Hannah, S.; Lynch, V.; Guldi, D.M.; Gerasimchuk, N.N.; MacDonald, C.B.; Magda, D.; Sessler, J.L. Late First-Row Transition-Metal Complexes of Texaphyrin. J. Am. Chem. Soc. 2002, 124, 8416–8427. [Google Scholar] [CrossRef] [PubMed]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).